

Abstract
We present a molecular dynamics study of cytochrome c oxidase from Paracoccus denitrificans in the fully oxidized state, embedded in a fully hydrated dimyristoylphosphatidylcholine lipid bilayer membrane. Parallel simulations with different levels of protein hydration, 1.125 ns each in length, were carried out under conditions of constant temperature and pressure using three-dimensional periodic boundary conditions and full electrostatics to investigate the distribution and dynamics of water molecules and their corresponding hydrogen-bonded networks inside cytochrome c oxidase. The majority of the water molecules had residence times shorter than 100 ps, but a few water molecules are fixed inside the protein for up to 1.125 ns. The hydrogen-bonded network in cytochrome c oxidase is not uniformly distributed, and the degree of water arrangement is variable. The average number of solvent sites in the proton-conducting K- and D-pathways was determined. In contrast to single water files in narrow geometries we observe significant diffusion of individual water molecules along these pathways. The highly fluctuating hydrogen-bonded networks, combined with the significant diffusion of individual water molecules, provide a basis for the transfer of protons in cytochrome c oxidase, therefore leading to a better understanding of the mechanism of proton pumping.
INTRODUCTION
Cytochrome c oxidase (COX), located in the inner membrane of mitochondria and many bacteria, is one of the most intensively studied membrane proteins. It catalyzes the terminal step in cellular respiration, a transfer of four electrons from cytochrome c to dioxygen (Babcock and Wikström, 1992). The reduction of dioxygen to water is accompanied by the translocation of four protons across the inner mitochondrial membrane or the bacterial cytoplasmic membrane (Wikström, 1977). The resulting electrochemical proton gradient can be used by ATP synthase to generate ATP. Atomic structures of cytochrome c oxidase from Paracoccus denitrificans (Iwata et al., 1995; Ostermeier et al., 1997), bovine heart mitochondria (Tshukihara et al., 1996), and Rhodobacter sphaeroides (Svensson-Ek et al., 2002) have been determined by x-ray crystallography. Subunit II of COX contains a bimetallic CuA center and subunit I contains two redox-active cofactors: a low spin heme a and a binuclear metal center formed by the heme a3 and CuB (Iwata et al., 1995; reviewed by Ferguson-Miller and Babcock, 1996; Michel, 1998). Electrons from cytochrome c are transferred to the CuA center and then further via heme a to the binuclear center (see Babcock and Wikström, 1992; Michel, 1998; Mills and Ferguson-Miller, 2003; Brzezinski and Larsson, 2003 for reviews). Within the binuclear center, molecular oxygen is bound to the heme a3 iron, then reduced, and two molecules of water are formed.
The results of site-directed mutagenesis experiments (Thomas et al., 1993; Fetter et al., 1995; Garcia-Horsman et al., 1995; Brzezinski and Adelroth, 1998; Mills and Ferguson-Miller, 1998; Aagaard et al., 2000; Zaslavsky and Gennis, 2000; Pfitzner et al., 2000) and the analysis of the structural data (Iwata et al., 1995) indicate two different proton transfer pathways. The K-pathway (for chemical protons) of proton transfer leads to the active site through the essential lysine residue Lys-354, and the D-pathway (for pumped protons) named after Asp-124 leads from the Asp-124 via a solvent-filled cavity to Glu-278. Beyond Glu-278 the pathway of protons is unclear. It may lead directly to the binuclear site via a temporarily established chain of water molecules or to the heme a3 propionate (Iwata et al., 1995), either by direct contacts upon conformational changes of Glu-278, or via unresolved intervening water molecules (reviewed by Michel, 1998). It has been suggested that these two proton pathways may be functionally associated with different parts of the catalytic cycle (Konstantinov et al., 1997). Despite recent advances in our structural knowledge (Ostermeier et al., 1997; Tshukihara et al., 1996; Svensson-Ek et al., 2002), the mechanism of coupling the redox processes to proton pumping in COX is largely unknown.
Proton transfer via a hydrogen-bonded network in a membrane protein was proposed several years ago (Nagle and Morowitz, 1978). An experimentally well-characterized example of such a network in a biomolecule is bacteriorhodopsin, where water molecules visible in the crystals seem to be involved in a hydrogen-bonding network (Le Coutre et al., 1995; reviewed by Luecke, 2000).
The literature dealing with hydration of both bacterial and mitochondrial cytochrome c oxidases focuses on the prediction of bound water molecules within the proton-conducting D- and K-pathways (Riistama et al., 1997; Hofacker and Schulten, 1998; Pomès et al., 1998; Zheng et al., 2003; Wikström et al., 2003). Riistama et al. (1997) used a statistical-mechanical “potential of mean force” methodology (Garcia et al., 1996) to place bound water molecules within proton pathways based on the x-ray coordinates of the mitochondrial cytochrome aa3 from bovine heart. Hofacker and Schulten (1998) and Zheng et al. (2003) determined likely water sites using the program DOWSER (Zhang and Hermans, 1996) and refined the water positions using energy minimization and molecular dynamics simulations. The large number of water molecules suggests an important contribution to the proton pathways studied—the pathways for transport of oxygen and protons—by predicting the distribution of water molecules in the protein and by molecular dynamics (MD) simulation of oxygen diffusion for COX from P. denitrificans.
To place water molecules in the two-subunit enzyme of cytochrome c oxidase we have used the program GRID (Goodford, 1985). This method has already been successful in locating water positions in a number of crystal structures, for example, in cytochrome P450cam (Wade, 1990; Helms and Wade, 1995), and in acetylcholinesterase (Henchman and McCammon, 2002). The program implements a computationally fast method of determining energetically favorable ligand binding sites on molecules of known structure. The probe molecule is moved through the protein matrix on a grid, and at each point, energies are calculated as a sum of Lennard-Jones, electrostatic, and hydrogen-bonding terms with explicit modeling of hydrogen-bond geometries. Since certain dynamic features of cytochrome c oxidase cannot be captured by crystallographic techniques, MD has been used to elucidate conformational fluctuations and COX-water mobility (Hofacker and Schulten, 1998; Backgren et al., 2000; Zheng et al., 2003; Wikström et al., 2003). There are some examples of studies where different redox states of protein systems have been analyzed using MD procedures (Hayashi et al., 2002; Bret et al., 2002; Wikström et al., 2003). However, fully atomic simulations of membrane proteins must include a lipid bilayer to model the natural environment. Given the irregular shape of membrane proteins, obtaining a correctly configured initial system is a nontrivial task, and yet the reliability of the subsequent simulation may depend on how carefully this is performed. To build these protein-lipid bilayer systems, two approaches have been reported in the literature. The first (Petrache et al, 2000; Woolf and Roux, 1994, 1996; Berneche et al., 1998; Bernache and Roux, 2000), which was used in this study, consists of building a bilayer around the protein lipid by lipid, each individual molecule being selected from a library of lipid conformers. The second approach (Shen at al., 1997; Tieleman and Berendsen, 1998) uses a previously equilibrated lipid bilayer, in which a cylindrical hole to accommodate the protein is created by the application of weak repulsive radial forces of the lipid atoms. In both cases the protein-lipid system is energy-minimized before the MD simulation.
The purpose of this work was to identify and characterize hydrogen-bonded networks inside the protein as possible pathways for proton transport in cytochrome c oxidase by predicting water binding sites and then characterizing the hydrogen-bonded networks during molecular dynamics simulations.
SIMULATION DETAILS
Coordinates
The starting configuration of the fully oxidized two-subunit COX from P. denitrificans at 2.7 Å resolution was the Protein Data Bank (PDB) entry 1AR1, designated post-1AR1 after further refinement (A. Harrenga and H. Michel, unpublished; the coordinate set is available upon request). This structure was obtained under oxidizing conditions. This set of coordinates contains a Mg2+ ion, located at the interface between subunits I and II, that is coordinated by His-403, Asp-404, Glu(II)218, a Ca2+ ion bound to His-59, Gly-61, Gln-63, and Glu-56, and 88 water molecules. A hydroxide ion OH− was modeled at a distance of 2.05 Å from the CuB, in agreement with previous theoretical calculations (Kannt et al., 1998). Hydrogens were added to the structure by using CHARMM's hbuild function (Brünger and Karplus, 1988).
Charges on the protein atoms and ionizable groups in different protonation states were taken from the CHARMM22 force field (MacKerell et al., 1995, 1998). Lys-354 was assigned as neutral because of its hydrophobic environment. Partial atomic charges for neutral Lys-354 as well as for protonated Asp-399, and for protonated Glu-278, were taken from the AMBER96 force field. Because all titrating protons have been positioned in the structure, we can describe the deprotonation/protonation of the particular residue as the addition of an explicit hydrogen atom.
Electrostatic calculations (in these calculations water molecules were not included explicitly) showed that Lys-354 is in its neutral state over a wide pH range (pH 4–11.5) (Kannt et al., 1998). However, this has been questioned recently because water molecules hydrogen-bonded to Lys-362 and Ser-299 were found in the Rh. sphaeroides COX structure (Svensson-Ek et al., 2002). This observation has led to the postulation that Lys-362 is protonated at pH = 7 (Rh. sphaeroides numbering) (Svensson-Ek et al., 2002). One heme a3 propionate shares a proton with Asp-399; all other propionates are deprotonated at pH = 7 (Kannt et al., 1998).
The atomic partial charges for the redox centers—the heme a and heme a3 sites, the His-276-to-Tyr-280 crosslink and for CuB and its three histidine ligands (His-276, His-325, and His-326) for the fully oxidized enzyme—were obtained from quantum chemical calculations (see Fig. 1, A–E). Atom-centered point charges for the heme a and heme a3 and the copper center were derived from restrained electrostatic potential fits on model systems using the ESP module of NWChem 4.1 employing the 6-31G* basis set (NWChem, A Computational Chemistry Package for Parallel Computers, Version 4.1., 2002). Computing partial atomic charges for unusual cofactors by the electrostatic potential (ESP) fit method is not the standard procedure in the CHARMM community and is therefore a potential source of error. However, electrostatic potential charges combined with CHARMM gave good agreement with experimental pKa values in electrostatic continuum calculations for bacterial photosynthetic reaction centers (Rabenstein et al., 1998). For protoporphyrin IX (heme) a 3-21G optimized structure as well as a high-resolution crystal structure geometry were used. However, the charges obtained for the hemes showed only little dependence on the molecular geometry employed. In particular we found a partial charge of 0.36 for Fe(III) only; the ligating nitrogens compensate most of the positive charge. The partial charge of NE2 decreases from −0.70 in a neutral histidine (HSD) to −0.14 in the case of a five-coordinated Fe(III) and to −0.42 for a six-coordinated Fe(III), whereas the partial charges of the nitrogens on the porphyrin ring change to −0.10 for Fe(III). The calcium ion was modeled with its formal −2 e charge. This is not believed very crucial because the ion is strongly coordinated by four residues (see above) (Ostermeier et al., 1997). Therefore, its partial atomic charge is shielded from the environment and it is bound in a stable geometry.
FIGURE 1.

