

Abstract
Motions through the energy landscape of proteins lead to biological function. At temperatures below a dynamical transition (150–250 K), some of these motions are arrested and the activity of some proteins ceases. Here, we introduce the technique of temperature-derivative fluorescence microspectrophotometry to investigate the dynamical behavior of single protein crystals. The observation of glass transitions in thin films of water/glycerol mixtures allowed us to demonstrate the potential of the technique. Then, protein crystals were investigated, after soaking the samples in a small amount of fluorescein. If the fluorophore resides within the crystal channels, temperature-dependent changes in solvent dynamics can be monitored. Alternatively, if the fluorophore binds to the protein, local dynamical transitions within the biomolecule can be probed directly. A clear dynamical transition was observed at 175 K in the active site of crystalline human butyrylcholinesterase. The results suggest that the dynamics of crystalline proteins is strongly dependent on solvent composition and confinement in the crystal channels. Beyond applications in the field of kinetic crystallography, the highly sensitive temperature-derivative fluorescence microspectrophotometry technique opens the way to many studies on the dynamics of biological nanosamples.
INTRODUCTION
Intramolecular motions are essential to the function of proteins. They are associated with transitions between substates representing local minima in the highly complex energy landscape of a protein (Frauenfelder et al., 1991). Motions occurring on the picosecond or slower timescales exhibit a so-called dynamical transition at a temperature within the range 150–250 K, below which they are “frozen out” (Parak et al., 1982; Doster et al., 1989; Ferrand et al., 1993; Réat et al., 1997; Fitter, 1999; Tsai et al., 2000; Paciaroni et al., 2002; Lee and Wand, 2001; Vitkup et al., 2000; Hayward and Smith, 2002). Abrupt changes in the amplitude and timescales of atomic fluctuations at the dynamical transition have been shown to mark the onset of activity for a number of proteins (Ding et al., 1994; Ferrand et al., 1993; Ostermann et al., 2000; Heyes et al., 2003). Besides its fundamental importance as an essential feature of protein dynamics, the dynamical transition is of considerable practical interest in structural cryoenzymology. In particular, it can be employed in temperature-controlled crystallographic experiments to trigger enzymatic activity within the crystal, allowing the visualization of conformational changes at the atomic level (Rasmussen et al., 1992; Ostermann et al., 2000; Royant et al., 2000; Schlichting et al., 2000a,b; Weik et al., 2001a; Ursby et al., 2002). However, molecular dynamics in protein crystals are affected by packing forces within the lattice and by the solvent arrangement, composition, and content (Doster et al., 1986; Miyazaki et al., 2000; Weik et al., 2001b). Thus, they may differ significantly from those measured in dilute solutions and require dedicated techniques to observe them.
Here we introduce the technique of temperature-derivative fluorescence microspectrophotometry (TDFM) as a powerful tool to investigate dynamical changes occurring within biological nanosamples such as single protein crystals. TDFM extends the panel of methods that have been used to monitor the dynamics of crystalline proteins, such as solid state NMR (Usha and Wittebort, 1989), Mössbauer spectroscopy (Parak, 1986; Chong et al., 2001), calorimetry (Doster et al., 1986; Miyazaki et al., 1993, 2000), mechanical experiments (Morozov and Gevorkian, 1985), and crystallography (Tilton et al., 1992; Teeter et al., 2001).
The difference in wavelength between absorbed and emitted photons (the Stokes shift) in a steady-state fluorescence emission spectrum is dependent on the motions of the molecules surrounding the fluorophore. If the corresponding dipolar relaxation time becomes comparable to the fluorescence lifetime, abrupt changes in Stokes shifts may appear in a temperature-dependent experiment. Since fluorescence lifetimes are typically of the order of nanoseconds, fluorescence spectroscopy is sensitive to molecular dynamics occurring on that timescale. Based on this principle, the dynamics of noncrystalline biological samples have been investigated (Vincent et al., 2000).
To validate the TDFM technique, we measured the glass transition and crystallization temperatures of thin films of water-glycerol mixtures to which a small amount of fluorescein was added. The study was then extended to protein crystals soaked in fluorescein. Depending on whether the fluorophore simply resides within the solvent channels or binds to the protein molecules, either solvent glass transitions or protein dynamical transitions, respectively, could be monitored. Crystals of three different proteins, TcAChE, HuBChE, and the well-known HEWL were employed. TcAChE belongs to the family of acetylcholinesterases, which terminate impulse transmission at cholinergic synapses by hydrolyzing the neurotransmitter acetylcholine at a diffusion-limited rate. The physiological role of the structurally similar human butyrylcholinesterase is unknown, yet it has been shown that the protein is capable of catalyzing similar hydrolysis reactions as TcAChE does. The secondary structure of all three proteins comprises α-helices as well as β-sheets.
MATERIALS AND METHODS
Sample preparation
Thin films of binary mixtures of water and glycerol (30–95% (v/v)) containing 0.3 mM fluorescein (Sigma Aldrich, St. Louis, MO; referred to as solution A when the glycerol content is 40%) were flash-cooled to 100 K using a cryoloop (Hampton Research, Laguna Niguel, CA) in the cryostream of a cooling device (600 series, Oxford Cryosystems, Oxford, UK).
Crystals of TcAChE (MW 65000, Sussman et al., 1991; Raves et al., 1997) were grown in space group P3121, using 32–34% (v/v) PEG 200 and 0.3 M MES at pH 6.0. These crystals contain 68% (v/v) solvent. They were soaked for 24 h in 34% (v/v) PEG 200, 0.3 M MES pH 6.0, 0.3% DMSO, 3% ethanol, and 0.3 mM fluorescein. Subsequently, they were transferred for 20 s into a drop containing 34% (v/v) PEG 200 and 0.3 M MES pH 6.0 (referred to as cryosolution B), and flash-cooled to 100 K in a cryoloop.
