

Abstract
The function of membrane proteins often depends on the proteins' interaction with their lipid environment, spectacularly so in the case of mechanosensitive channels, which are gated through tension mediated by the surrounding lipids. Lipid bilayer tension is distributed quite inhomogeneously, but neither the scale at which relevant variation takes place nor the effect of varying lipid composition or tension has yet been investigated in atomic detail. We calculated lateral pressure profile distributions in lipid bilayers of various composition from all-atom molecular dynamics simulations totaling 110.5 ns in length. Reproducible pressure profile features at the 1 Å length scale were determined. Lipids with phosphatidylcholine headgroups were found to shift the lateral pressure out of the hydrophobic core and into the headgroup region by an amount that is independent of area per lipid. POPE bilayers simulated at areas smaller than optimal exerted dramatically higher lateral pressure in a narrow region at the start of the aliphatic chain. Stretching of POPC bilayers increased tension predominantly in the same region. A simple geometric analysis for the gating of the mechanosensitive channel MscL suggests that pressure profiles affect its gating through the second moment of the profile in a tension-independent manner.
INTRODUCTION
Far from playing an inert role as a mere divider between parts of the cell, lipid bilayers are an active, tightly regulated cellular component whose physical properties are critical for the proper function of the membrane proteins contained within them. Rhodopsin, which is picky about the lipid composition of its environment, can be fooled into functioning in a bilayer containing none of the lipids native to the rod outer segment where rhodopsin is found (Botelho et al., 2002); the alien membrane must only have the correct balance of pressures in the headgroup and tail region. Ion channels have played a central role as reporters of membrane properties, most notably gramicidin A (Elliot et al., 1983; Huang, 1986; Lundbaek and Andersen, 1994), a small dimeric channel whose kinetics and stability have been shown to be sensitive to membrane thickness, tension, and monolayer intrinsic curvature. Mechanosensitive (MS) channels play important biological roles in nearly all forms of life, from bacteria to eukaryotes (Sukharev et al., 1997; Hamill and Martinac, 2001); the mechanism of MS channel response to tension has, therefore, been studied extensively. Bacterial MS channels such as MscL can be gated solely by membrane tension in a patch-clamp apparatus (Sukharev et al., 1994). However, incorporation of MscL in thin bilayers, or the introduction of cone-shaped lysophospholipids into one of the monolayers, appears to lower the tension threshold of MscL enough in some cases to open the channel spontaneously (Perozo et al., 2002b; Martinac et al., 1990).
Efforts to study the effect of membrane strain on protein function has fallen into two broad categories. Both sets of approaches attempt to calculate the change in free energy due to protein-membrane interactions associated with the conformational change of a membrane protein. In the first category are calculations of the energetic cost of bilayer deformation, given a specified change in the shape of a membrane inclusion, such as a membrane protein (Dan et al., 1993; Dan and Safran, 1998; Nielsen et al., 1998; Nielsen and Andersen, 2000). In this approach, the shape of the protein is treated as one boundary condition for the solution of the shape of the membrane (the other usually being the assumption of an unperturbed bilayer sufficiently far from the protein), and the membrane itself is treated using continuum mechanics methods. In the second category are approaches that seek to calculate the inhomogeneous lateral pressure within a membrane to find the work done on the bilayer by a protein in the course of its conformational change. This approach has been extensively employed by Cantor (1997, 1999). The inhomogeneous lateral pressure profile can affect the equilibrium distribution of conformations of membrane proteins if the difference in cross-sectional area between two conformations varies with depth; for example, a conical expansion, but not a cylindrical expansion, would be affected by the lateral pressure profile. In fact, even nonmechanosensitive channels can be sensitive to changes in membrane composition that alter the distribution of lateral pressures (Lundbaek and Andersen, 1994; Casado and Ascher, 1998; Botelho et al., 2002; Bezrukov, 2000).
The pressure profile in bilayers arises due to the amphipathic nature of the lipids composing it: the hydrophilic headgroups are squeezed together to prevent exposure of the hydrophobic tails to solvent, while maintaining a nearly constant volume due to attractive dispersion forces and entropic repulsion between the lipid tails. A typical value for the surface tension in each monolayer is 50 dyn/cm. In a bilayer at equilibrium, the contracting influence of the headgroups must be balanced by the tendency of the lipid tails to maximize their entropy by occupying a greater volume, since the net tension in a free bilayer is zero. If the entropic tail pressure is evenly distributed throughout the 30 Å thick hydrophobic core of the bilayer, the lateral pressures in the core will be ∼350 atm. By the same token, since the contracting surface tension in each monolayer is localized in a thin, 5 Å slab around the boundary between hydrophilic and hydrophobic groups in each lipid, constricting lateral pressures on the order of 1000 atm can be expected here.