(A) Partial atomic charges for the fully oxidized heme a/heme a3 derived from quantum chemical calculations. (B) Partial atomic charges for Tyr-280–His-276 crosslink (charges for His-276 are listed in C); (C) partial atomic charges for the histidine ligands to CuB. The charges on His-326 were restrained to be identical to those of His-325 and therefore not shown. (D) Partial atomic charges for the histidine ligand to Fe atom of heme a3. (E) Partial atomic charges for histidine ligands to Fe atom of heme a; the two His ligands carry the same charges.
Internal water modeling
The program GRID (Goodford, 1985) was run with a water probe over the two-subunit structure of cytochrome c oxidase to identify possible water binding sites. Due to the large size of ∼13,000 atoms, the system was split into eight parts, and the GRID program was run for each part independently. The largest region with a favorable binding energy in the GRID energy map was in an internal cavity close to Glu-278 where a structural water molecule, WS, was detected. Although this water molecule is clearly defined in the electron density map, GRID was used to position it in the cavity without using the information from the experimental coordinates. GRID calculations over the whole internal cavity region gave an energy of −14 kcal/mol for this water probe, which indicates favorable hydration. After locating the energy minimum, its position was refined and an internal GRID water molecule, WG, was then assigned to it. GRID was then run again with input coordinates containing this water molecule. A second energy minimum was found and a second water molecule assigned to it. The criteria for water placement in a cavity were that the energy was <−8 kcal/mol for the first set of coordinates (W8 water molecules), and <−12 kcal/mol for the second set of coordinates (W12 water molecules), and that each water molecule generates at least two hydrogen bonds (Helms and Wade, 1995). The GRID procedure was repeated four times keeping all previously located water molecules until further waters could be added in suitably deep energy minima (<−8 kcal/mol or <−12 kcal/mol) in the last round, indicating that all cavities were fully solvated for the chosen energy criteria (Wade and Goodford, 1993). The GRID energy function is very short-ranged. Electrostatic interactions are computed using a cutoff of only 4.5 Å. Therefore, although potentially of significance, the order of the parts is irrelevant because water molecules in the boxes hardly interact with those in other boxes.
Initial setup of protein-membrane-water system
As described in the following, a proper membrane environment of the cytochrome c oxidase was provided by constructing a dimyristoylphosphatidylcholine (DMPC) lipid bilayer. We followed the general protocol for molecular dynamics (MD) simulations (Berneche et al., 1998; Berneche and Roux, 2000) to construct the initial configuration of a protein-membrane-water system. The microscopic system consists of cytochrome c oxidase (two subunits of 549 and 252 amino acids, respectively), 181 DMPC lipids (90 in the top and 91 in the bottom layer), 88 internal crystal waters WS, and 24,323 bulk water molecules (W0, W1, W2, W3, and W4). Additionally, 176 internal water molecules (W12 water molecules WG) or 755 internal water molecules (W8 water molecules WG) constructed using the GRID method were included in the calculations, and 42 Na+ and 30 Cl− ions were randomly inserted to simulate a 100 mM aqueous salt solution (ions were positioned >6 Å away from the bilayer and protein, and no ion pairs were allowed to form). After solvation the entire system consisted of 101,852 atoms (the W12 set of coordinates) and 103,589 atoms (the W8 set of coordinates).
The membrane normal is oriented along the z axis, and the center of the bilayer is at z = 0. The protein was oriented along the z axis, in a position that left the hydrophilic residues in contact with the W0 water, and the hydrophobic residues in contact with lipid acyl chains. The ratio of oxygen and carbon atoms of <0.2 along the membrane normal for z values from −17 Å to +17 Å, corresponds to the hydrophobic part of the protein surface (Lancaster and Michel, 1997). Periodic boundary conditions were applied in the x,y directions to simulate an infinite planar layer and in the z direction to simulate a bilayer system; the periodic system has the dimensions 90 × 90 × 125 Å3. Whereas the x,y dimensions are kept constant, the z dimension of the unitary cell was allowed to vary according to the constant pressure and temperature thermodynamic ensemble with a fixed surface area (Berneche et al., 1998, Berneche and Roux, 2000). To surround the protein by a complete lipid environment the dimension of the system in the x,y plane was set at 90 Å × 90 Å, corresponding to an area of 8100 Å2. Since the average cross-sectional area of a DMPC molecule is 64 Å2 (Gennis, 1989; Nagle, 1993), the difference in the cross-sectional area of COX between the periplasmic side (2319 Å2) and the cytoplasmic side (2247 Å2) corresponds to ∼1 lipid. The appropriate number of lipids can be determined to be 90 for the upper layer and 91 for the lower layer. The DMPC molecules were taken from a library of 2000 preequilibrated and prehydrated DMPCs (Pastor et al., 1991; Venable et al., 1993). The resulting configuration of the W12 is shown in Fig. 2.
FIGURE 2.

Simulation system setup (side view): two-subunits of cytochrome c oxidase embedded in a DMPC membrane solvated by a 100 mM NaCl aqueous salt solution (the Na+ ions are yellow and the Cl− ions are blue). The snapshot was taken at the beginning of the molecular dynamics production phase after 575 ps of equilibration for the W12 set.
Equilibration and dynamics
The minimization and dynamics simulations were performed using the academic version c28b2 of the biomolecular simulation package CHARMM (Brooks et al., 1983). The protein was initially fixed and the system was minimized by 1000 steps of steepest descent followed by 1000 steps of adopted-basis Newton-Raphson. A number of energy restraints were used during the minimization and at the beginning of the equilibration period to ensure a smooth relaxation of the system toward an equilibrated configuration (Berneche et al., 1998; Berneche and Roux, 2000). A harmonic potential of 10 kcal mol−1 Å−2 was applied to the backbone atoms to prevent large spurious motions, the center of mass of the lipid polar heads was kept at ∼z = ± 17 Å by planar harmonic constraints to maintain the planarity of the membrane, and the penetration of water into the bilayer region was prevented by the use of planar potentials in the z direction. The protein and water constraints were then decreased to 5 kcal mol−1 Å−2 and were gradually reduced to get a free system after 575 ps of equilibration. The only remaining constraints are those on lipid headgroups (see below) and harmonic distance restraints between CuB and its ligands His-276, His-325, His-326, OH− and CuB, OH−, and the Fe atom of heme a3, the Fe atom of heme a3 and His-411, the Fe atom of heme a and His-413, the Fe atom of heme a and His-94, CuA(I) and Cys(II)220, CuA(I) and His(II)181, CuA(II) and Cys(II)216, CuA(II) and Met(II)227, Mg and Glu(II)78, Mg and His-403, and Mg and Asp-404. The simulations were performed under three-dimensional periodic boundary conditions, with constant temperature of T = 330 K and pressure. The average temperature was 330 K, above the gel-liquid crystal phase transition temperature, and consistent with experimental conditions (Woolf and Roux, 1996). The system was equilibrated for 250 ps by molecular dynamics simulation. The system was coupled to a heat bath at 330 K by the use of Verlet dynamics for the first 575 ps; the time steps were 2 fs. The coordinates were saved every 5 ps and the nonbonded and image lists were updated every 20 steps. The list of nonbonded interactions was truncated at 12 Å, using a group-based cutoff. The nonbonded van der Waals interaction was switched off at 10–12 Å. The electrostatic interactions were computed without truncation, using the particle-mesh Ewald algorithm (Essmann et al., 1995) with an order of 4; FFT grid points for the charge mesh per Å were 90 × 90 × 125. In the particle-mesh Ewald method implemented in CHARMM the electrostatic energy is split into a direct and a reciprocal Ewald sum. A real space Gaussian-width κ of 0.3 Å−1 was used. All bonds involving hydrogen atoms were constrained by applying the SHAKE algorithm (Ryckaert et al., 1977).
The all-atom potential energy functions of PARAM-22 for protein (MacKerrell et al., 1995, 1998) and phospholipids (Schlenkrich et al., 1996) were used. The TIP3P potential was used for the water molecules (Jorgensen et al., 1983). During the production trajectory, the center of mass of the protein was restrained to the center of the x,y plane. The overall simulation time was ∼1125 ps.
All molecular structures were drawn using the Visual Molecular Dynamic Software VMD 1.8 (Humphrey et al., 1996).
It is important to consider possible methodological limitations of our simulation protocol. The main limitation is the harmonic restraint potential constantly applied to the headgroups of the DMPC lipids during the molecular dynamics production phase after equilibration to avoid a structural disorder of the lipid phase observed without restraints. Although the times of the present simulation were nearly 600 ps for equilibration and 1125 ps for the molecular dynamics production run, they seem still not long enough for a full equilibration of the lipid phase. The restraints will not, however, influence the energetics and dynamics of the internal water molecules in cytochrome c oxidase that was the main focus of this work.
ANALYSIS OF THE TRAJECTORIES FROM THE SIMULATIONS
Hydrogen bonds
The hydrogen-bond patterns were analyzed from the production trajectories with 0.15 ps time resolution. The criteria for a hydrogen bond (A…H – D) was that the distance between the acceptor and the hydrogen atom (A … H) should be <2.5 Å and an A … H – D angle of >120° (Eriksson et al., 1995; Eriksson and Nilsson, 1995; Tang and Nilsson, 1999). The percentage of occupancy of a hydrogen bond was defined as the number of frames with the hydrogen bond present divided by the total number of frames used for analysis. The lifetime of a hydrogen bond was calculated as the time elapsed from its first appearance until it was first broken. The average lifetime of a hydrogen bond during the simulation was then calculated as the average of all of its occurrences excluding those with a lifetime <1 ps.
Root mean-square deviations (RMSD) and atomic fluctuations (RMSF)
The coordinate sets from every 0.15 ps of the production run were superimposed on the initial structure of the system by minimizing the mass-weighted root mean-square deviations (RMSD) of the heavy atoms from the initial structure. The average RMSD values of the Cα atoms, side chains, and some amino acids were then calculated for the entire MD trajectory.
B-factors (Debye-Waller factor) from the x-ray structure of the two-subunit structure of cytochrome c oxidase were compared with the atomic fluctuations (RMSF) in the simulation using the relation
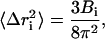 |
(1) |
where Δri is the atomic displacement for atom i and Bi is the corresponding B-factor.
Diffusion coefficient
The diffusion coefficients for water molecules (DH2O) in the simulated system were estimated using the Einstein relation (Eriksson et al., 1995a),
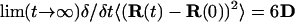 |
(2) |
where 〈 〉 denotes an average over the water molecules and R(t) the position of a water molecule at time t.
COMPUTATIONAL DETAILS
Internal water modeling was done on a one-processor Alpha machine using the GRID program (Goodford, 1985). Energy minimization, membrane modeling, and molecular dynamics simulations were performed in parallel with 32 processors, using version c28b2 of the biomolecular simulation program CHARMM (Brooks et al., 1983) on an IBM RS/6000 PS3 Regatta supercomputer at the Max Planck Society Rechenzentrum in Garching. Forty picoseconds of simulation took 10 h of CPU time on 32 Power4 processors.