Crystals of HEWL (MW 14000) were grown in space group P43212, using 0.8 M NaCl and 0.1 M sodium acetate at pH 4.5. These crystals contain 39% (v/v) solvent. They were soaked for 72 h in 1 M NaCl, 0.1 M sodium acetate at pH 4.5, and 0.3 mM fluorescein. Subsequently, they were transferred for 10 min into a drop of cryosolution containing 1 M NaCl, 0.1 M sodium acetate at pH 4.5, and 30% (v/v) glycerol (cryosolution C), and flash-cooled to 100 K.
Crystals of HuBChE (MW of the recombinant protein is 70000) were grown in space group I422, using 2.1 M ammonium sulfate, 0.1 M MES pH 6.5, and 1 mM butyrylcholine (Nachon et al., 2002). These crystals contain 60% (v/v) solvent. They were soaked for 96 h in a solution containing 2.5 M ammonium sulfate, 0.1 M MES pH 6.5, and 0.3 mM fluorescein. Subsequently, they were transferred for 10 min into a drop of cryosolution containing 2.5 M ammonium sulfate, 0.1 M MES pH 6.5, and 20% (v/v) glycerol (cryosolution D), and flash-cooled to 100 K.
Transfer to cryosolution B, C, or D allowed the removal of the fluorophore from the solvent surrounding crystals of TcAChE, HEWL, and HuBChE, respectively.
Temperature derivative fluorescence microspectrophotometry
Fluorescence emission spectra were recorded with a CCD-based microspectrophotometer (Figs. 1 and 2) dedicated to the study of protein crystals at cryotemperatures (Bourgeois et al., 2002). Fluorescence excitation was provided at 455 nm by an argon ion laser (Melles Griot, Carlsbad, CA). Short excitation pulses (10–30 ms) were delivered at a frequency of 0.1 Hz to minimize photobleaching. Steady-state single-shot spectra were collected at this frequency throughout the application of a temperature profile consisting of a heating phase from 100 to 220 K at a rate of 180 Kh−1 and a 10 min plateau at 220 K. Temperatures corresponding to each spectrum were recorded online via a Labview-based graphical interface (National Instruments, Austin, TX) controlling the synchronization of the experiment. The center of mass of emission spectra, which is closely linked to the Stokes shift, was calculated as a function of temperature with the software IDL (Research Systems, Boulder, CO). Temperature-dependent shifts in pH (Yguerabide et al., 1994) may slightly change the ratio between the monoanionic and the dianionic forms of fluorescein, possibly leading to an additional small shift in the center of mass of the total (monoanionic and dianionic) fluorescence spectrum. To minimize this potential bias, the centers of mass were calculated within a fixed wavelength range delimiting the emission peak from the dianionic form of fluorescein only, as recorded at 100 K.
FIGURE 1.

Schematic view (a) and picture (b) of the microspectrophotometer setup. Three mirror objectives (in orange) focus the ultraviolet-visible light beam onto a 50 μm diameter spot. The sample mounted in a nylon loop is cooled by a cryostream system delivering cooled gaseous nitrogen. The sample is positioned at the intersecting focal points of the mirror objectives with a one-circle goniometer and can be visually inspected with a microscope.
FIGURE 2.

Photo of a flash-cooled HEWL crystal (300 μm × 300 μm) soaked in fluorescein and mounted in a cryoloop at 100 K. Fluorescence is observed upon continuous laser illumination with blue light. Only the crystal, not the vitreous cryosolvent around it, contains fluorescein, which is at the origin of the green emitted light.
The concentration of fluorescein was chosen so that the absorbance of typical loop-mounted samples did not exceed 0.3 OD at the absorption maximum, so as to probe the sample homogeneously and minimize inner filtering effects. However, in the case of HuBChE, the affinity of fluorescein for the enzyme considerably increased the concentration of the fluorophore within the sample whose OD at the peak absorbance exceeded 2.0. As a consequence, only a shallow volume of HuBChE crystals could be probed.
RESULTS AND DISCUSSION
Amorphous mixtures of glycerol, water, and fluorescein
Steady-state fluorescence emission spectra at 100 and 220 K from a flash-cooled aqueous solution containing 40% (v/v) glycerol and 0.3 mM fluorescein (solution A, see Materials and Methods) are shown in Fig. 3. Upon raising the temperature, the peak maximum undergoes a red shift from 500 to 510 nm and the spectrum broadens. Fluorescence from solution A was continuously monitored during heating from 100 to 220 K. The center of mass of the emission spectrum from fluorescein in the dianionic state is shown as a function of temperature in Fig. 4. Three main temperature windows can be delimited, i.e., 100–158 K, 158–179 K, and 179–220 K. In the range 100–158 K, a continuous red shift is observed and the sample remains macroscopically transparent, as assessed by visual inspection (Fig. 4, inset a). At 158 K, a transient blue shift sets in, followed by a drastic red shift and the sample becomes slightly opaque (Fig. 4, inset b). At 179 K, a second blue shift is observed and the sample rapidly becomes opaque (Fig. 4, inset c). Above 192 K, the fluorescence spectrum is red shifted again, and from 207 K the rate of this red shift decreases markedly.
FIGURE 3.