Stress distributions within the bilayer are difficult to measure directly, though some successes have been reported (Templer et al., 1998). Corresponding calculations can be obtained in various ways, namely through mean-field theories (Xiang and Anderson, 1994; Harries and Ben-Shaul, 1997), Monte Carlo models of simplified lipids (Harries and Ben-Shaul, 1997; Cantor, 1997), and molecular dynamics (MD) simulations of coarse-grained (Goetz and Lipowsky, 1998; Venturoli and Smit, 1999) and all-atom (Lindahl and Edholm, 2000) models of lipids. Mean-field approaches have the advantage of not being necessarily subject to the statistical errors due to inadequate sampling that are inherent in MD approaches. However, mean-field approaches, as well as lattice models employed by Cantor (1997), typically invoke uniform packing in the hydrophobic core, an assumption that is quite clearly invalid; electron density in the center of the bilayer is a factor of two or more smaller than at the edge (Nagle et al., 1996), and MD simulations report a similar distribution (Schneider and Feller, 2001; Heller et al., 1993). This inhomogeneous mass distribution is acknowledged to have potentially large effects on the calculated pressure distribution (Harries and Ben-Shaul, 1997).
MD simulations of lipid bilayers are still computationally demanding, and recently, coarse-grained models of lipids and water have been invoked to accelerate calculations (Goetz and Lipowsky, 1998; Shelley et al., 2001; Marrink and Mark, 2003). Goetz and Lipowsky (1998) found that a coarse-grained model of lipids composed simply of hydrophobic and hydrophilic beads interacting through van der Waals-type potentials can self-assemble and exhibit a nonuniform pressure profile distribution. Naturally, the most precise calculations are furnished by all-atom MD simulations. As a step to this goal, Lindahl and Edholm (2000) studied the pressure profile of dipalmitoylphosphatidylcholine using an all-atom representation of the lipid headgroup, and a united atom model for the CH2 groups of the aliphatic carbon tails (Bekker et al., 1993; Lindahl et al., 2001). The use of MD simulations for the study of membrane pressure profiles seems to be well justified, as long as 1), the membrane is self-assembled, so there is no need to apply restraints on the headgroups to impose a bilayer condition; 2), model parameters for the lipid tails have been well optimized to reproduce the observed lipid order parameters, and 3), it is straightforward to incorporate the effect of varying lipid composition into the calculation. The variations in the lateral pressure both within and just outside a lipid bilayer are by no means obvious; we are therefore led to use the most accurate model possible for the lipid and surrounding solvent to study the dependence of the pressure profile on the composition and physical state of the bilayer.
In this article we report the most extensive MD investigations to date of pressure profiles in lipid bilayers. We present results of all-atom MD simulations of bilayers of varying composition and tension. Since pressure profiles computed from simulations are subject to statistical errors caused by incomplete sampling, particular attention will be paid to the sources and magnitudes of errors in the calculation. We then examine how the calculated pressure profiles may be of use in understanding MscL gating.
METHODS
Membrane assembly and equilibration
Membranes studied in this article are summarized in Table 1 and include dilaurylphosphatidylethanolamine (DLPE), dilaurylphosphatidylcholine (DLPC), palmitoyloleoylphosphatidylethanolamine (POPE), and palmitoyloleoylphosphatidylcholine (POPC). All membranes were constructed using the structure-building tools of VMD (Humphrey et al., 1996). Initial coordinates for individual lipids were taken from the ideal molecular geometry, replicated to create the desired number of lipids, and arranged in a bilayer with the appropriate area per lipid and bilayer thickness. For the structures employed in simulations B–E, each lipid was also rotated by a random angle about its primary axis to reduce artificial long-range order. Periodic boundary conditions in all three dimensions were employed to minimize edge effects.
TABLE 1.
Summary of pressure profile simulations. Thickness is measured as the distance between the peaks of the mass distribution
| Simulation | Lipid | Number of lipids | Simulation time/ns (equilibration time) | Area/lipid/Å2 | Thickness/Å |
|---|---|---|---|---|---|
| A1 | DLPE | 200 | 8.7 (0.5) | 57.0 | 32.8 |
| A2 | DLPE | 200 | 7.8 (0.5) | 57.0 | 32.8 |
| B | DLPC | 200 | 6.5 (1.0) | 59.8 | 32.8 |
| C1 | POPE | 200 | 4.2 (4.2) | 48.5 | 48.3 |
| C2 | POPE | 200 | 4.4 (4.2) | 53.3 | 44.0 |
| C3 | POPE | 200 | 4.4 (4.4) | 59.1 | 40.2 |
| C4 | POPE | 200 | 4.4 (4.1) | 64.9 | 38.0 |
| D1 | POPC | 128 | 10.7 (2.0) | 64.0 | 39.2 |
| D2 | POPC | 128 | 10.2 (1.5) | 67.24 | 37.5 |
| D3 | POPC | 128 | 10.2 (1.5) | 70.56 | 35.8 |
| D4 | POPC | 128 | 10.8 (1.5) | 73.96 | 34.3 |
After this initial construction stage, the headgroup atoms were fixed and the rest of the atoms heated to 300 K for 500 ps to “melt” the aliphatic carbon tails. Fixing the headgroups prevented oppositely charged groups in neighboring lipids from bonding with each other, a condition that would prevent proper solvation of the interface. The system was then solvated using the SOLVATE program (Grubmüller, 1996) so that water molecules were distributed down to the hydrophobic core of the bilayer, at the position of the ester-oxygens. Water molecules were observed to retain this distribution even after multi-nanosecond equilibration times. Sufficient bulk water was included in each simulation to ensure full lipid hydration. Finally, the entire systems were minimized and heated to 310 K, except for simulations C1–C4 where a temperature of 320 K was employed.