RESULTS AND DISCUSSION
Predicted water binding regions
Because GRID energies are only effective energies, not true Gibbs free enthalpies, it is not quite clear which value to choose for deciding whether predicted hydration sites are occupied or not. Based on a comparison with free energy perturbation calculations, GRID energies <−6 kcal/mol were suggested as a criterion in a previous study on cytochrome P450cam (Helms and Wade, 1995). Here, we tested two levels of hydration, a looser criterion of accepting all GRID waters with energies of <−8 kcal/mol and a tighter criterion by requiring all waters to be more favorable than −12 kcal/mol. After four GRID cycles for the two-subunit structure of COX a total of 755 water molecules was found: 176 molecules with interaction energies <−12 kcal/mol, 267 molecules with interaction energies between −12 kcal/mol and −10 kcal/mol, and 314 molecules with interaction energies between −10 kcal/mol and −8 kcal/mol. We refer to the 176 water molecules with interaction energies <−12 kcal/mol as the W12 set of water molecules, and to the 755 water molecules with interaction energies <−8 kcal/mol as the W8 set. The GRID calculations showed that the 88 crystallographically observed water molecules are all located in energetically favorable positions.
The general distribution of internal water molecules detected by x-ray crystallography and found by GRID is in agreement. Fewer water molecules were found near the center of the protein, and more were identified in the regions close to the surface. The results from the GRID calculations were also compared with the positions of the observed water molecules in the crystal structure (Svensson-Ek et al., 2002) of cytochrome c oxidase from Rh. sphaeroides. The comparison showed that the water molecules resolved in the crystal structure are contained in the W12 set of water molecules.
Average structural properties
After constructing an initial water hydrogen network, MD simulations were carried out to study the distribution and dynamics of water molecules. The atomic system W12 at the beginning of the equilibration procedure is shown in Fig. 1. Analyzing the deviation of the structure from the initial crystal structure can assess the stability of the simulated protein. Fig. 3 presents the root mean-square deviation (RMSD) of the Cα atoms, side-chain atoms, and heme a/heme a3 atoms from the corresponding x-ray structure with respect to the simulation time. The coordinate sets after every 0.15 ps of the production run were superimposed onto the initial structure of the system. From the beginning of the dynamics run, the heme clusters seem to have reached a rather stable state characterized by an average RMSD value from the crystal structures of 1.6 Å. The RMSD values of Cα atoms and side-chain atoms are higher. After 575 ps the average deviation remains at ∼1.8 Å for the Cα atoms, and at 2.5 Å for the side-chain atoms. The first 575 ps were therefore considered as equilibration and not used for analysis.
FIGURE 3.

Root mean-square deviation (RMSD) relative to the x-ray structure as a function of time, calculated over all backbone atoms (black line), side-chain atoms (gray line), and heme a/a3 atoms (light gray). (A) RMSD from the W12 set of coordinates; (B) RMSD from the W8 set.
To study flexibility along the backbone, the root mean-square atom-positional fluctuations of the Cα atoms during the production trajectories were compared with fluctuations of the crystal structure, derived from the experimental B-factors (Fig. 4). The MD fluctuations are smaller than the experimental B-factors. They lack the contributions for the static lattice disorder of a 2.7 Å crystal structure. However, the MD and x-ray maxima and minima correlate well with one another. Regions with high flexibility are N- and C-termini. This difference may be due to the missing residues in the simulations. The simulated atomic system includes residues 17–545 for subunit I of the enzyme, and 1–252 for the subunit II of the enzyme (Ostermeier et al., 1997). However, the missing residues 1–16 that were not resolved in the crystallographic structure could nevertheless have an influence on the structure and dynamics of the rest of the protein. Fluctuations of the W12 and W8 systems are compared in Fig. 5. No significant trend is observed and neither simulation showed a higher fluctuation than the other; the fluctuations are ∼0.1 Å smaller in the W8 simulations, at later stages again reflecting the denser packing.
FIGURE 4.

RMSF of the backbone atoms calculated from the molecular dynamics trajectories at 1–45 ps (black line), 450–495 ps (red line), 1080–1125 ps (green line), and from the experimental B-factor (blue line). All values are averaged over the individual amino acids. (a) RMSF from the W12 set. (b) RMSF from the W8 set.
FIGURE 5.

RMSF of the backbone atoms calculated from the molecular dynamics trajectories for the W12 and W8 sets of water molecules during 1–45 ps (a), 450–495 ps (b), and 1080–1125 ps (c). All values are averaged over the individual amino acids; the backbone atoms from the W12 set of coordinates are shown in black; the backbone atoms from the W8 set of coordinates are shown in red.
Water distribution in cytochrome c oxidase and its dynamical properties
The identification of protein-bound water molecules and of hydrogen-bonded networks allows us to study their dynamic behavior. The water molecules in cytochrome c oxidase for the W12 set at the start of the simulation are shown in Fig. 6. Since some of the hydrogen-bonded pathways are assumed to constitute proton pathways, it is important to estimate their lifetimes, as that can provide some insights into the efficiency of proton translocation. The average hydrogen-bonding statistics for COX were computed from the 1125 ps W12 and W8 simulations. Table 1 shows the time evolution of the number of hydrogen bonds between water oxygens and protein residues. During the first 270 ps of molecular dynamics production the number increased rapidly, and reached a steady state. This fact can be ascribed to the penetration and escape of water molecules into and out of the enzyme; an imbalance between these two processes causes the fluctuation. As the MD simulation proceeds, the system relaxes, and some water molecules get access to the protein interior, making favorable interactions with residues.
FIGURE 6.

The distribution of water molecules in the COX during the simulation.
TABLE 1.
Statistics of hydrogen-bond network for the W12 and W8 systems during MD production run
| W12
|
W8
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time, ps | ≥90% | ≥75% | ≥50% | ≥25% | Total | ≥90% | ≥75% | ≥50% | ≥25% | Total |
| 45 | 23 | 47 | 96 | 266 | 1455 | 23 | 39 | 107 | 265 | 1348 |
| 90 | 31 | 56 | 119 | 289 | 1417 | 30 | 53 | 112 | 278 | 1313 |
| 135 | 29 | 65 | 122 | 301 | 1325 | 32 | 59 | 120 | 272 | 1285 |
| 180 | 36 | 62 | 110 | 304 | 1353 | 27 | 53 | 110 | 277 | 1288 |
| 225 | 33 | 53 | 116 | 313 | 1440 | 33 | 72 | 128 | 279 | 1292 |
| 270 | 37 | 63 | 126 | 381 | 1400 | 32 | 62 | 132 | 278 | 1255 |
| 315 | 37 | 65 | 132 | 306 | 1358 | 28 | 61 | 131 | 276 | 1248 |
| 360 | 44 | 67 | 133 | 311 | 1449 | 24 | 59 | 125 | 288 | 1240 |
| 405 | 39 | 63 | 138 | 292 | 1316 | 33 | 57 | 132 | 306 | 1294 |
| 450 | 37 | 66 | 115 | 296 | 1406 | 31 | 57 | 117 | 291 | 1297 |
| 495 | 34 | 58 | 132 | 293 | 1427 | 46 | 71 | 137 | 280 | 1228 |
| 540 | 33 | 59 | 128 | 309 | 1379 | 33 | 63 | 129 | 283 | 1239 |
| 585 | 33 | 61 | 134 | 310 | 1335 | 26 | 58 | 130 | 277 | 1280 |
| 630 | 38 | 69 | 130 | 291 | 1335 | 27 | 62 | 128 | 281 | 1282 |
| 675 | 37 | 61 | 133 | 292 | 1345 | 33 | 64 | 129 | 290 | 1249 |
| 720 | 45 | 67 | 130 | 306 | 1358 | 30 | 59 | 127 | 299 | 1284 |
| 765 | 40 | 72 | 130 | 290 | 1363 | 31 | 59 | 118 | 309 | 1321 |
| 810 | 37 | 78 | 138 | 315 | 1321 | 36 | 57 | 120 | 288 | 1290 |
| 855 | 37 | 76 | 142 | 314 | 1374 | 28 | 55 | 125 | 293 | 1270 |
| 900 | 42 | 72 | 128 | 309 | 1358 | 36 | 63 | 133 | 289 | 1262 |
| 945 | 44 | 75 | 139 | 302 | 1407 | 34 | 70 | 136 | 300 | 1254 |
| 990 | 33 | 68 | 131 | 300 | 1411 | 42 | 67 | 135 | 280 | 1323 |
| 1035 | 37 | 70 | 135 | 313 | 1434 | 37 | 55 | 118 | 305 | 1352 |
| 1080 | 33 | 69 | 136 | 324 | 1447 | 36 | 75 | 131 | 281 | 1271 |
| 1125 | 40 | 66 | 126 | 310 | 1458 | 40 | 66 | 128 | 290 | 1264 |
Of the total of (176 + 88) water molecules in the W12 system, 92% had lifetimes of hydrogen-bonding of <100 ps, and <2% had lifetimes of >1 ns. In the W8 system, 93% of the total (755 + 88) water molecules had lifetimes of hydrogen-bonding <100 ps, and <1% had lifetimes of >1 ns. For a further analysis of the dynamic properties of the internal water molecules in the cytochrome c oxidase, we calculated the self-diffusion coefficient of water for both the W12 and the W8 sets of coordinates. The diffusion coefficients (D) of the water molecules were estimated from the mean-square displacement obtained from the molecular dynamics trajectories (Eq. 2). In our simulations, water molecules were allowed to leave the protein during the period analyzed. For the 88 water molecules found in the crystal structure plus the 176 water molecules from the GRID calculations (W12 set of coordinates), DH2O was (3.0 ± 0.005) × 10−9 m2/s, and for the 88 structural water molecules plus the 755 water molecules from the W8 set of coordinates, DH2O was (3.1 ± 0.005) × 10−9 m2/s. Experimental values for DH2O at 300 K are 2.3 × 10−9 m2/s (Mills, 1973), and in pure TIP3P water (Jorgensen et al., 1983) at 300 K DH2O has been estimated to be 1.3–4.2 × 10−9 m2/s (Norberg and Nilsson, 1994).
The simulations show interesting features concerning the water distribution around the hemes. The hemes and the axial histidine ligands His-94, His-413, and His-411 were constructed to be in similar conformations as in the post-1AR1 structure. In our MD simulations we find that water molecules form hydrogen bonds with the ND1 atom of the axial His-413. For the whole period of the simulations, His-411 formed a hydrogen bond with the O atom of Tyr-391 with an occupancy of 60% and a residence time of 691 ps, and with water molecule WG114 with an occupancy of 46% and a residence time of 513 ps.