Fluorescence emission spectra of a flash-cooled film of an aqueous solution containing 40% (v/v) glycerol and 0.3 mM fluorescein at 100 K and after heating at 220 K. The main peak and the red-shifted shoulder in both spectra are due to fluorescence from the dianionic and monoanionic states of fluorescein, respectively. The signal at 455 nm originates from scattering of laser light that is more pronounced at 220 K due to crystalline ice formation in the sample.
FIGURE 4.

Transitions in a glycerol/water mixture as seen by TDFM. Center-of-mass of the dianionic peak (solid triangles) in the fluorescence spectrum of an amorphous film of an aqueous solution containing 40% (v/v) glycerol and 0.3 mM fluorescein as a function of experimental time. After flash cooling, the temperature (solid line) was raised from 100 K to 220 K. The insets show optical images of the fluorescent sample illuminated with blue light and recorded at different temperatures. The film of solution is held in a nylon loop of typical size 100 × 300 μm2. The probed volume is ≈1 nL.
The temperature, i.e., 158 K, at which the red shift deviates from a nearly linear behavior in Fig. 4, is very close to the glass transition temperature (160 K) of a similar sample, as determined by calorimetry (Harran, 1978). Corroborative evidence that this deviation from linearity indeed originates from a glass transition is provided by the excellent agreement (Fig. 5) of the corresponding temperatures with glass-transition temperatures of aqueous solutions of glycerol at various concentrations as determined by calorimetry (Harran, 1978).
FIGURE 5.

Glass transition temperatures for mixtures of glycerol and water as determined by calorimetry (open squares, Harran, 1978) and TDFM (solid squares) as a function of glycerol concentration.
We attribute the red shift of the emission spectrum in the temperature range between 165 and 179 K (Fig. 4) to increased solvent mobility above the glass transition. Since solvent molecules gain rotational freedom at the glass transition (Fisher and Devlin, 1995), the rate of dipolar relaxation of the excited fluorophores is increased, inducing a red shift of the emission spectrum according to:
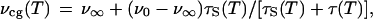 |
where νcg(T) is the wavelength corresponding to the center of mass of the emission spectrum observed at temperature T, ν0 and ν∞ are the centers of mass in the absence and after completion of solvent relaxation, respectively, and τS(T) and τ(T) are the lifetimes of the solvent relaxation processes and of the fluorescence, respectively (Lakowicz, 1999). Such a red shift has been observed in early studies of liquid-state dynamics (Yu et al., 1992).
The transient blue shift between 158 K and 165 K is consistent with a temporary increase in sample rigidity accompanying the process of enthalpy relaxation reported to occur near the glass transition of amorphous samples (Johari et al., 1991; Mayer, 1991). However, the magnitude of this blue shift varies between identically prepared samples, which may be related to small differences in sample volumes.
The onset of the second blue shift at 179 K (Fig. 4) reflects the formation of crystalline ice. This assignment is supported by the strong and rapid opacification of the sample at 180 K (Fig. 4, inset c) due to scattering from ice clusters that become comparable in size with the laser wavelength. Upon crystallization, dipolar relaxation is slowed down due to an increased rigidity of the solvent molecules, which shifts the emission maximum to the blue. However, crystalline ice also reabsorbs the emitted fluorescence light. This reabsorption is stronger at shorter wavelengths, resulting in a competing red shift of the observed emission maximum. This latter effect becomes dominant from 192 K onward, as confirmed by simulations (not shown). At 207 K, the decrease in slope and return to a more linear regime (Fig. 4) mark the completion of ice formation.
Vitrified bulk water has been argued to undergo a glass transition at 136 K upon warming (Johari et al., 1987). As the temperature is increased, it crystallizes at ∼150 K into cubic ice, which transforms into ordinary hexagonal ice at 186 K (McMillan and Los, 1965). Amorphous water in the temperature window between the glass transition and crystallization has been described as being “ultraviscous” (Mishima and Stanley, 1998), but the existence of this window is still controversial (Velikov et al., 2001). Our results on water-glycerol mixtures show a temperature window that corroborates the findings of Mishima and Stanley (1998). However, in our case, a possible “ultraviscous” phase seems to coexist with the presence of slowly growing, small ice crystals, as suggested by the slight opacity of the sample (Fig. 4, inset b) observed throughout the 158–179 K temperature window.
To determine the molecular relaxation time in the “ultraviscous” phase, the temperature of a flash-cooled solution A was increased from 100 to 170 K, held constant at 170 K for ∼16 h, and then increased further to 220 K (Fig. 6). Similarly to data shown in Fig. 4, a transient blue shift in the emission spectrum is observed between 160 K and 165 K, marking the occurrence of a glass transition. A red shift follows, which continues to develop while the temperature is maintained at 170 K, with an associated monoexponential relaxation time of 205 s. This relaxation time is comparable to the one attributed to molecular relaxation at the glass transition (100 s, Angell, 1995), although it may include a small contribution from slowly growing ice crystals. Further heating leads to massive crystallization, as monitored by the appearance of a characteristic blue shift at 177 K (Fig. 6 b).
FIGURE 6.

Molecular relaxation at the glass transition. Center-of-mass of the dianionic peak (solid triangles) in the fluorescence spectrum of a film of an amorphous solution containing 40% (v/v) glycerol and 0.3 mM fluorescein as a function of experimental time. The temperature profile is represented as a solid line. Data corresponding to different time windows are shown, i.e., between 0 min and 80 min (a) and between 980 min and 1010 min (b).
In summary, TDFM provides the glass transition temperature of flash-cooled water-glycerol mixtures as well as the crystallization temperature of the water fraction. Consequently, the temperature window in which water is likely to be ultraviscous (Mishima and Stanley, 1998) can be detected. In the following, we extend these studies to the case of protein crystals.