All simulations were conducted using the parallel molecular dynamics program NAMD (Kalé et al., 1999). The CHARMM22 force field for lipids was employed (MacKerell Jr. et al., 1992, 1998; Schlenkrich et al., 1996), using a cutoff of 10 Å for van der Waals (VDW) interactions and the particle mesh Ewald method (PME) (Essmann et al., 1995) for full long-range electrostatics. The density of grid points for PME was at least 1/Å in all cases. Although the cutoff of 10 Å is somewhat smaller than the value of 12 Å more commonly employed, no difference in the lipid order parameter or membrane thickness was observed in a comparison of POPC simulated for 1 ns under otherwise identical conditions. Constant pressure simulations were performed using the hybrid Nose-Hoover Langevin piston method of NAMD (Feller et al., 1995), with a decay period of 200 fs and an oscillation time of 100 fs. Velocities were reassigned every picosecond to maintain constant temperature during equilibration. A time step of 1 fs was used, permitting a multiple time-stepping algorithm to be employed in which medium-range nonbonded interactions were computed every two time steps, whereas PME electrostatic forces were computed every four time steps. The simulations described here extended typically over a volume of ∼70 × 70 × 70 Å3, contained 30–40,000 atoms, and altogether lasted 110.5 ns. They were carried out at the Pittsburgh Supercomputer Center and required ∼400 CPU h/ns of simulation time on 64 1-GHz quad-processor Alpha CPUs.
Pressure profile calculations
All pressure profile calculations were performed at constant volume, using the unit cell dimensions at the end of the respective equilibration periods. A constant temperature ensemble was maintained with a weak Langevin coupling constant of 1/ps.
At each time step of the simulation, the instantaneous pressure was computed from all kinetic energies and pairwise interparticle (virial) interactions. Pairwise interactions include both nonbonded forces, i.e., electrostatic and van der Waals terms, as well as two-, three-, and four-body terms representing the covalent interactions between bonded atoms. The three- and four-body terms can be decomposed into simple two-body forces for the purpose of virial decomposition. Full electrostatics using PME cannot be so simply decomposed, as discussed below. Summing all contributions to the pressure gives the total pressure tensor P:
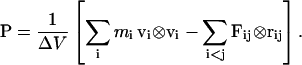 |
(1) |
The off-diagonal elements of P vanish in equilibrium, and for an isotropic system, the diagonal elements are expected to be equal. For an anisotropic system such as a lipid bilayer, the diagonal elements need not be equal, leading to a finite surface tension given by
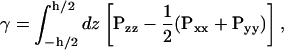 |
(2) |
which we write
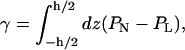 |
(3) |
where h is the height of the simulation volume and the normal and lateral pressures are given by PN = Pzz and PL = 1/2(Pxx + Pyy), respectively, for a bilayer whose normal is parallel to the z axis. If sufficient water is placed outside the bilayer, the integrand vanishes at the endpoints, since bulk water is tension-free; we may therefore integrate over the entire simulation volume without making any arbitrary definition of the interface.
To study spatial variation in P, we must define a local pressure. Following Lindahl and Edholm (2000), we partition the simulation space into slabs in the x, y plane and compute the contribution to the pressure due to particles interacting within or through a given slab:
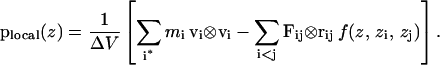 |
(4) |
Here plocal(z) is the local pressure tensor in the slab centered on the coordinate z normal to the membrane, and zi and zj are the z coordinate of particles i and j, respectively. The sum over i* in the kinetic contribution runs over all atoms in the slab at the current time step. The weighting function f(z, zi, zj) of Eq. 4 is chosen such that the fractional contribution of the interaction between a pair of particles i and j to the pressure in the slab centered at z is 1.0 if both particles are in the slab, dz/|z1 − z2| if one particle is in the slab, and Δz/|z1 − z2| if neither particle is in the slab, but the slab lies between the two particles (taking periodic boundary conditions into account). Here dz is the distance of the particle in the slab to the boundary of the slab closest to the other particle, again taking periodic boundary conditions into account, and Δz is the (uniform) slab thickness. It can be seen that the function f(z, z1, z2) smoothly distributes the virial with a total weight of 1, and is free from divergences if one particle is near the edge of a slab.