The water molecules bound to the axial histidines are not the only water molecules fixed inside the protein for quite a long time. The majority of water molecules had shorter residence times than 100 ps, but a few water molecules in the binuclear center had longer residence times, up to the whole simulation time (Tables 2–7). In Fig. 9, A and B, and in Tables 6 and 7, we show that the structural water WS69 in both the W12 and the W8 sets and calculated water WG7 in the W8 set remained bound to CuB-ligated OH− during 1125 ps of simulation. There are quite a number of water molecules trapped within cavities, although in certain cases there is exchange with the W0 water. The behavior of this cavity water is like that of mobile water molecules in W0 water with many alternating hydrogen-bond partners. Table 6 lists waters with three, four, and five partners, and we could only list the interactions with >20% occupancy.
TABLE 2.
Hydrogen bonds between internal water molecules and amino acids in the K-pathway during 1125 ps of molecular dynamics production for the W12 set of water molecules binding sites (with occupancy ≥20%; please note all listed protein residues belong to subunit I)
| Hydrogen bonds | Occup. (%) | Average lifetime | Events | ||||||
|---|---|---|---|---|---|---|---|---|---|
| OH− | OH2 | — | WS | 69 | H2 | 100 | 1125.0 | 1 | |
| OH− | OH2 | — | WG | 7 | H1 | 100 | 1125.0 | 1 | |
| TRP | 358 | HN | — | LYS | 354 | O | 98 | 12.6 | 87 |
| HIS | 276 | HE1 | — | WS | 69 | OH2 | 98 | 13.1 | 84 |
| THR | 361 | HG1 | — | SER | 357 | O | 97 | 52.2 | 21 |
| SER | 357 | HN | — | ILE | 353 | O | 94 | 5.9 | 180 |
| TYR | 280 | O | — | LEU | 284 | HN | 92 | 7.0 | 148 |
| TRP | 358 | HE1 | — | SER | 291 | OG | 86 | 4.8 | 201 |
| SER | 291 | HN | — | PHE | 287 | O | 86 | 4.9 | 197 |
| THR | 351 | HG1 | — | ILE | 347 | O | 85 | 4.8 | 199 |
| HEMA3 | HO11 | — | WS | 69 | OH2 | 74 | 13.2 | 63 | |
| HIS | 326 | HE1 | — | WG | 7 | OH2 | 73 | 2.8 | 296 |
| TYR | 280 | HD2 | — | HIS | 276 | O | 70 | 2.4 | 327 |
| SER | 357 | OG | — | GLY | 304 | HN | 68 | 2.6 | 295 |
| THR | 361 | HN | — | SER | 357 | O | 64 | 2.4 | 302 |
| HIS | 276 | HE1 | — | WG | 7 | OH2 | 59 | 2.1 | 315 |
| SER | 295 | HN | — | SER | 291 | O | 58 | 2.8 | 234 |
| TYR | 280 | HH | — | Ws | 69 | OH2 | 49 | 9.7 | 57 |
| SER | 291 | HG1 | — | PHE | 287 | O | 47 | 5.5 | 96 |
| HIS | 325 | HE1 | — | WS | 69 | OH2 | 46 | 2.9 | 177 |
| TRP | 272 | O | — | HIS | 276 | HN | 44 | 2.9 | 171 |
| SER | 295 | HG1 | — | SER | 291 | O | 42 | 3.6 | 129 |
| TYR | 280 | HN | — | HIS | 276 | O | 41 | 2.3 | 205 |
| WS | 6 | OH2 | — | WG | 161 | H1 | 27 | 4.1 | 75 |
| GLY | 319 | O | — | WS | 81 | H2 | 21 | 79.2 | 3 |
| WG | 161 | OH2 | — | WG | 149 | H2 | 21 | 3.5 | 68 |
TABLE 7.
Hydrogen bonds between internal water molecules and amino acids in the heme a3/CuB region during 1125 ps of molecular dynamics production for the W8 set of water molecules binding sites (with occupancy ≥20%; please note all listed protein residues belong to subunit I)
| Hydrogen bonds | Occup. (%) | Average lifetime | Events | ||||||
|---|---|---|---|---|---|---|---|---|---|
| OH− | OH2 | — | WS | 69 | H2 | 100 | 1125.0 | 1 | |
| HIS | 413 | HN | — | VAL | 409 | O | 97 | 13.7 | 80 |
| HIS | 411 | HD1 | — | VAL | 408 | O | 92 | 9.3 | 111 |
| ASP | 399 | HN | — | GLN | 395 | O | 91 | 5.2 | 198 |
| HIS | 325 | HE1 | — | OH− | OH2 | 82 | 20.5 | 45 | |
| HIS | 94 | HN | — | MET | 90 | O | 81 | 3.6 | 253 |
| HIS | 276 | HN | — | TRP | 272 | O | 75 | 3.5 | 243 |
| HIS | 325 | HD1 | — | WG | 158 | OH2 | 74 | 13.7 | 61 |
| HIS | 413 | O | — | SER | 417 | HN | 67 | 2.3 | 327 |
| HIS | 326 | HN | — | W0 | 1385 | OH2 | 66 | 3.3 | 222 |
| HEMA3 | OMA | — | WG | 158 | H2 | 65 | 9.1 | 81 | |
| HEMA3 | O1D | — | ARG | 473 | HH21 | 63 | 7.1 | 100 | |
| W0 | 1385 | OH2 | — | WG | 158 | H1 | 60 | 5.9 | 115 |
| HIS | 94 | O | — | MET | 98 | HN | 55 | 2.3 | 269 |
| SER | 394 | HG1 | — | WG | 491 | OH2 | 54 | 6.7 | 90 |
| HIS | 276 | O | — | TYR | 280 | HD2 | 53 | 1.9 | 306 |
| ARG | 473 | HE | — | WG | 51 | OH2 | 53 | 7.2 | 83 |
| HIS | 413 | HD1 | — | WS | 42 | OH2 | 50 | 6.2 | 91 |
| HEMA3 | O2A | — | ASP | 399 | HD2 | 49 | 91.6 | 6 | |
| WS | 69 | OH2 | — | WG | 453 | H1 | 47 | 4.2 | 124 |
| HIS | 276 | O | — | TYR | 280 | HN | 43 | 2.1 | 225 |
| ARG | 473 | HE | — | WS | 13 | OH2 | 42 | 8.7 | 55 |
| HIS | 326 | HE1 | — | WG | 453 | OH2 | 39 | 3.0 | 146 |
| HEMA3 | O1D | — | ARG | 473 | HH12 | 37 | 8.9 | 47 | |
| OH− | OH2 | — | WG | 453 | H2 | 34 | 385.8 | 1 | |
| TYR | 280 | HH | — | WG | 188 | OH2 | 33 | 6.9 | 54 |
| ARG | 473 | HH12 | — | W4 | 178 | OH2 | 29 | 11.5 | 28 |
| WG | 177 | OH2 | — | WG | 189 | H2 | 29 | 6.6 | 49 |
| TYR | 406 | HH | — | WG | 46 | OH2 | 26 | 9.7 | 30 |
| SER | 394 | HG1 | — | WG | 188 | OH2 | 26 | 10.0 | 29 |
| ARG | 474 | HH11 | — | WG | 31 | OH2 | 26 | 9.0 | 33 |
| HEMA3 | O2D | — | ARG | 473 | HH21 | 25 | 4.3 | 66 | |
| HEMA3 | O2D | — | W4 | 182 | H2 | 25 | 11.3 | 25 | |
| HIS | 413 | HD1 | — | W4 | 219 | OH2 | 23 | 7.8 | 34 |
| HIS | 325 | HN | — | WG | 326 | OH2 | 23 | 3.0 | 87 |
| TYR | 406 | HH | — | W4 | 224 | OH2 | 22 | 3.8 | 65 |
| TYR | 406 | HH | — | W1 | 3872 | OH2 | 22 | 8.1 | 30 |
| HIS | 276 | HE1 | — | WS | 69 | OH2 | 22 | 2.7 | 90 |
| GLY | 390 | HN | — | WG | 395 | OH2 | 22 | 2.5 | 101 |
| GLY | 352 | HN | — | W2 | 6384 | OH2 | 22 | 2.4 | 106 |
| WG | 515 | OH2 | — | WG | 178 | H1 | 22 | 6.0 | 41 |
| HIS | 411 | HN | — | TYR | 407 | O | 21 | 1.9 | 125 |
| HIS | 236 | O | — | W3 | 799 | H2 | 21 | 14.2 | 17 |
| ARG | 473 | HH11 | — | WG | 51 | OH2 | 21 | 2.3 | 106 |
| HEMA3 | OMA | — | WG | 158 | H1 | 20 | 5.3 | 43 | |
| HEMA3 | O1D | — | W4 | 191 | H1 | 20 | 6.4 | 35 | |
| HEMA | O11 | — | WG | 48 | H1 | 20 | 2.7 | 83 | |
| WS | 81 | H2 | — | W2 | 7182 | OH2 | 20 | 9.7 | 23 |
FIGURE 9.

Multiple configurations of selected residues and water molecules close to the heme a/heme a3–CuB region for the W12 (A) and W8 (B) set of coordinates observed from MD trajectories. Initial coordinates are shown with the thick licorice. Positions of the selected residues after 1125 ps of MD run are shown with the thin licorice. Snapshots for selected water molecules are taken after 225, 450, 675, 900, and 1125 ps of simulations. For the W12 set of coordinates water molecule WS13 is represented in blue, WS69 in red, WG5 in yellow, WG7 in green, WG30 in pink, WG144 in magenta, WG173 in orange, W4172 in gold, and W4214 in rose. For the W8 set of coordinates water molecule WS13 is represented in blue, WS14 in yellow, WS69 in red, WG31 in light green, WG42 in orange, WG46 in magenta, WG51 in pink, WG157 in green, WG188 in rose, WG189 in black, and WG453 in gold.
TABLE 6.