Solvent behavior in crystals of Torpedo californica acetylcholinesterase with large solvent channels
A single crystal of TcAChE, soaked in 0.3 mM fluorescein and cryoprotected with solution B (see Materials and Methods), was flash-cooled to 100 K. The optical densities at 490 nm and fluorescence Stokes shifts at 100 K of such crystals were comparable to those of a flash-cooled cryosolution B alone to which 0.3 mM fluorescein has been added. This indicates that the fluorophore did not specifically bind to TcAChE and resides in the crystal channels where it can probe dynamical changes of the solvent. Upon raising the temperature, two transitions are observed, at 158 K and 188 K (Fig. 7 a). We attribute the former to a glass transition of the crystal solvent and the latter to its massive crystallization. A comparative experiment performed on cryosolution B alone shows two characteristic transitions at 151 K and 172 K (Fig. 7 b).
FIGURE 7.

Transitions in a TcAChE crystal as seen by TDFM. Center-of-mass of the dianionic peak (solid triangles) in the fluorescence spectrum of fluorescein soaked into TcAChE crystals (a) and of fluorescein in the cryoprotective solution B of TcAChE crystals (b) as a function of experimental time. After flash cooling, the temperature (solid line) was raised from 100 to 220 K.
Given the large solvent channels in trigonal TcAChE crystals (65 Å diameter), the observation of a solvent glass transition at a temperature close to the one seen in bulk cryosolution is consistent with the idea that solvent in large channels behaves similarly to bulk solvent (Weik et al., 2001b). The temperatures of the glass transition measured from four similarly prepared crystals varied from 151 to 164 K, implicating that more than one sample should always be investigated by TDFM before drawing detailed conclusions. We attribute this variation to small differences in the initial flash-cooling process, in crystal size, crystal shape, or other parameters (Garman, 2003). Therefore, the 7 K difference between the glass transition temperatures in Fig. 7, a and b, may not be unambiguously attributed to confinement of the crystal solvent. Interestingly, x-ray diffraction experiments on an 80-fold increased timescale (several tens of hours) showed that crystalline ice in trigonal TcAChE crystals starts to form at ∼155 K (Weik et al., 2001b). This temperature is close to the glass transition temperature observed here (Fig. 7 a), suggesting that ice may form slowly as soon as the glass transition is passed, in line with the observations on flash-cooled mixtures of glycerol and water described above.
The temperature at which massive crystallization is observed by TDFM is expected to depend on the kinetics of this process relative to the timescale of the experiment. Here, crystallization of unconfined bulk solvent, starting at 172 K, is particularly rapid, occurring on a timescale of a few seconds (Fig. 7 b). On the contrary, the kinetics of ice formation within the crystal channels appears to be much slower (Fig. 7 a), which may explain the 16 K difference between the crystallization temperatures observed in Fig. 7, a and b.
In summary, TDFM allowed the detection of a glass transition and massive crystallization of solvent in a protein crystal with large channels. The results also suggest that confinement in such channels significantly slows down the kinetics of ice formation. In the following section, experiments on protein crystals with narrow channels are described in which the solvent neither undergoes a glass transition nor crystallizes.
Solvent behavior in crystals of hen egg white lysozyme with narrow solvent channels
A single crystal of HEWL, soaked in 0.3 mM fluorescein and cryoprotected with solution C (see Materials and Methods) was flash-cooled to 100 K. The temperature was subsequently raised from 100 to 220 K. Inspection of electron density maps in an x-ray crystallographic study of HEWL revealed that fluorescein does not bind to the enzyme (not shown). Again, the fluorophores reside in the solvent channels. The evolution of the Stokes shift is essentially featureless up to 190 K, where a small red shift appears (Fig. 8 a). Investigation of a second crystal led to the same result. In contrast, a similar experiment performed on cryosolution C alone to which 2.1 mM fluorescein was added reveals marked transitions at 160 and 177 K (Fig. 8 b), which are assigned to a glass transition and the onset of ice formation, respectively. Comparison of Fig. 8, a and b, suggests that a glass transition does not occur in the solvent channels of tetragonal crystals of HEWL. Furthermore, the transition at 190 K in Fig. 8 a originates from the massive formation of ice outside the crystal, in the cryosolution embedding it, as substantiated in the following. First, temperature derivative absorption spectra (not shown) revealed massive ice formation at 190 K, resulting in a significant reabsorption of the emitted light (see section about glycerol/water mixtures) at the origin of the observed red shift. Second, x-ray diffraction experiments based on cell volume measurements (Weik et al., 2001b) indicated that the solvent within HEWL crystals does not crystallize in the temperature range 100–220 K. Third, the TDFM profile from 190 K onward in Fig. 8, a and b, are similar, consistent with massive crystallization in the range 190–208 K in cryosolution C.
FIGURE 8.

Transitions in a HEWL crystal as seen by TDFM. Center-of-mass of the dianionic peak (solid triangles) in the fluorescence spectrum of fluorescein soaked into HEWL crystals (a) and of fluorescein in the cryoprotective solution C of HEWL crystals (b) as a function of experimental time. After flash cooling, the temperature (solid line) was raised from 100 to 220 K.
The absences of a glass transition and of ice formation in the solvent channels of HEWL crystals may be explained by the very small size of these channels (14 Å in diameter), resulting from the tight packing of the macromolecules in the lattice. A substantial fraction of water molecules are ordered in the crystal channels (>70% of them is required for monolayer coverage of the protein molecules (Usha et al., 1991)) and an extended three-dimensional hydrate lattice may not form. Viscous drag forces are generated, hindering unordered water molecules from freely reorienting as the temperature is elevated.