The contribution to the local pressure tensor from the kinetic energy and the covalent interactions was computed at each 1 fs time step and saved every 100 fs. It was observed that strong fluctuations arising from the multiple time-stepping algorithm employed increased the variance in the calculated pressure components. This artificial fluctuation could be discounted by using a block averaging scheme: in addition to the instantaneous local pressure every 100 fs, the pressures at each time step were summed and averaged over the entire 100 time-step block. Mean local pressures in each slab could then be calculated using the block-averaged data, whereas the autocorrelation function and corresponding autocorrelation time could be computed from the nonaveraged data. Block-averaged data were collected and analyzed for simulations D1–D4 (see Table 1).
Since the contribution to the pressure from electrostatic forces computed by the PME method cannot be readily decomposed into pairwise interactions (Lindahl and Edholm, 2000), the contribution to the pressure from VDW and electrostatic interactions was instead computed using a cutoff of 18 Å. Pressure profile simulations were conducted using PME as described above, with coordinates saved every 500 fs. These saved coordinates were then analyzed using the direct Coulomb interaction, truncated at the cutoff distance.
The simulations described were conducted using a multiple time-stepping algorithm to reduce the amount of computation required by the molecular dynamics algorithm (Kalé et al., 1999). Covalent bonds and short-range VDW and electrostatic interactions were computed every time step. Intermediate-range nonbonded interactions were computed and added to the total force only every two time steps, albeit with a factor of two increase in the size of the impulse (Leimkuhler et al., 1996). Long-range electrostatic forces from PME were computed only every four time steps. As described in Results, the use of multiple time stepping for the intermediate-range forces shifts the pressure profile by a constant amount in the hydrophobic core region of the bilayer; this artifact cannot be removed through the use of block averages. On the basis of a second round of simulations in which intermediate-range forces were computed every time step, we show that our results pertaining to bilayer stretching and pressure profile moments are essentially the same with either methodology.
In the following we shall only consider the difference between lateral and normal pressure, and refer to this difference simply as the lateral pressure.
Analysis of pressure profile results
The statistical error in the computed pressure profiles was analyzed using standard methods. In particular, the error in the estimate of the mean was adjusted using the estimated correlation time at each slab. This correlation time was found to vary substantially in space, with the headgroup regions showing the longest times, though in no case was any correlation time found to be >1/10th of the length of the simulation. Correlation times were estimated by summing all terms in the autocorrelation function up to, but not including, the first nonnegative term. Thus, the error bars reported here may be regarded as upper bounds on the true statistical error. No attempt was made to compensate for very slow undulations or deformations of the bilayer.
RESULTS
Structures and simulation times for all simulated membranes are summarized in Table 1. The simulations conducted cover biologically and experimentally relevant lipids, short (DLPE) and long (POPE) amphipathic tails, as well as small (phosphatidylethanolamine (PE)) and large (phosphatidylcholine (PC)) lipid headgroups.
Equilibration proceeded with an initial simulation at constant area and constant normal pressure. In simulations B and C, a period of constant area equilibration was performed, at which point the membrane thickness had stabilized. The systems were subsequently allowed to relax in all three dimensions independently; this always resulted in a membrane with a smaller area per lipid than the nominal values taken from experiment. In simulation B, the DLPC bilayer was constructed with an area per lipid of 62.9 Å2; the area fell to 59.78 Å2 after 500 ps of equilibration in a fully flexible unit cell. In simulation C, the POPE bilayer was constructed at 57 Å2; after constant-area equilibration for 1 ns and fully flexible equilibration for 3.2 ns, the area fell to 53.3 Å2. Free equilibration proceeded until the unit cell dimensions appeared to be stable. Longer simulations would likely have resulted in even smaller membrane areas, as has been observed previously (Feller and Pastor, 1999); this likely reflects a deficiency in the force field employed, as very long simulations (on the order of tens of nanoseconds) are required to fully relax the area of a membrane. Simulations A and D were equilibrated at constant area for the entire duration indicated in Table 1.
Pressure profile discretization
A question that must be addressed at the outset of the pressure profile calculation is how finely to discretize the simulation space. A bin thickness of ∼1 Å seems reasonable since lipid order parameters vary little over this distance, but one might still wonder if reproducible structures might be observed if a finer discretization were chosen. Lindahl and Edholm (2000) used 60 bins with a thickness of ∼1 Å, but the smoothing function (a running average of five bins) applied to the computed pressure profile would have masked peaks in the pressure profile that were narrower than this width.