Hydrogen bonds between internal water molecules and amino acids in the heme a3/CuB region during 1125 ps of molecular dynamics production for the W12 set of water molecules binding sites (with occupancy ≥20%; please note all listed protein residues belong to subunit I)
| Hydrogen bonds | Occup. (%) | Average lifetime | Events | ||||||
|---|---|---|---|---|---|---|---|---|---|
| HEMA3 | O1A | — | ASP | 399 | HD2 | 100 | 140.4 | 8 | |
| OH− | OH2 | — | WS | 69 | H2 | 100 | 1125.0 | 1 | |
| OH− | OH2 | — | WG | 7 | H1 | 100 | 1125.0 | 1 | |
| HIS | 276 | HE1 | — | WS | 69 | OH2 | 98 | 13.1 | 84 |
| ARG | 473 | HE | — | WS | 13 | OH2 | 98 | 15.3 | 72 |
| HEMA3 | O1A | — | HSD | 403 | HD1 | 96 | 12.6 | 86 | |
| HIS | 413 | O | — | SER | 417 | HN | 91 | 4.5 | 230 |
| ASP | 399 | O | — | HSD | 403 | HN | 89 | 5.3 | 189 |
| ASP | 399 | HN | — | GLN | 395 | O | 83 | 4.3 | 218 |
| HEMA3 | O2A | — | WG | 5 | H1 | 74 | 23.0 | 36 | |
| HEMA3 | HO11 | — | WS | 69 | OH2 | 74 | 13.2 | 63 | |
| HIS | 326 | HE1 | — | WG | 7 | OH2 | 73 | 2.8 | 296 |
| HIS | 413 | HN | — | VAL | 409 | O | 70 | 2.9 | 271 |
| HIS | 276 | O | — | TYR | 280 | HD2 | 70 | 2.4 | 327 |
| HEMA3 | O2D | — | ARG | 473 | HH12 | 69 | 17.9 | 43 | |
| HEMA3 | O2A | — | HSD | 403 | HD1 | 65 | 2.8 | 261 | |
| HIS | 94 | O | — | MET | 98 | HN | 64 | 3.0 | 243 |
| HIS | 94 | HD1 | — | SER | 46 | O | 61 | 2.7 | 252 |
| HIS | 411 | HN | — | TYR | 407 | O | 60 | 2.6 | 266 |
| HIS | 276 | HE1 | — | WG | 7 | OH2 | 59 | 2.1 | 315 |
| VAL | 408 | O | — | WG | 214 | H2 | 54 | 9.9 | 61 |
| HEMA3 | O1D | — | ARG | 473 | HH21 | 49 | 21.0 | 26 | |
| TYR | 280 | HH | — | WS | 69 | OH2 | 49 | 9.7 | 57 |
| HIS | 411 | HE1 | — | W4 | 114 | OH2 | 48 | 2.6 | 208 |
| HIS | 325 | HE1 | — | WS | 69 | OH2 | 46 | 2.9 | 177 |
| HIS | 276 | HN | — | TRP | 272 | O | 44 | 2.9 | 171 |
| TYR | 328 | HN | — | W4 | 172 | OH2 | 43 | 3.9 | 124 |
| GLY | 319 | O | — | W3 | 5248 | H1 | 43 | 22.0 | 22 |
| GLN | 463 | OE1 | — | WG | 30 | H2 | 43 | 3.1 | 159 |
| TYR | 280 | HN | — | HIS | 276 | O | 41 | 2.3 | 205 |
| HIS | 325 | HD1 | — | VAL | 322 | O | 41 | 8.2 | 57 |
| HIS | 276 | O | — | TYR | 280 | HN | 41 | 2.3 | 205 |
| ARG | 473 | HH22 | — | WS | 13 | OH2 | 41 | 3.3 | 140 |
| W0 | 2000 | H1 | — | W3 | 5248 | OH2 | 41 | 8.3 | 55 |
| WG | 5 | OH2 | — | W4 | 214 | H1 | 41 | 6.3 | 73 |
| GLN | 463 | OE1 | — | WG | 30 | H1 | 40 | 3.0 | 151 |
| VAL | 408 | O | — | W4 | 214 | H1 | 39 | 10.5 | 42 |
| THR | 389 | OG1 | — | HIS | 411 | HD1 | 35 | 2.5 | 159 |
| HEMA3 | O2A | — | W3 | 1204 | H1 | 34 | 26.9 | 14 | |
| HEMA | O2D | — | ARG | 474 | HE | 34 | 6.9 | 56 | |
| HEMA3 | O1D | — | ARG | 473 | HH12 | 32 | 7.8 | 46 | |
| HEMA | OMA | — | GLN | 463 | HE21 | 32 | 2.0 | 184 | |
| TYR | 475 | HN | — | WG | 173 | OH2 | 31 | 3.7 | 95 |
| ASP | 399 | OD1 | — | W4 | 172 | H1 | 31 | 6.5 | 53 |
| WG | 130 | OH2 | — | WG | 114 | H2 | 29 | 4.2 | 79 |
| HEMA3 | O1A | — | W4 | 172 | H2 | 28 | 4.2 | 75 | |
| TYR | 475 | HN | — | WS | 42 | OH2 | 28 | 2.5 | 130 |
| HEMA3 | O1D | — | W3 | 1204 | H2 | 27 | 20.0 | 15 | |
| HIS | 94 | HN | — | MET | 90 | O | 27 | 2.2 | 135 |
| ARG | 54 | HE | — | WG | 31 | OH2 | 26 | 11.2 | 26 |
| HEMA3 | O1A | — | W4 | 172 | H1 | 25 | 4.8 | 60 | |
| PHE | 383 | O | — | WG | 129 | H2 | 23 | 4.0 | 65 |
| GLY | 387 | O | — | WG | 114 | H1 | 23 | 4.0 | 64 |
| ARG | 54 | HE | — | W4 | 220 | OH2 | 23 | 14.9 | 17 |
| WG | 130 | OH2 | — | WG | 114 | H1 | 23 | 3.7 | 69 |
| THR | 50 | OG1 | — | W4 | 220 | H2 | 22 | 2.6 | 97 |
| WS | 13 | OH2 | — | WG | 31 | H1 | 22 | 2.6 | 94 |
| WG | 130 | H1 | — | WG | 129 | OH2 | 22 | 6.7 | 37 |
| TRP | 164 | HE1 | — | WG | 4 | OH2 | 21 | 2.4 | 98 |
| GLY | 319 | O | — | WS | 81 | H2 | 21 | 79.2 | 3 |
| WG | 99 | H1 | — | WG | 28 | OH2 | 21 | 2.9 | 82 |
| WG | 5 | OH2 | — | W4 | 214 | H2 | 21 | 6.5 | 37 |
| THR | 50 | OG1 | — | W4 | 220 | H1 | 20 | 2.5 | 91 |
| WS | 81 | OH2 | — | W3 | 5248 | H2 | 20 | 10.2 | 22 |
Depending on the definition of the hydrogen-bond geometry used, our analysis sometimes shows that a contact between a water molecule and a residue breaks while at the same time a new interaction between the same water molecule and another neighboring residue develops. The water molecule changes its orientation only, and, as a result, remains trapped in that region for quite a long time, but the average lifetime of the hydrogen bond is smaller than the overall residence time of this water molecule. An example of such a water molecule is water WG158 (Table 7), which can form hydrogen bonds with His-325 and with water W01385.
The reason for performing two parallel simulations that only differ by the amount of internal water molecules added, is, of course, to determine the level of hydration of the protein interior. All water molecules of the W8 set had GRID energies <−8 kcal/mol indicating high occupancy. The hydrogen-bond lifetimes are longer for the W12 set than for the W8 set. This is due to a greater number of hydrogen-bonding possibilities in the W8 system that is filled with water more densely. The MD simulations indicate that the solvent distribution is more diffuse for higher hydration states (Helms and Wade, 1998). According to our observations, the hydrogen-bonded network in cytochrome c oxidase is not uniformly distributed, and the degree of water arrangement is variable, inasmuch as side chains induce variable level of the water ordering (Bret et al., 2002).
The distribution of water in the K- and D-pathways
Detailed information about energetically favorable water binding sites along the K- and D-pathways is essential for understanding the mechanism of proton transfer in COX. By examining the MD trajectories for water molecules and using the definition of hydrogen-bonding by Tang and Nilsson (1999), the number of water molecules can be obtained, which are likely to form the hydrogen-bonded network. The numbers of water molecules in the K- and D-pathways at different times are given in Table 8. The GRID method placed seven water molecules in the K-pathway for the beginning of the simulations. It can be seen that the number of solvents is not static, but ranges quite substantially from 8 to 3 with an average of 6.5 for the W12 set of coordinates, and from 5 to 15 with an average of 9.5 for the W8 set. These sites participate in the formation of a reasonable hydrogen-bonded network in the K-pathway. This network is not continuous (Fig. 7, A and B). Tables 2 and 3 list the hydrogen bonds inside the K-pathway with occupancies >20%. A hydrogen-bonded connection can be seen at the beginning of the K-pathway and also at the end, leading up to heme a3, but there is no direct or water-mediated connection between Lys-354 and Thr-351 at the beginning of the W12 simulations. After 500 ps of the MD run we observe a significant reorientation of the side chain of neutral Lys-354 and formation of a hydrogen bond between the O atom of water WG149 and the HZ1 atom of Lys-354 with an occupancy of 6% (data not shown). Due to the low occupancy, we can conclude that Lys-354 has little effect on water ordering, perhaps because of its long flexible side chain. For the W8 simulations, the hydrogen-bonded network was continuous after the placement of GRID water molecules. However, after the first 45 ps of dynamics many water molecules had left their positions and the hydrogen-bonded network did not remain continuous.
TABLE 8.
The average number of water molecules at instantaneous snapshots in the K- and D-pathways of cytochrome c oxidase from Paracoccus denitrificans
| Average number of water molecules
|
||||
|---|---|---|---|---|
| Time, ps | K-path W12 | K-path W8 | D-path W12 | D-path W8 |
| 0 | ||||
| 45 | 8 | 15 | 23 | 29 |
| 225 | 8 | 5 | 20 | 25 |
| 450 | 6 | 10 | 18 | 27 |
| 675 | 9 | 5 | 22 | 24 |
| 900 | 4 | 9 | 22 | 27 |
| 1125 | 3 | 13 | 24 | 26 |
FIGURE 7.

Multiple configurations of selected residues and water molecules in the K-pathway for the W12 (A) and W8 (B) set of coordinates observed from MD trajectories. Initial coordinates are shown with the thick licorice. Positions of the selected residues after 1125 ps are shown with the thin licorice. Snapshots for selected water molecules are taken after 225, 450, 675, 900, and 1125 ps of simulations. For the W12 set of coordinates water molecule WS6 (structural water) is represented in blue, WS9 in yellow, WS69 in red, WS81 in rose, WG7 (internal GRID water) in light green, WG149 in pink, and WG161 in magenta. For the W8 set of coordinates water molecule WS6 is represented in blue, WS69 in red, WS88 in yellow, WG13 in rose, WG188 in light green, WG194 in gold, WG379 in pink, WG491 in magenta, and W26484 in orange.
TABLE 3.