Protein dynamics are acknowledged to be considerably influenced by the surrounding solvent (Beece et al., 1980; Walser and van Gunsteren, 2001). It has been proposed that solvent glass transitions trigger dynamical transitions in proteins (Vitkup et al., 2000; Réat et al., 2000). Based on this assumption, our results suggest that a dynamical transition may not occur in tetragonal crystals of HEWL in the temperature range 100–220 K.
Interestingly, a glass transition at 150 K was observed in similar HEWL crystals by calorimetric measurements (Miyazaki et al., 2000). However, calorimetric glass transitions do not only relate to molecular dynamics on the nanosecond timescale, in contrast to the transitions probed here. The glass transition detected by calorimetry may thus concern faster motions that are not observable by fluorimetry. However, protein movements developing on the nanosecond timescale have been described as determinant in controlling biological activity (Brooks et al., 1988). Therefore, our findings indicate that some or all conformational changes associated with enzymatic activity of HEWL, and that are “slaved” to solvent dynamics (Iben et al., 1989), may not take place at cryotemperatures in tetragonal crystals. Nevertheless, other so-called “nonslaved” protein motions whose temperature dependence does not correlate with solvent fluctuations (Fenimore et al., 2002) may in principle still take place.
Dynamical transition in crystalline human butyrylcholinesterase
The structure of crystalline HuBChE has been solved recently (Nicolet et al., 2003). HuBChE crystals soaked in 0.3 mM fluorescein and cryoprotected with solution D (see Materials and Methods) were flash-cooled to 100 K. A crystallographic study revealed that fluorescein binds to the active site of the enzyme as evidenced by the inspection of electron density maps (not shown). This was confirmed by the observation of a shift of the fluorescence spectrum by ∼15 nm as compared to the spectrum of fluorescein in cryosolution D alone, and by the high optical density (OD > 2.0 at 490 nm) of representative crystals, suggestive of a concentration of fluorescein in the crystal similar to the HuBChE concentration (8.7 mM). The evolution of the fluorescence peak emission as a function of temperature (Fig. 9 a) is, therefore, a marker of the active site dynamics rather than of the solvent dynamics.
FIGURE 9.

Transitions in a HuBChE crystal as seen by TDFM. Center-of-mass of the dianionic peak (solid triangles) in the fluorescence spectrum of fluorescein bound to the active site of crystalline HuBChE (a) and of fluorescein in the cryoprotective solution D of HuBChE crystals (b) as a function of experimental time. After flash cooling, the temperature (solid line) was raised from 100 to 220 K. Oscillations seen around 200 K in b are attributed to the crystallization of ammonium sulfate, which has been identified visually.
Upon heating, the slope of the Stokes shift starts to increase significantly at 175 K. We assign this observation to the onset of a dynamical transition in the active site of HuBChE. This increase is consistent with the model of a higher flexibility of protein residues above the transition (Parak et al., 1982; Doster et al., 1989; Ferrand et al., 1993). The study of five similar crystals employing the same protocol showed that this transition varied from 175 to 183 K.
Remarkably, a glass transition is observed at 153 K in the corresponding cryosolution D alone to which 2.1 mM fluorescein was added, 22 K below the dynamical transition in the active site (Fig. 9 b). However, firm conclusions about the origin of this shift may not be drawn at this point. Given the moderately large channels in HuBChE crystals (42 Å in diameter), the crystal solvent is expected to undergo a glass transition upon heating, although being somewhat more confined than in the case of TcAChE. To investigate whether or not this transition directly triggers the dynamical transition observed in the active site (Fenimore et al., 2002), its temperature must be measured. To this end, new experiments can be envisaged where an irreversible HuBChE inhibitor prevents the fluorophore from binding to the active site, which would then only reside in the crystal channels.
Massive crystallization of water in cryosolution D and within or around HuBChE crystals occurs at ∼200 K (Fig. 9, a and b), as confirmed by visual inspection and by the appearance of strong ice rings in x-ray diffraction patterns (not shown). The progressive onset of a blue shift seen in Fig. 9, a and b, from ∼200 K onward is a manifestation of an increased rigidity of the fluorophore environment and correlates well with the idea that the kinetics of water crystallization within the crystal channels and within cryosolution D are much slower than in the case of TcAChE. In the latter case, a red shift owing to reabsorption of the emitted fluorescence light by crystalline ice masked a blue shift due to a crystallization-related increase in rigidity of the fluorophore's environment.
The evolution of the Stokes shift in Fig. 9 a defines a temperature window (175–200 K) between a dynamical transition of the protein and crystallization of its surrounding solvent. In this window, the protein gains conformational flexibility and may find itself in a partially or fully functional state although reaction rates are expected to be considerably lower than at physiological temperatures. The identification of this window is especially pertinent to the design of kinetic-crystallography experiments aimed at trapping functional intermediate states (Ursby et al., 2002). In the case of crystalline HuBChE, the following experimental strategy can be devised based on our TDFM results (Fig. 10). A photolabile precursor of the enzymatic product (caged compound), i.e., 1-(2-nitrophenyl)ethyl-caged choline (Peng et al., 1998), is soaked into HuBChE crystals at room temperature and inhibits the enzyme by binding to the active site. Subsequent to flash-cooling the crystal to 100 K, the caged compound is cleaved by laser cryophotolysis (Specht et al., 2001), liberating a choline molecule in the active site. The crystal is then warmed to a temperature above the dynamical transition of the active site (i.e., to above 175 K), yet still below the ice-formation temperature (200 K), to allow the build-up of a putative enzymatic reaction intermediate associated with choline exit from the active site. A second cooling process traps the intermediate state, which then can be studied by conventional monochromatic x-ray crystallography. This strategy is likely to be applicable to fast enzymes other than cholinesterases if the enzymatic reaction can be initiated at 100 K.