For the pressure profile methodology described above, in which the virial contribution from each pair interaction is uniformly distributed between bins, there is no a priori reason to use a large bin size, even if one believes that no reproducible pressure peak will be observed that is smaller than the bin size chosen. This is because a running average of size n is exactly equivalent to reducing the number of bins by a factor of n. One should therefore choose the bin size fine enough at the outset of the simulation; an overly fine discretization can be compensated for by post-simulation averaging.
Fig. 1 shows the effect of discretization for a DLPE membrane simulated once with 60 bins and once with 120 bins. The two simulations began from the same equilibrated starting point but were otherwise independent, running under identical conditions. The two simulations contain identical peaks at the center of the bilayer, at ±10 Å near the edge of the hydrophobic core, and at ±22 Å in the aqueous region. The high-tension troughs at ±14 Å are similar in size and width. In the less well-converged region 15–20 Å from the center, the differences between the profiles is most evident. No reproducible features in simulation A2 seem to have been averaged out in simulation A1. The results in Fig. 1 clearly suggest that a 1 Å resolution is sufficient for calculation of pressure profiles.
FIGURE 1.

Effect of pressure profile discretization for DLPE. (Solid line) Simulation A1, computed with 60 bins of thickness 1.09 Å. (Dashed line) Simulation A2, computed with 120 bins of thickness 0.55 Å.
Error and correlation time analysis
Fig. 2 a shows the contribution to the pressure profile from the various force field terms. The kinetic and bonded components (i.e., two-, three-, and four-body terms) together contribute a large positive term to the pressure profile, whereas the nonbonded components of the force field (the VDW and electrostatic forces) tend to provide a negative contribution. The two large dips in the nonbonded contribution are located in a region 11.9–15.5 Å from the center of the bilayer, and are equal in size to, but slightly offset from, the corresponding peak in the bonded component, which is more tightly localized in a region 11.9–14.2 Å from the center. This slight offset yields the minima in the lateral pressure seen in Fig. 2 c.
FIGURE 2.

Pressure profile analysis of POPC membrane from simulation D1. (a) Bonded and kinetic component (solid line) and nonbonded component (dashed line) of the total pressure profile; (b) autocorrelation time of bonded and kinetic component (solid line) and nonbonded component (dashed line); (c) total pressure profile (thick line), bracketed by one standard deviation (thin solid lines).
Fig. 2 b shows that the autocorrelation times of the nonbonded component of the profile are much longer than that of the kinetic and bonded component. The standard deviation of the calculated nonbonded component is actually highest in the bulk solvent (≈30 bar), and drops monotonically to ≈3.5 bar in the hydrophobic tail region. However, due to the long correlation times in the layer of ester oxygens, the statistical errors are highest in this region. Statistical errors in the bonded component are approximately one quarter as great as the errors in the nonbonded component.
Fig. 2 c presents the total pressure profile for POPC from simulation D1, with error bars indicated by the bracketing lines. It can be seen that the profile declines to zero in the bulk solvent, as one would expect for a fully hydrated bilayer. The hydrophobic region of the bilayer is well resolved, with a peak in the lateral pressure at the center and two smaller peaks at the edge of the hydrophobic core. Minima in the lateral pressure are symmetrically located 15–17 Å outside of the center of the bilayer. Also well resolved are the two outermost peaks, which are located entirely in the aqueous region of the system. Autocorrelation times of the water-water contribution to the local pressure could not be calculated, but both the total pressure autocorrelation time as well as the local water diffusion coefficient were found to be diminished by roughly half at the locations of the outermost pressure profile peaks, relative to bulk water.
Between the aqueous peaks and the deep tension troughs, a secondary peak is evident; though the statistical errors are high in this region, the peak is found in the identical place in each monolayer, suggesting that it is not a statistical artifact. The location of the peak corresponds to the peak density of phosphate groups (see Fig. 7 b); however, further study will be required to ascertain that interactions involving the phosphate groups are indeed the source of the pressure profile peaks.
FIGURE 7.

(a) Mechanosensitive channel MscL from Mycobacterium tuberculosis (Chang et al., 1998) superimposed on a snapshot from POPC membrane simulation D1 (see Table 1). Left, distribution of water (blue), lipid acyl chains (cyan), phosphate atoms (yellow), and ester oxygens (green). Right, pressure profile calculated from simulation D1. Regions of positive surface tension are colored red, negative tension is colored blue, and zero tension is colored white. (b) Time-averaged mass density from simulation D1, relative to bulk water density. Black, total density; yellow, phosphate atom density; green, ester oxygen density; cyan, lipid density; blue, water density. (c) Pressure profile from simulation D1, smoothed using a five-point running average. The net area under the pressure profile curve is nonzero due to the finite surface tension present in the system.