Hydrogen bonds between internal water molecules and amino acids in the K-pathway during 1125 ps of molecular dynamics production for the W8 set of water molecules binding sites (with occupancy ≥20%; please note all listed protein residues belong to subunit I)
| Hydrogen bonds | Occup. (%) | Average lifetime | Events | ||||||
|---|---|---|---|---|---|---|---|---|---|
| OH− | OH2 | — | WS | 69 | H2 | 100 | 1125.0 | 1 | |
| TYR | 280 | O | — | LEU | 284 | HN | 93 | 6.1 | 171 |
| TRP | 358 | HN | — | LYS | 354 | O | 77 | 4.6 | 191 |
| SER | 357 | HN | — | ILE | 353 | O | 77 | 3.4 | 256 |
| TRP | 272 | O | — | HIS | 276 | HN | 75 | 3.5 | 243 |
| TRP | 358 | HE1 | — | SER | 291 | OG | 66 | 2.9 | 259 |
| SER | 291 | HN | — | PHE | 287 | O | 61 | 2.6 | 266 |
| SER | 295 | HG1 | — | SER | 291 | O | 59 | 4.8 | 137 |
| SER | 291 | HG1 | — | PHE | 287 | O | 54 | 9.6 | 63 |
| TYR | 280 | HD2 | — | HIS | 276 | O | 53 | 1.9 | 306 |
| THR | 361 | HN | — | SER | 357 | O | 53 | 3.4 | 175 |
| THR | 351 | HG1 | — | ALA | 348 | O | 50 | 7.2 | 78 |
| THR | 361 | HG1 | — | SER | 357 | O | 48 | 11.8 | 46 |
| TYR | 280 | HN | — | HIS | 276 | O | 43 | 2.1 | 225 |
| SER | 357 | HG1 | — | ILE | 353 | O | 39 | 9.2 | 48 |
| TYR | 280 | HH | — | WG | 188 | OH2 | 33 | 6.9 | 54 |
| SER | 295 | HN | — | SER | 291 | O | 30 | 1.8 | 187 |
| PRO | 350 | O | — | WG | 379 | H1 | 28 | 5.1 | 61 |
| VAL | 349 | O | — | W2 | 6384 | H2 | 27 | 4.0 | 76 |
| THR | 351 | HN | — | ALA | 348 | O | 27 | 2.5 | 121 |
| WS | 6 | H2 | — | WG | 194 | OH2 | 25 | 6.9 | 40 |
| SER | 357 | OG | — | GLY | 304 | HN | 24 | 2.4 | 114 |
| WS | 6 | OH2 | — | WG | 164 | H1 | 23 | 4.3 | 61 |
| HIS | 276 | HE1 | — | WS | 69 | OH2 | 22 | 2.7 | 90 |
| W2 | 6384 | OH2 | — | GLY | 352 | HN | 22 | 2.4 | 106 |
| VAL | 349 | O | — | W2 | 6384 | H1 | 21 | 3.4 | 71 |
| GLY | 304 | O | — | WG | 194 | H2 | 21 | 4.0 | 61 |
| WS | 88 | OH2 | — | WG | 491 | H1 | 20 | 3.4 | 66 |
For the D-pathway, the number of solvent molecules ranged from 18 to 24 with an average of 21.5 for the W12 set, and from 24 to 29 with an average of 26.33 for the W8 set. Sixteen sites took part in the formation of a quite stable continuous hydrogen-bonded chain leading from Asp-124 to the region close to Glu-278, but only four waters in the hydrophobic pore between Ser-193 and Glu-278 were found to be required to form a hydrogen-bonded chain, which could be used for proton transfer (Fig. 8, A and B). The average lifetime of the hydrogen-bonded chain formed by these four water molecules is much shorter than the lifetime of hydrogen bonds formed by each individual pair of water molecules. Some of these waters have a high mobility during the MD simulations. The change in the number of water molecules arises either from water exchanging with W0 water or with the interior of protein (Henchman and McCammon, 2002). As an example, we examined the trajectory of water W4172 from the W12 set and found that this water molecule was able to traverse a distance of ∼3.5 Å during ∼0.3 ns. Although the placement of water molecules identifies an efficient pathway for protons up to Glu-278, the connection of the D-pathway with other protonatable sites beyond Glu-278 is not clear. One possibility is that the protonated glutamic acid side chain could flip upward and deliver its proton either to the binuclear center or to a heme a3 propionate group as proposed (Iwata et al., 1995). The space between Glu-278 and the binuclear center/heme a3 propionate group is proposed to be a part of the oxygen diffusion channel (Svensson-Ek et al., 2002), which is hydrophobic and does not contain any crystallographically identifiable water molecules. This area may be filled with mobile water molecules as has been suggested by Riistama et al. (1997). Within the time of 1.125 ns of MD simulations, such a conformational change of the protonated Glu-278 was not observed. Our data do not show that there is a connection between the protonated Glu-278 and the O1A propionate group of heme a3 for the COX in a fully oxidized state. However, an additional short simulation of COX in a fully oxidized state with deprotonated Glu-278 shows a significant reorientation of the side chain of Glu-278 and the formation of a hydrogen-bond chain between Glu-278 via water molecules up to the O1A atom of the heme a3 propionate (these data we had presented at the 12th European Bioenergetics Conference in 2002; referenced by Zheng et al., 2003). This difference may be of interest for routing protons in different parts of the catalytic cycle. Locally, the hydration network follows the reorientation of particular residues that change their conformation. Examples are shown in Fig. 7, A and B; Fig. 8, A and B; and Fig. 9, A and B.
FIGURE 8.

Multiple configurations of selected residues and water molecules in the D-pathway for the W12 (A) and W8 (B) set of coordinates observed from MD trajectories. Initial coordinates are shown with the thick licorice. Positions of the selected residues after 1125 ps of MD run are shown with the thin licorice. Snapshots for selected water molecules are taken after 225, 450, 675, 900, and 1125 ps of simulations. For the W12 set of coordinates water molecule WS38 is represented in blue, WS40 in yellow, WS80 in red, WG8 in light green, WG12 in gold, WG80 in light blue, WG131 in magenta, and WG144 in pink. For the W8 set of coordinates water molecule WS3 is represented in rose, WS38 in blue, WG9 in light blue, WG11 in orange, WG180 in light green, WG198 in magenta, WG220 in green, WG238 in red, WG540 in gray, and W1232 in black.
A large number of water molecules was observed in the gap between subunit I and II and in the hydrophilic cavity above heme a and heme a3. This result is in agreement with previous studies (Hofacker and Schulten, 1998; Zheng et al., 2003). These water molecules form a network of hydrogen bonds and connect the propionate groups of heme a and heme a3 to the external aqueous phase in many ways. Since it has been suggested that the propionate groups are involved in proton pumping (Michel, 1998; Puustinen and Wikström, 1999), this network could be a possible pathway for proton exit. In Fig. 9, A and B, we show that the cavities above heme a and heme a3 contain a number of positionally stable, but also highly mobile water molecules with hydrogen-bond occupancies of <30%. For example, the modeled internal water WG5 is present and essentially always hydrogen-bonded to the O2A atom of the heme a3 propionate for 828 ps in the W12 set. Noteworthy are the hydrogen bonds between some water molecules and the propionate groups of the hemes, which may be of structural and energetic importance. In agreement with previous electrostatics calculations (Kannt et al., 1998), it was found that Asp-399 forms a hydrogen bond with the O1A heme a3 propionate with an occupancy of 100% and a residence time of 1123.2 ps, and also forms a hydrogen bond with water W4172 with an occupancy of 31% and a residence time of 344.5 ps (Fig. 9, A and B) for the W12 set.
Another interesting observation is that Arg-473 and Arg-474 seem to play an important role in directing the water network orientation in the region above heme a/heme a3. Their highly mobile side chains significantly influence the reorientations of water molecules in both the electron transfer and proton exit pathways (Puustinen and Wikström, 1999). The HH21 atom of Arg-473 forms a hydrogen bond with the O1D atom of the heme a3 propionate with an occupancy of 63% and a residence time of 710 ps. The HH21 atom of Arg-474 forms a hydrogen bond with the O2D atom of the heme a propionate with an occupancy of 87% and a residence time of 990.6 ps. These results show that the Δ-propionate of the heme a is more strongly stabilized by charge interactions with this arginine, in agreement with previous electrostatics calculations (Kannt et al., 1998). The simulation with the W8 set shows a significant reorientation of Tyr-167 in the direction away from the hemes. Although there is no experimental evidence concerning the role of this residue, it is very likely that Tyr-167, which is located close to Arg-474, plays a role in the formation of a hydrogen-bonded connection to Arg-474 via mobile water molecules, and to the proton exit or electron transfer pathways.
CONCLUSION
The main results of the GRID calculations and dynamic simulations can be summarized as follows:
A much higher average number of internal water molecules than observed in crystal structures (Iwata et al., 1995; Ostermeier et al., 1997; Tshukihara et al., 1996; Svensson-Ek et al., 2002) was found in our simulations.
The dynamic model leads to a stable system. The MD simulation requires ∼1 ns to reach a dynamic equilibrium, due to the effect of protein-membrane and protein-water interactions. Also, water-protein interactions take a long time to reach equilibrium. Similar observations were reported for other systems (Garcia and Hummer, 2000; Bret et al., 2002).
The hydrogen-bonded network in cytochrome c oxidase is not uniformly distributed, and the degree of water arrangement is variable. The protein-membrane-water model provides a detailed description of the structure and dynamics of a hydrogen-bonded network and identifies a number of permanent water molecules sites in the K- and D-pathways. Networks of hydrogen-bonded water molecules are found that extend in many directions above the heme a and heme a3. Some of the water molecules in the vicinity of the binuclear center have lifetimes ∼1 ns, but in general the majority of internal water molecules are mobile. The presence of mobile water is indicated in regions of the protein where no water has been found by x-ray crystallography. The explanation for the large discrepancy of the number of internal water molecules as found during simulation and as compared to the crystallographically identified internal water molecules is the high mobility and exchange rate of water molecules between internal and external locations.
Although our simulations agree with previous theoretical studies in general (Riistama et al., 1997; Hofacker and Schulten, 1998; Backgren et al., 2000; Zheng et al., 2003), there are some differences. The main one concerns the significant diffusion of individual water molecules in the D- and K-pathways as well as in the region connecting to the binuclear center. Proton transfer along these pathways may be different in character than that along narrow water files as described in gramicidin A (Pomès and Roux, 1996) and carbon nanotubes (Hummer et al., 2001). We initially planned to identify a small number of unique hydrogen-bond networks in the D- and K-pathways as described, for example, for crystals of vitamin B12 (Savage, 1986). However, the hydrogen-bonded network in cytochrome c oxidase is so dynamic and of such a high dimensionality that it cannot be characterized by pictures and tables. Each hydrogen-bonded pathway is transient and exists only for a certain period of time. Proton flow should likely be thought of as a superimposition of many fluctuating alternative transport pathways, which are the result of a reorientation and exchange of the participating water molecules. The challenge for the future is to investigate how water permeation and diffusion in the proton transfer pathways are quantitatively related to the geometry, charge distribution, and oxidation state of the protein.
TABLE 4.