FIGURE 10.

Experimental strategy aimed at trapping putative intermediate states in HuBChE crystals. RT: room temperature.
CONCLUSION
We have demonstrated that temperature-derivative fluorescence microspectrophotometry is a simple and reliable tool to monitor dynamical changes in protein crystals in complement to other established methods. Depending on the size and composition of the solvent channels, a temperature window may be identified in which the protein molecules gain conformational flexibility by passing a dynamical transition while the solvent does not yet form crystalline ice. More fundamental aspects of protein dynamics may also be investigated such as gaining insight into the coupling between the dynamics of solvent and biological macromolecules. Although the use of the cheap and commonly available fluorophore fluorescein appears adequate in many cases, fluorescent amino acids, cofactors, ligands, or inhibitors can in principle also be used to investigate the local dynamics of active and/or binding sites. Furthermore, specific exogenous fluorophores targeted to residues of interest in engineered proteins (Boyd et al., 2000) may yield information on spatially resolved protein dynamics. Although we have developed the technique in the context of protein crystallography, a large number of applications in the fields of biophysics or physical chemistry can be envisaged due to the possibility of working with nanovolumes that can be readily flash-cooled.
Acknowledgments
We thank F. Nachon for providing us with purified HuBChE, I. Silman and L. Toker for purifying TcAChE, J. Colletier for crystallization of HuBChE and TcAChE, and the European Synchrotron Radiation Facility (Grenoble, France) for providing x-ray beam time. We are grateful to J. Zaccai for careful reading of the manuscript.
Abbreviations used: TDFM, temperature-derivative fluorescence microspectrophotometry; HEWL, hen egg white lysozyme; TcAChE, Torpedo californica acetylcholinesterase; HuBChE, human butyrylcholinesterase; PEG, polyethyleneglycol; MES, 2-morpholinoethanesulfonic acid; DMSO, dimethyl sulfoxide; MW, molecular weight; OD, optical density.
References
- Angell, C. A. 1995. Formation of glasses from liquids and biopolymers. Science. 267:1924–1935. [DOI] [PubMed] [Google Scholar]
- Beece, D., L. Eisenstein, H. Frauenfelder, D. Good, M. C. Marden, L. Reinisch, A. H. Reynolds, L. B. Sorensen, and K. T. Yue. 1980. Solvent viscosity and protein dynamics. Biochemistry. 19:5147–5157. [DOI] [PubMed] [Google Scholar]
- Bourgeois, D., X. Vernede, V. Adam, E. Fioravanti, and T. Ursby. 2002. A microspectrophotometer for UV-visible absorption and fluorescence studies of protein crystals. J. Appl. Crystallogr. 35:319–326. [Google Scholar]
- Boyd, A. E., A. B. Marnett, L. Wong, and P. Taylor. 2000. Probing the active center gorge of acetylcholinesterase by fluorophores linked to substituted cysteines. J. Biol. Chem. 275:22401–22408. [DOI] [PubMed] [Google Scholar]
- Brooks, C. L., M. Karplus, and B. M. Pettitt. 1988. Proteins. A Theoretical Perspective of Dynamics, Structure and Thermodynamics. I. Prigogine and S. Rice, editors. Wiley, New York. (A complete volume in Adv. Chem. Phys. 71:1–259.)
- Chong, S. H., Y. Joti, A. Kidera, N. Go, A. Ostermann, A. Gassmann, and F. Parak. 2001. Dynamical transition of myoglobin in a crystal: comparative studies of X-ray crystallography and Mossbauer spectroscopy. Eur. Biophys. J. 30:319–329. [DOI] [PubMed] [Google Scholar]
- Ding, X., B. F. Rasmussen, G. A. Petsko, and D. Ringe. 1994. Direct structural observation of an acyl-enzyme intermediate in the hydrolysis of an ester substrate by elastase. Biochemistry. 33:9285–9293. [PubMed] [Google Scholar]
- Doster, W., A. Bachleitner, R. Dunau, M. Hiebl, and E. Lüscher. 1986. Thermal properties of water in myoglobin crystals and solutions at subzero temperatures. Biophys. J. 50:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doster, W., S. Cusack, and W. Petry. 1989. Dynamical transition of myoglobin revealed by inelastic neutron scattering. Nature. 337:754–756. [DOI] [PubMed] [Google Scholar]
- Fenimore, P. W., H. Frauenfelder, B. H. McMahon, and F. G. Parak. 2002. Slaving: solvent fluctuations dominate protein dynamics and functions. Proc. Natl. Acad. Sci. USA. 99:16047–16051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrand, M., A. J. Dianoux, W. Petry, and G. Zaccai. 1993. Thermal motions and function of bacteriorhodopsin in purple membranes: effects of temperature and hydration studied by neutron scattering. Proc. Natl. Acad. Sci. USA. 90:9668–9672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, M., and J. P. Devlin. 1995. Defect activity in amorphous ice from isotopic exchange data: insight into the glass transition. J. Phys. Chem. 99:11584–11590. [Google Scholar]
- Fitter, J. 1999. The temperature dependence of internal molecular motions in hydrated and dry α-amylase: the role of hydration water in the dynamical transition of proteins. Biophys. J. 76:1034–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauenfelder, H., S. G. Sligar, and P. G. Wolynes. 1991. The energy landscapes and motions of proteins. Science. 254:1598–1603. [DOI] [PubMed] [Google Scholar]