Effect of lipid headgroup
To test the effect of varying the lipid headgroup in an otherwise identical bilayer, a comparison was made of the pressure profiles from membranes containing PE headgroups with those containing PC headgroups. Fig. 3 shows the difference in the pressure profile between a DLPE and a DLPC bilayer, and Fig. 4 shows the difference between a POPC bilayer and a POPE bilayer at two different lipid areal densities.
FIGURE 3.

Comparison of DLPE (solid line) and DLPC (dashed line) bilayers. (a) Lipid order parameter. (b) Pressure profile from simulations A1 (DLPE) and B (DLPC). Data are smoothed using a five-point running average. (c) Difference between A1 and B in part b.
FIGURE 4.

Comparison of POPC from simulation D1 (64 Å2/lipid) and POPE from simulations C3 and C4 (59 and 64 Å2/lipid, respectively). (a) Lipid order parameters for simulations D1 (circles), C3 (diamonds) and C4 (triangles). (b) Pressure profile for simulation D1 (thick solid line), C3 (dashed line), and C4 (thin solid line). Data are smoothed using a five-point running average. (c) Difference between POPE and POPC pressure profiles. Dashed line, C3–D1; solid line, C4–D1.
Lipid order parameters presented in part a of Figs. 3–6 are calculated from the expression 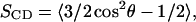 where θ is the angle between each C-H bond and the bilayer normal. SCD quantifies the degree of orientational order of the lipid tails and can be compared directly with experimentally determined values obtained through quadrupole splitting measurements. The value of SCD for POPC has been measured at 0.2 in the “plateau” region of the order parameter profile (Seelig, 1977; Seelig and Seelig, 1977), and good agreement between simulation and experiment should be expected when the appropriate area per lipid is chosen (Heller et al., 1993; Feller, 2000).
where θ is the angle between each C-H bond and the bilayer normal. SCD quantifies the degree of orientational order of the lipid tails and can be compared directly with experimentally determined values obtained through quadrupole splitting measurements. The value of SCD for POPC has been measured at 0.2 in the “plateau” region of the order parameter profile (Seelig, 1977; Seelig and Seelig, 1977), and good agreement between simulation and experiment should be expected when the appropriate area per lipid is chosen (Heller et al., 1993; Feller, 2000).
FIGURE 6.

Effect of stretching a POPC bilayer. Black, simulation D1; red, D2; green, D3; blue, D4. (a) Lipid order parameter for simulations D1–D4. (b) Pressure profiles for simulations D1–D4. Data have been smoothed using a five-point running average. (c) Difference in pressure profile between simulations D2–D4 and simulation D1, using the smoothed data from part b.
Examining first Fig. 3, the central pressure peak is identical for the DLPE and DLPC bilayers, but the DLPC bilayer has a much smaller pressure peak next to the central peak. Fig. 3 c shows that this drop in lateral pressure at the hydrophobic core is compensated by an increase in lateral pressure in the headgroup region. The shift is easily rationalized by observing that the bulkier PC headgroups make room for the lipid tails in the well-ordered part of the hydrophobic core. However, examination of the lipid order parameters in Fig. 3 a shows that the DLPC lipid tails are more disordered, suggesting that the lower internal pressure in the DLPC bilayer could be due to differences in lipid area, rather than the identity of the headgroup.
To clarify the situation we turn to Fig. 4, where again the POPC bilayer, with its larger headgroup, has a lower pressure peak at the hydrophobic core, as seen in parts b and c. The POPE bilayer from simulation C4 has a lower lipid order parameter than the POPC bilayer of simulation D1, as seen in Fig. 4 a; nonetheless, as seen in Fig. 4 b, the lateral pressure peak at ±18 Å from the center is larger in the POPE than the POPC bilayer.
Fig. 4 c also shows that the pressure shift attributable to the change in headgroup is independent of the area per lipid. The positive peaks in Fig. 4 c are higher for simulation C3 than for C4 due to the tighter lipid packing in simulation C3. However, the negative peaks in part c are the same size for C3 and C4, which indicates that the same amount of lateral pressure has been shifted out of the core and into the headgroup for the two simulations.
Effect of membrane stretching
The effect of varying the area per lipid of a membrane was studied in simulations of POPE and POPC. For POPE, the area per lipid was varied by −9%, 11%, and 21.8% in simulations C1, C3, and C4, respectively, relative to the area density of 53.3 Å2/lipid in the original equilibrated membrane patch; for POPC the membrane was stretched areawise by 5%, 10%, and 16% in simulations D2, D3, and D4, relative to the original area density of 64 Å2/lipid. The POPE simulations thus represent overcompressed bilayers, which unfortunately can easily result from zero-tension simulations using the Charmm force field (Feller and Pastor, 1999). It is thus of practical interest to study the pressure profile of these bilayers to determine what effect compression has on simulations of membranes or protein-membrane systems.