Hydrogen bonds between internal water molecules and amino acids in the D-pathway during 1125 ps of molecular dynamics production for the W12 set of water molecules binding sites (with occupancy ≥20%; please note all listed protein residues belong to subunit I)
| Hydrogen bonds | Occup. (%) | Average lifetime | Events | ||||||
|---|---|---|---|---|---|---|---|---|---|
| TYR | 35 | O | — | ALA | 39 | HN | 100 | 40.0 | 28 |
| ASN | 199 | O | — | THR | 203 | HG1 | 100 | 70.1 | 16 |
| TYR | 35 | HN | — | ILE | 31 | O | 98 | 17.3 | 64 |
| SER | 134 | HN | — | LEU | 130 | O | 98 | 15.2 | 73 |
| ASN | 199 | O | — | THR | 203 | HN | 97 | 8.8 | 123 |
| SER | 193 | HN | — | SER | 183 | O | 95 | 6.6 | 163 |
| ASN | 199 | OD1 | — | PHE | 127 | HN | 95 | 5.9 | 181 |
| GLE | 278 | HN | — | PHE | 274 | O | 94 | 8.4 | 126 |
| SER | 193 | O | — | ALA | 197 | HN | 90 | 5.5 | 183 |
| THR | 26 | O | — | WG | 144 | H1 | 86 | 10.9 | 89 |
| ASN | 131 | HD22 | — | WG | 144 | OH2 | 78 | 4.3 | 205 |
| ASP | 124 | O | — | ASP | 124 | HN | 76 | 3.0 | 281 |
| SER | 134 | O | — | TYR | 138 | HN | 74 | 2.6 | 318 |
| SER | 134 | O | — | SER | 192 | HG1 | 73 | 6.9 | 118 |
| TYR | 35 | HH | — | WG | 131 | OH2 | 64 | 18.4 | 39 |
| ASN | 113 | OD1 | — | ASN | 131 | HD21 | 64 | 3.6 | 202 |
| ASP | 124 | OD1 | — | WG | 144 | H2 | 53 | 85.7 | 7 |
| ASP | 124 | OD1 | — | MET | 125 | HN | 52 | 6.5 | 90 |
| ASP | 124 | OD1 | — | HSD | 28 | HD1 | 51 | 8.4 | 69 |
| ASN | 113 | HN | — | GLY | 109 | O | 49 | 2.5 | 221 |
| ASN | 199 | HN | — | LEU | 195 | O | 49 | 2.3 | 242 |
| ASN | 113 | HD22 | — | GLY | 109 | O | 41 | 3.2 | 147 |
| ASN | 199 | HD22 | — | WG | 143 | OH2 | 41 | 3.5 | 130 |
| ASP | 124 | OD2 | — | MET | 125 | HN | 40 | 9.9 | 45 |
| ASN | 113 | HD21 | — | ASN | 131 | OD1 | 40 | 5.5 | 82 |
| ASN | 131 | O | — | TYR | 135 | HN | 39 | 1.9 | 225 |
| SER | 134 | OG | — | WG | 131 | H1 | 38 | 4.2 | 103 |
| SER | 134 | HG1 | — | LEU | 130 | O | 34 | 3.1 | 125 |
| ASP | 124 | OD2 | — | WG | 144 | H2 | 34 | 47.4 | 8 |
| ASN | 131 | OD1 | — | WG | 131 | H2 | 32 | 3.3 | 107 |
| ASP | 124 | OD2 | — | HSD | 28 | HD1 | 29 | 4.9 | 68 |
| ASN | 199 | HD22 | — | WG | 80 | OH2 | 21 | 2.7 | 87 |
| SER | 134 | OG | — | WG | 131 | H2 | 20 | 3.5 | 63 |
| ASN | 131 | OD1 | — | WG | 131 | H1 | 20 | 3.2 | 70 |
| WG | 8 | H1 | — | WG | 12 | OH2 | 20 | 10.5 | 22 |
TABLE 5.
Hydrogen bonds between internal water molecules and amino acids in the D-pathway during 1125 ps of molecular dynamics production for the W8 set of water molecules binding sites (with occupancy ≥20%; please note all listed protein residues belong to subunit I)
| Hydrogen bonds | Occup. (%) | Average lifetime | Events | ||||||
|---|---|---|---|---|---|---|---|---|---|
| TYR | 35 | O | — | ALA | 39 | HN | 99 | 22.3 | 50 |
| ASN | 199 | O | — | THR | 203 | HN | 96 | 8.6 | 125 |
| TYR | 35 | HN | — | ILE | 31 | O | 93 | 6.4 | 162 |
| ASN | 199 | OD1 | — | PHE | 127 | HN | 93 | 7.8 | 134 |
| SER | 193 | O | — | ALA | 197 | HN | 87 | 4.6 | 215 |
| SER | 134 | HN | — | LEU | 130 | O | 81 | 4.7 | 195 |
| ASN | 199 | O | — | THR | 203 | HG1 | 78 | 11.6 | 76 |
| SER | 134 | O | — | TYR | 138 | HN | 65 | 2.7 | 275 |
| SER | 193 | HN | — | SER | 189 | O | 50 | 2.8 | 202 |
| ASN | 131 | HN | — | PHE | 127 | O | 46 | 2.2 | 234 |
| TYR | 35 | HH | — | WG | 540 | OH2 | 43 | 10.0 | 49 |
| TYR | 114 | O | — | WG | 198 | H1 | 41 | 5.0 | 91 |
| ASP | 124 | O | — | ASP | 124 | HN | 38 | 1.9 | 225 |
| ASP | 124 | OD1 | — | MET | 125 | HN | 37 | 4.8 | 86 |
| PHE | 274 | O | — | GLE | 278 | HN | 33 | 3.8 | 99 |
| GLE | 278 | HN | — | PHE | 274 | O | 33 | 3.8 | 99 |
| ASN | 113 | HD22 | — | GLY | 109 | O | 32 | 2.6 | 140 |
| ASP | 124 | OD2 | — | MET | 125 | HN | 31 | 4.4 | 79 |
| ASN | 199 | HN | — | LEU | 195 | O | 27 | 1.7 | 177 |
| ASN | 113 | HN | — | GLY | 109 | O | 24 | 2.4 | 110 |
| SER | 193 | HG1 | — | WG | 220 | OH2 | 23 | 9.2 | 28 |
| SER | 134 | HG1 | — | LEU | 130 | O | 23 | 4.8 | 55 |
| TYR | 35 | HH | — | W1 | 232 | OH2 | 22 | 15.4 | 16 |
| ASN | 131 | OD1 | — | WG | 540 | H1 | 20 | 3.0 | 76 |
Acknowledgments
We thank Prof. Peter Goodford for providing the GRID program, and Dr. Mike Crowley and Dr. Thomas Soddemann (Rechenzentrum Garching Max-Planck-Gesellschaft and the Max-Planck-Institut für Plasmaphysik) for help with parallelization of the CHARMM version c28b2 program for the IBM Power4 machine. We are also thankful to Dr. C. Roy D. Lancaster, Elena Herzog, Dr. Stephen Marino, and Hildur Palsdottir for useful discussions and reading the manuscript. E.O. is grateful to Barbara Schiller for the computational support.
This work was supported by the Deutsche Forschungsgemeinschaft (SFB 472), the Fonds der Chemischen Industrie, and the Max-Planck-Gesellschaft. Computing time was generously provided by the Rechenzentrum Garching Max-Planck-Gesellschaft and the Max-Planck-Institut für Plasmaphysik (Dr. I. Weidl and Dr. H. Lederer).
Present address for Michael C. Hutter and Volkhard Helms is Universität des Saarlandes, Zentrum für Bioinformatik, Postfach 15 11 50, 66041, Saarbrücken, Germany.
References
- Aagaard, A., G. Gilderson, D. A. Mills, S. Ferguson-Miller, and P. Brzezinski. 2000. Redesign of the proton-pumping machinery of cytochrome c oxidase: proton pumping does not require Glu(I-286). Biochemistry. 39:15847–15850. [DOI] [PubMed] [Google Scholar]
- Babcock, G. T., and M. Wikström. 1992. Oxygen activation and the conservation of energy in cell respiration. Nature. 356:301–309. [DOI] [PubMed] [Google Scholar]
- Backgren, C., G. Hummer, M. Wikström, and A. Puustinen. 2000. Proton translocation by cytochrome c oxidase can take place without the conserved glutamic acid in subunit I. Biochemistry. 39:7863–7867. [DOI] [PubMed] [Google Scholar]
- Berneche, S., M. Nina, and B. Roux. 1998. Molecular dynamics simulation of melittin in a dimyristoylphosphatidylcholine bilayer membrane. Biophys. J. 75:1603–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berneche, S., and B. Roux. 2000. Molecular dynamics of the KcsA K+ channel in a bilayer membrane. Biophys. J. 78:2900–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bret, S., M. Roth, S. Nørager, E. C. Hatchikian, and M. J. Field. 2002. Molecular dynamics study of Desulfovibrio africanus cytochrome c3 in oxidized and reduced forms. Biophys. J. 83:3049–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy minimization and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- Brünger, A., and M. Karplus. 1988. Polar hydrogen positions in proteins: empirical energy placement and neutron diffraction comparison. Proteins. 4:148–156. [DOI] [PubMed] [Google Scholar]
- Brzezinski, P., and P. Adelroth. 1998. Proton-controlled electron transfer in cytochrome c oxidase: functional role of the pathways through Glu 286 and Lys 362. Acta Physiol. Scand. Suppl. 643:7–16. [PubMed] [Google Scholar]
- Brzezinski, P., and G. Larsson. 2003. Redox-driven proton pumping by heme-copper oxidases. Biochim. Biophys. Acta. 1605:1–13. [DOI] [PubMed] [Google Scholar]
- Eriksson, M., T. Härd, and L. Nilsson. 1995. Molecular dynamics simulations of the glucocorticoid receptor DNA-binding domain in complex with DNA and free in solution. Biophys. J. 68:402–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson, M., and L. Nilsson. 1995. Structure, thermodynamics and cooperativity of glucocorticoid receptor DNA-DNA-binding domain in complex with different response elements: molecular dynamics and free energy perturbation study. J. Mol. Biol. 253:453–472. [DOI] [PubMed] [Google Scholar]
- Essmann, U., L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- Ferguson-Miller, S., and G. T. Babcock. 1996. Heme-copper terminal oxidases. Chem. Rev. 96:2889–2908. [DOI] [PubMed] [Google Scholar]
- Fetter, J. R., J. Quian, J. Shapleigh, J. W. Thomas, J. A. Garcia-Horsman, E. Schmidt, J. Hosler, G. T. Babcock, R. B. Gennis, and S. Ferguson-Miller. 1995. Possible proton relay pathways in cytochrome c oxidase. Proc. Natl. Acad. Sci. USA. 92:1604–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Horsman, J. A., A. Puustinen, R. B. Gennis, and M. Wikström. 1995. Proton transfer in cytochrome bo3 ubiquinol oxidase of Escherichia coli: second-site mutations in subunit I that restore proton pumping in the mutant Asp-135→Asn. Biochemistry. 34:4428–4433. [DOI] [PubMed] [Google Scholar]
- Garcia, A. E., G. Hummer, and D. M. Soumpasis. 1996. Theoretical description of biomolecular hydration. Application to a-DNA. Basic Life Sci. 64:299–308. [DOI] [PubMed] [Google Scholar]
- Garcia, A. E., and G. Hummer. 2000. Water penetration and escape in proteins. Proteins. 38:261–272. [PubMed] [Google Scholar]
- Gennis, R. B. 1989. Biomembranes: Biomolecular Structure and Functions. Springer-Verlag, New-York.