- Garman, E. 2003. ‘Cool’ crystals: macromolecular cryocrystallography and radiation damage. Curr. Opin. Struct. Biol. 13:545–551. [DOI] [PubMed] [Google Scholar]
- Harran, D. 1978. Thermal behavior and glass transition curve of glycerol-water binary mixture. I. Bull. Soc. Chim. Fr. 1–2:40–44. [Google Scholar]
- Hayward, J. A., and J. C. Smith. 2002. Temperature dependence of protein dynamics: computer simulation analysis of neutron scattering properties. Biophys. J. 82:1216–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyes, D. J., A. V. Ruban, and C. N. Hunter. 2003. Protochlorophyllide oxidoreductase: “dark” reactions of a light-driven enzyme. Biochemistry. 42:523–528. [DOI] [PubMed] [Google Scholar]
- Iben, I. E., D. Braunstein, W. Doster, H. Frauenfelder, M. K. Hong, J. B. Johnson, S. Luck, P. Ormos, A. Schulte, P. J. Steinbach, A. H. Xie, and R. D. Young. 1989. Glassy behavior of a protein. Phys. Rev. Lett. 62:1916–1919. [DOI] [PubMed] [Google Scholar]
- Johari, G. P., A. Hallbrucker, and E. Mayer. 1987. The glass-liquid transition of hyperquenched water. Nature. 330:552–553. [Google Scholar]
- Johari, G. P., A. Hallbrucker, and E. Mayer. 1991. The dielectric behavior of vapor-deposited amorphous solid water and of its crystalline forms. J. Chem. Phys. 95:2955–2964. [Google Scholar]
- Lakowicz, J. R. 1999. Principles of Fluorescence Spectroscopy, 2nd ed. Kluwer Academic/Plenum Publishers, New York.
- Lee, A. L., and A. J. Wand. 2001. Microscopic origins of entropy, heat capacity and the glass transition in proteins. Nature. 411:501–504. [DOI] [PubMed] [Google Scholar]
- Mayer, E. 1991. Calorimetric glass transition in the amorphous forms of water: a comparison. J. Mol. Struct. 250:403–411. [Google Scholar]
- McMillan, J. A., and S. C. Los. 1965. Vitreous ice: irreversible transformations during warm-up. Nature. 206:806–807. [Google Scholar]
- Mishima, A., and H. E. Stanley. 1998. The relationship between liquid, supercooled and glassy water. Nature. 396:329–335. [Google Scholar]
- Miyazaki, Y., T. Matsuo, and H. Suga. 1993. Glass transition of myoglobin crystal. Chem. Phys. Lett. 213:303–307. [Google Scholar]
- Miyazaki, Y., T. Matsuo, and H. Suga. 2000. Low-temperature heat capacity and glassy behavior of lysozyme crystal. J. Phys. Chem. B. 104:8044–8052. [Google Scholar]
- Morozov, V. N., and S. G. Gevorkian. 1985. Low-temperature glass transition in proteins. Biopolymers. 24:1785–1799. [Google Scholar]
- Nachon, F., Y. Nicolet, N. Viguie, P. Masson, J. C. Fontecilla-Camps, and O. Lockridge. 2002. Engineering of a monomeric and low-glycosylated form of human butyrylcholinesterase: expression, purification, characterization and crystallization. Eur. J. Biochem. 269:630–637. [DOI] [PubMed] [Google Scholar]
- Nicolet, Y., O. Lockridge, P. Masson, J. C. Fontecilla-Camps, and F. Nachon. 2003. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 278:41141–41147. [DOI] [PubMed] [Google Scholar]
- Ostermann, A., R. Waschipky, F. G. Parak, and G. U. Nienhaus. 2000. Ligand binding and conformational motions in myoglobin. Nature. 404:205–208. [DOI] [PubMed] [Google Scholar]
- Paciaroni, A., S. Cinelli, and G. Onori. 2002. Effect of the environment on the protein dynamical transition: a neutron scattering study. Biophys. J. 83:1157–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parak, F., E. W. Knapp, and D. Kucheida. 1982. Protein dynamics. Mossbauer spectroscopy on deoxymyoglobin crystals. J. Mol. Biol. 161:177–194. [DOI] [PubMed] [Google Scholar]
- Parak, F. 1986. Correlation of protein dynamics with water mobility: Mossbauer spectroscopy and microwave absorption methods. Methods Enzymol. 127:196–206. [DOI] [PubMed] [Google Scholar]
- Peng, L., F. Nachon, J. Wirz, and M. Goeldner. 1998. 2-nitrobenzylarsonium compounds that photorelease heavy-atom cholinergic ligands for time-resolved crystallographic studies on cholinesterases. Angew. Chem. Int. Ed. 37:2691–2693. [DOI] [PubMed] [Google Scholar]
- Rasmussen, B. F., A. M. Stock, D. Ringe, and G. A. Petsko. 1992. Crystalline ribonuclease A loses function below the dynamical transition at 220 K. Nature. 357:423–424. [DOI] [PubMed] [Google Scholar]
- Raves, M. L., M. Harel, Y.-P. Pang, I. Silman, A. P. Kozikowski, and J. L. Sussman. 1997. 3D structure of acetylcholinesterase complexed with the nootropic alkaloid (-)-huperzine. Nat. Struct. Biol. 4:57–63. [DOI] [PubMed] [Google Scholar]
- Réat, V., R. Dunn, M. Ferrand, J. L. Finney, R. M. Daniel, and J. C. Smith. 2000. Solvent dependence of dynamic transitions in protein solutions. Proc. Natl. Acad. Sci. USA. 97:9961–9966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Réat, V., G. Zaccai, M. Ferrand, and C. Pfister. 1997. Functional dynamics in purple membrane. In Biological Macromolecular Dynamics. S. Cusack, H. Büttner, M. Ferrand, P. Langan, and P. Timmins, editors. Adenine, Guilderland, NY. 117–122.