Since lipid bilayers on the macroscopic length scales studied in experiments typically expand no more than 5% before tearing, one cannot relate the much larger area expansions studied in these simulations to any macroscopic value of applied tension. Microscopic bilayers such as those studied here support much higher values of surface tension and wider ranges of area per lipid than what is measured experimentally; for example, Marrink and Mark (2001) obtained surface tension values of −40 to 97 dyn/cm over a corresponding area per lipid range of 24–42 Å2 using glycerolmonoolein lipids. The microscopic surface tensions reported here should not, therefore, be compared directly to experimental values.
Results for POPE are presented in Fig. 5. It is clear from Fig. 5 a that the lipid order parameters are much too high in simulations C1 and C2, and possibly also C3; this means that the lipids are more tightly packed than in their native state. The pressure profiles of simulations C1–C4 are shown in Fig. 5 b. For simulations C3 and C4, in which the order parameter is close to its nominal value, the central peak in the pressure profile is of the same size as the secondary peak at the edge of the hydrophobic core. One notices from Fig. 5 b that all the pressure profile peaks are shifted inward as the membrane stretches and thins. The shift in the bilayer thickness exactly tracks the shift in the location of the peaks in the pressure profile. The central pressure peak decreases in size with membrane stretch, indicating that, even as the bilayer thins, the lipids are becoming more loosely packed; Fig. 5 a shows that the lipid tails become progressively more disordered as well. There is no change at all in the aqueous peak at the outermost edge of the pressure profile. This suggests that there is no significant change in water-headgroup interactions with the change in lipid areal density within this range of lipid areas.
FIGURE 5.

Effect of stretching a POPE bilayer. Black, simulation C1; red, C2; green, C3; blue, C4. (a) Lipid order parameter for simulations C1–C4. (b) Pressure profiles for simulations C1–C4. Data have been smoothed using a five-point running average. (c) Difference in pressure profile between simulations C2–C4 and simulation C1, using the smoothed data from part b.
The most prominent change in the POPE pressure profile due to stretching, besides the shift in the location of the pressure peaks, is the reduction in size of the secondary tension peak and corresponding increase in the depth of the tension trough. Though the positions of these peaks shift along with the thickness of the bilayer as the membrane thins, Fig. 5 c shows that this shift can actually be understood as an increase in the tension of the bilayer localized between the pressure peak and the tension trough.
Results of varying the area per lipid of POPC are presented in Fig. 6. The lipid order parameters in Fig. 6 a show that, in contrast to the POPE bilayer of simulations C1–C4, the POPC simulations D1–D4 represent a bilayer near the native physical state that is then stretched through application of tension. The pressure profiles shown in Fig. 6 b are much more similar to each other than are the POPE profiles of C1–C4; in particular, there are no large secondary pressure peaks flanking the central peak. The width of the central pressure peak is independent of the area per lipid, whereas the positions of the other pressure profile features scale with the membrane thickness as the latter decreases with increasing area per lipid. A small tension increase with increasing area per lipid (negative lateral pressure) is evident in Fig. 6 c at ±12 Å from the center of the bilayer, the same location where an increase in tension is seen for overcompressed POPE bilayers in Fig. 5 c, though of much lower magnitude.
A better understanding of the features observed in the pressure profile may be gained by comparison with the mass distribution of water and lipids, as shown in Fig. 7 for simulation D1. The minimum lateral pressure, corresponding to the region of maximum surface tension, occurs in all simulations at the maximum of the lipid density. The density peak is in turn composed of contributions from the phosphate group and the ester oxygen groups of the lipids, which are the most highly polarized groups. Water density in this region is still significant, ∼25% of its bulk value. Strong attractions between polar lipid groups therefore are the cause of the peak in the tension.
The small pressure peaks flanking the central peak at ±7 Å in Fig. 7 c correspond to the location in the membrane where the water density falls to zero. The single unsaturated bond in the oleic acid group of POPC is also located in this region; however, since secondary peaks are also seen in DLPE and DLPC (see Fig. 3), these peaks must be a generic effect of lipid packing, rather than a localized effect of the cis bond in the lipid tail.
The effect of the molecular dynamics integrator
Multiple time-stepping algorithms for MD are an important, well-established method for reducing the computational requirements of a simulation by performing the expensive calculation of nonbonded interactions every two or four time steps, rather than on each time step (Grubmüller et al., 1991; Schlick et al., 1999). To explore the possibility that the multiple time-stepping algorithm employed for the MD integrator may have affected our computed pressure profiles, we repeated all simulations described in Table 1 with the same run parameters as before, but with short-range nonbonded forces computed every time step, rather than every other time step. In addition, full electrostatics were computed every two time steps rather than every four time steps to halve the size of the impulse (Leimkuhler et al., 1996). Since nonbonded force calculations comprise the bulk of the necessary computation, these modifications caused the simulation to run at about half the speed as before; as a result, we could not afford to run any longer than ∼2.5 ns for each new simulation. However, as a further test, simulation D1 was repeated for 5.5 ns with full electrostatics computed every time step, i.e., without multiple time stepping.