- Goodford, P. J. 1985. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 28:849–857. [DOI] [PubMed] [Google Scholar]
- Hayashi, S., E. Tajkhorshid, and K. Schulten. 2002. Structural changes during the formation of early intermediates in the bacteriorhodopsin photocycle. Biophys. J. 83:1281–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms, V., and R. C. Wade. 1995. Thermodynamics of water mediating protein-ligand interactions in cytochrome P450cam: a molecular dynamics study. Biophys. J. 69:810–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms, V., and R. C. Wade. 1998. Hydration energy landscape of the active site cavity in cytochrome P450cam. Proteins. 32:381–396. [PubMed] [Google Scholar]
- Henchman, R. H., and J. A. McCammon. 2002. Structural and dynamic properties of water around acetylcholinesterase. Protein Sci. 11:2080–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker, I., and K. Schulten. 1998. Oxygen and proton pathways in cytochrome c oxidase. Proteins. 30:100–107. [PubMed] [Google Scholar]
- Hummer, G., J. C. Rasaiah, and J. P. Noworyta. 2001. Water conduction through the hydrophobic channel of a carbon nanotube. Nature. 414:188–190. [DOI] [PubMed] [Google Scholar]
- Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- Iwata, S., C. Ostermeier, B. Ludwig, and H. Michel. 1995. Structure at 2.8 Å resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature. 376:660–669. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- Kannt, A., C. R. D. Lancaster, and H. Michel. 1998. The coupling of electron transfer and proton translocation: electrostatic calculations on Paracoccus denitrificans cytochrome c oxidase. Biophys. J. 74:708–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinov, A. A., S. Siletsky, D. Mitchell, A. Kaulen, and R. B. Gennis. 1997. The roles of the two proton input channels in cytochrome c oxidase from Rhodobacter spheroides probed by the effects of site-directed mutations on time-resolved electrogenic intraprotein proton transfer. Proc. Natl. Acad. Sci. USA. 94:9085–9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster, C. R. D., and H. Michel. 1997. The coupling of light-induced electron transfer and proton uptake as derived from crystal structures of reaction centres from Rhodopseudomonas viridis modified at the binding site of the secondary quinone, QB. Structure. 5:1339–1359. [DOI] [PubMed] [Google Scholar]
- Le Coutre, J., J. Tittor, D. Oesterhelt, and K. Gerwert. 1995. Experimental evidence for hydrogen-bonded network proton transfer in bacteriorhodopsin shown by Fourier-transform infrared spectroscopy using azide as catalyst. Proc. Natl. Acad. Sci. USA. 92:4962–4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luecke, H. 2000. Atomic resolution structures of bacteriorhodopsin photocycle intermediates: the role of discrete water molecules in the function of this light-driven ion pump. Biochim. Biophys. Acta. 1460:133–156. [DOI] [PubMed] [Google Scholar]
- MacKerell, A., Jr., J. Wiorkiewicz-Kuczera, and M. Karplus. 1995. An all-atom empirical energy function for the simulation of nucleic acids. J. Am. Chem. Soc. 117:11946–11975. [Google Scholar]
- MacKerell, A. D., Jr., D. Bashford, M. Bellot, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, D. Joseph-McCarthy, S. Ha, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, and M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- Michel, H. 1998. The mechanism of proton pumping by cytochrome c oxidase. Proc. Natl. Acad. Sci. USA. 95:12819–12824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills, D. A., and S. Ferguson-Miller. 1998. Proton uptake and release in cytochrome c oxidase: separate pathways in time and space? Biochim. Biophys. Acta. 1365:46–52. [DOI] [PubMed] [Google Scholar]
- Mills, D. A., and S. Ferguson-Miller. 2003. Understanding the mechanism of proton movement linked to oxygen reduction in cytochrome c oxidase: lessons from other proteins. FEBS Lett. 545:47–51. [DOI] [PubMed] [Google Scholar]
- Mills, R. 1973. Self-diffusion in normal and heavy water in the range 1–45°. J. Phys. Chem. 77:685–688. [Google Scholar]
- Nagle, J. F., and H. J. Morowitz. 1978. Molecular mechanism for proton transport in membranes. Proc. Natl. Acad. Sci. USA. 75:298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle, J. F. 1993. Area/lipid of bilayers from NMR. Biophys. J. 64:1476–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norberg, J., and L. Nilsson. 1994. Stacking-unstacking of the dinucleoside monophosphate guanylyl-3′,5′-uridine studied with molecular dynamics. Biophys. J. 67:812–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostermeier, C., A. Harrenga, U. Ermler, and H. Michel. 1997. Structure at 2.7 Å resolution of the Paracoccus denitrificans two-subunit cytochrome c oxidase complexed with an antibody FV fragment. Proc. Natl. Acad. Sci. USA. 94:10547–10553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor, R. W., R. M. Venable, and M. Karplus. 1991. Model for the structure of the lipid bilayers. Proc. Natl. Acad. Sci. USA. 88:892–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrache, H. I., A. Grossfield, K. R. MacKenzie, D. M. Engelman, and T. B. Woolf. 2000. Modulation of glycophorin A transmembrane helix interactions by lipid bilayers: molecular dynamics calculations. J. Mol. Biol. 302:727–746. [DOI] [PubMed] [Google Scholar]
- Pfitzner, U., K. Hoffmeier, A. Harrenga, A. Kannt, H. Michel, E. Bamberg, O. M. Richter, and B. Ludwig. 2000. Tracing the D-pathway in reconstituted site-directed mutants of cytochrome c oxidase from Paracoccus denitrificans. Biochemistry. 39:6756–6762. [DOI] [PubMed] [Google Scholar]
- Pomès, R., and B. Roux. 1996. Structure and dynamics of a proton wire: a theoretical study of H+ translocation along the single-file water chain in the gramicidin A channel. Biophys. J. 71:19–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomès, R., G. Hummer, and M. Wikström. 1998. Structure and dynamics of a proton shuttle in cytochrome c oxidase. Biochim. Biophys. Acta. 1365:255–260. [Google Scholar]
- Puustinen, A., and M. Wikström. 1999. Proton exit from the heme-copper oxidase of Escherichia coli. Proc. Natl. Acad. Sci. USA. 96:35–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabenstein, B., G. M. Ullmann, and E.-W. Knapp. 1998. Calculation of protonation patterns in proteins with structural relaxation and molecular ensembles—application to the photosynthetic reaction center. Eur. Biophys. J. 27:626–637. [Google Scholar]
- Riistama, S., G. Hummer, A. Puustinen, R. B. Dyer, W. H. Woodruff, and M. Wikström. 1997. Bound water in the protein translocation mechanism of the heme-copper oxidases. FEBS Lett. 414:275–280. [DOI] [PubMed] [Google Scholar]
- Ryckaert, J. P., G. Ciccotti, and H. J. C. Berendsen. 1977. Numerical integration of the Cartesian equation of motions of a system with constraints: molecular dynamics of n-alkanes. J. Comp. Chem. 23:327–341. [Google Scholar]
- Savage, H. 1986. Water structure in vitamin B12 coenzyme crystals. I. Analysis of the neutron and x-ray solvent densities. Biophys. J. 50:947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, L., D. Bassolino, and T. Stouch. 1997. Transmembrane helix structure, dynamics, and interactions: multi-nanosecond molecular dynamics simulations. Biophys. J. 73:3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlenkrich, M. J., J. Brickmann, Jr., A. D. MacKerell, and M. Karplus. 1996. An empirical potential energy function for phospholipids: criteria for parameter optimization and applications. In Biological Membranes. A Molecular Perspective from Computation and Experiment. K. M. Merz and B. Roux, editors. Birkhäuser, Boston, MA. 31–81
- Svensson-Ek, M., J. Abramson, G. Larsson, S. Tornroth, P. Brzezinski, and S. Iwata. 2002. The x-ray crystal structures of wild-type and EQ(I-286) mutant cytochrome c oxidases from Rhodobacter sphaeroides. J. Mol. Biol. 321:329–339. [DOI] [PubMed] [Google Scholar]
- Tang, Y., and L. Nilsson. 1999. Molecular dynamics simulations of the complex between human U1A protein and hairpin II of U1 small nuclear RNA and of free RNA in solution. Biophys. J. 77:1284–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, J. W., A. Puustinen, J. O. Alben, R. B. Gennis, and M. Wikström. 1993. Substitution of asparagine for aspartate-135 in subunit I of the cytochrome bo ubiquinol oxidase of Escherichia coli eliminates proton-pumping activity. Biochemistry. 32:10923–10928. [DOI] [PubMed] [Google Scholar]
- Tieleman, D. P., and H. J. C. Berendsen. 1998. A molecular dynamics study of the pores formed by Escherichia coli OmpF porin in a fully hydrated palmitoyloleoylphosphatidylcholine bilayer. Biophys. J. 74:2786–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tshukihara, T., H. Aoyama, E. Yamashita, T. Tomizaki, H. Yamaguchi, K. Shinzawa-Itoh, R. Nakashima, R. Yaono, and S. Yoshikawa. 1996. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 Å. Science. 272:1136–1144. [DOI] [PubMed] [Google Scholar]
- Venable, R. M., Y. Zhang, B. J. Hardy, and R. W. Pastor. 1993. Molecular dynamics simulations of a lipid bilayer and of hexadecane: an investigation of membrane fluidity. Science. 262:223–226. [DOI] [PubMed] [Google Scholar]
- Wade, R. C. 1990. Solvation of the active site of cytochrome P450 cam. J. Comput. Aided Mol. Des. 4:199–204. [DOI] [PubMed] [Google Scholar]
- Wade, R. C., and P. J. Goodford. 1993. Further development of hydrogen bond functions for use in determining energetically favorable binding sites on molecules of known structure. 2. Ligand probe groups with the ability to form more than two hydrogen bonds. J. Med. Chem. 36:148–156. [DOI] [PubMed] [Google Scholar]
- Wikström, M. K. F. 1977. Proton pump coupled to cytochrome c oxidase in mitochondria. Nature. 266:271–273. [DOI] [PubMed] [Google Scholar]
- Wikström, M., M. I. Verkhovsky, and G. Hummer. 2003. Water-gated mechanism of proton translocation by cytochrome c oxidase. Biochim. Biophys. Acta. 1604:61–65. [DOI] [PubMed] [Google Scholar]
- Woolf, T. B., and B. Roux. 1994. Molecular dynamics simulation of the gramicidin channel in a phospholipid bilayer. Proc. Natl. Acad. Sci. USA. 91:11631–11635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf, T. B., and B. Roux. 1996. Structure, energetics, and dynamics of lipid-protein interactions: a molecular dynamics study of the gramicidin A channel in a DMPC bilayer. Proteins. 24:92–114. [DOI] [PubMed] [Google Scholar]
- Zaslavsky, D., and R. B. Gennis. 2000. Proton pumping by cytochrome oxidase: progress, problems and postulates. Biochim. Biophys. Acta. 1458:164–179. [DOI] [PubMed] [Google Scholar]
- Zheng, X., D. M. Medvedev, J. Swanson, and A. A. Stuchebrukhov. 2003. Computer simulation of water in cytochrome c oxidase. Biochim. Biophys. Acta. 1557:99–107. [DOI] [PubMed] [Google Scholar]
- Zhang, L., and J. Hermans. 1996. Hydrophilicity of cavities in proteins. Proteins. 24:433–438. [DOI] [PubMed] [Google Scholar]