- Royant, A., K. Edman, T. Ursby, E. Pebay-Peyroula, E. M. Landau, and R. Neutze. 2000. Helix deformation is coupled to vectorial proton transport in the photocycle of bacteriorhodopsin. Nature. 406:645–648. [DOI] [PubMed] [Google Scholar]
- Schlichting, I., J. Berendzen, K. Chu, A. M. Stock, S. A. Maves, D. E. Benson, R. M. Sweet, D. Ringe, G. A. Petsko, and S. G. Sligar. 2000a. The catalytic pathway of cytochrome p450cam at atomic resolution. Science. 287:1615–1622. [DOI] [PubMed] [Google Scholar]
- Schlichting, I., K. Chu, J. Berendzen, A. M. Stock, S. A. Maves, D. E. Benson, R. M. Sweet, D. Ringe, G. A. Petsko, and S. G. Sligar. 2000b. Trapping intermediates in the crystal: ligand binding to myoglobin. Curr. Opin. Struct. Biol. 10:744–752. [DOI] [PubMed] [Google Scholar]
- Specht, A., T. Ursby, M. Weik, L. Peng, J. Kroon, D. Bourgeois, and M. Goeldner. 2001. Cryophotolysis of ortho-nitrobenzyl derivatives of enzyme ligands for the potential kinetic crystallography of macromolecules. Chembiochem. 2:845–848. [DOI] [PubMed] [Google Scholar]
- Sussman, J. L., M. Harel, F. Frolow, C. Oefner, A. Goldman, L. Toker, and I. Silman. 1991. Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine-binding protein. Science. 253:872–879. [DOI] [PubMed] [Google Scholar]
- Teeter, M. M., A. Yamano, B. Stec, and U. Mohanty. 2001. On the nature of a glassy state of matter in a hydrated protein: relation to protein function. Proc. Natl. Acad. Sci. USA. 98:11242–11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilton, R. F., Jr., J. C. Dewan, and G. A. Petsko. 1992. Effects of temperature on protein structure and dynamics: X-ray crystallographic studies of the protein ribonuclease-A at nine different temperatures from 98 to 320 K. Biochemistry. 31:2469–2481. [DOI] [PubMed] [Google Scholar]
- Tsai, A. M., D. A. Neumann, and L. N. Bell. 2000. Molecular dynamics of solid-state lysozyme as affected by glycerol and water: a neutron scattering study. Biophys. J. 79:2728–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursby, T., M. Weik, E. Fioravanti, M. Delarue, M. Goeldner, and D. Bourgeois. 2002. Cryophotolysis of caged compounds: a technique for trapping intermediate states in protein crystals. Acta Crystallogr. D Biol. Crystallogr. 58:607–614. [DOI] [PubMed] [Google Scholar]
- Usha, M. G., J. Speyer, and R. J. Wittebort. 1991. Dynamics of the hydrate and amide groups of crystalline ribonuclease and lysozyme. Chem. Phys. 158:487–500. [Google Scholar]
- Usha, M. G., and R. J. Wittebort. 1989. Orientational ordering and dynamics of the hydrate and exchangeable hydrogen atoms in crystalline crambin. J. Mol. Biol. 208:669–678. [DOI] [PubMed] [Google Scholar]
- Velikov, V., S. Borick, and C. A. Angell. 2001. The glass transition of water, based on hyperquenching experiments. Science. 294:2335–2338. [DOI] [PubMed] [Google Scholar]
- Vincent, M., A.-M. Gilles, I. M. Li de la Sierra, P. Briozzo, O. Bârzu, and J. Gallay. 2000. Nanosecond fluorescence dynamic stokes shift of tryptophan in a protein matrix. J. Phys. Chem. B. 104:11286–11295. [Google Scholar]
- Vitkup, D., D. Ringe, G. A. Petsko, and M. Karplus. 2000. Solvent mobility and the protein ‘glass’ transition. Nat. Struct. Biol. 7:34–38. [DOI] [PubMed] [Google Scholar]
- Walser, R., and W. F. van Gunsteren. 2001. Viscosity dependence of protein dynamics. Proteins. 42:414–421. [PubMed] [Google Scholar]
- Weik, M., R. B. Ravelli, I. Silman, J. L. Sussman, P. Gros, and J. Kroon. 2001a. Specific protein dynamics near the solvent glass transition assayed by radiation-induced structural changes. Protein Sci. 10:1953–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weik, M., G. Kryger, A. M. Schreurs, B. Bouma, I. Silman, J. L. Sussman, P. Gros, and J. Kroon. 2001b. Solvent behaviour in flash-cooled protein crystals at cryogenic temperatures. Acta Crystallogr. D Biol. Crystallogr. 57:566–573. [DOI] [PubMed] [Google Scholar]
- Yguerabide, J., E. Talavera, J. M. Alvarez, and B. Quintero. 1994. Steady-state fluorescence method for evaluating excited state proton reactions: application to fluorescein. Photochem. Photobiol. 60:435–441. [Google Scholar]
- Yu, J., P. Earvolino, and M. Berg. 1992. Solvent-electronic state interactions measured from the glassy to the liquid state. II. Fluorescence line narrowing spectroscopy in glycerol. J. Chem. Phys. 96:8750–8756. [Google Scholar]