The described simulations are summarized in Table 2. The large error estimates for the pressure profile moments indicate that the simulation time was too short to obtain well-converged values; however, the computed surface tension is found to be significantly higher in all cases, by around a factor of 2. The computed pressure profile moments are also consistently greater in magnitude, as discussed below.
TABLE 2.
Pressure profile moments for all simulations performed with short-range nonbonded forces calculated every time step
| Simulation | Description | Simulation time (ns) | γ (dyn/cm) | M1/kBT (Å−1) | M2/kBT |
|---|---|---|---|---|---|
| R-A1 | DLPE (60 bins) | 1.4 | 34.2 (2.6) | −1.1 (0.1) | −12.7 (3.4) |
| R-A2 | DLPE (120 bins) | 2.1 | 21.8 (4.7) | −0.6 (0.1) | −3.8 (2.6) |
| R-B | DLPC | 1.6 | 40.36 (4.7) | −0.8 (0.4) | −2.7 (9.3) |
| R-C1 | POPE (48.5 Å2) | 1.9 | −24.1 (8.5) | 0.8 (0.3) | 14.6 (8.9) |
| R-C2 | POPE (53.3 Å2) | 1.9 | 46.6 (7.0) | −1.8 (0.5) | −29.3 (14.7) |
| R-C3 | POPE (59.1 Å2) | 1.9 | 43.9 (1.3) | −1.5 (0.1) | −19.9 (2.7) |
| R-C4 | POPE (64.9 Å2) | 1.9 | 48.8 (4.3) | −1.6 (0.3) | −22.7 (8.7) |
| R-D1 | POPC (64.0 Å2) | 2.4 | 37.2 (6.0) | −1.0 (0.1) | −8.6 (0.3) |
| R2-D1 | POPC (64.0 Å2) | 5.5 | 39.6 (1.8) | −1.0 (0.1) | −8.8 (2.3) |
| R-D2 | POPC (67.24 Å2) | 2.6 | 27.6 (10.2) | −0.5 (0.4) | 3.1 (5.6) |
| R-D3 | POPC (70.56 Å2) | 2.5 | 35.7 (2.4) | −0.9 (0.1) | −5.3 (4.3) |
| R-D4 | POPC (73.96 Å2) | 2.5 | 38.2 (7.4) | −1.0 (0.3) | −9.1 (6.7) |
In simulation R2-D1, full electrostatics were also calculated every time step. For the sake of accuracy, values are averaged over the two monolayers; statistical errors are given in parentheses.
All of these shifts can be explained in view of the shift in the pressure profile evident in Fig. 8. We note first that the lipid order parameter shown in Fig. 8 a is unaffected by the particular integration scheme employed. The lipid tail distribution is therefore similar in all three simulations, so that differences in the pressure profile must result from differences in the computational approach, rather than the physical state of the bilayer. Fig. 8 b compares the computed pressure profile for simulations D1, R-D1, and R2-D1. Our results show that the position of pressure profile features was not affected by the use of multiple time stepping, and that reliable results may be obtained as long as short-range nonbonded forces are computed on each MD time step. The pressure profiles from simulations without intermediate-range multiple time stepping (R-D1 and R2-D1) are shifted downward in the hydrophobic region of the bilayer. The shift is essentially identical in simulations R-D1 and R2-D1, suggesting that the shift in the pressure profile is due to intermediate-range forces being applied only every two time steps; long-range electrostatic forces may be safely computed every two time steps without adversely affecting the computed pressure profile.
FIGURE 8.

Effect of multiple time-stepping algorithm on calculated pressure profiles for simulations D1 (thick solid line), R-D1 (thin solid line), and R2-D1 (dashed line). (a) Lipid order parameter; (b) pressure profile, smoothed using a five-point running average.
The central downward shift in the pressure profile arises from the nonbonded contribution to the pressure; the bonded component is mostly unchanged in this region (data not shown). In the interfacial region, both bonded and nonbonded components are affected by the use of multiple time stepping, but their combined contributions cancel so that no net effect is seen in this region. Neither the bonded nor the nonbonded components are affected by multiple time stepping in the aqueous region.
The downward shift in the central part of the pressure profile explains the increase in computed surface tension (since tension carries the opposite sign as the lateral pressure) as well as the apparent shift in the higher-order pressure profile moments toward more negative values. The effect on the higher-order moments is less pronounced than the effect on the tension since the moments are weighted by the distance from the center of the bilayer; there is no significant change in the pressure profile outside of the hydrophobic core.
Models of MscL-bilayer interactions
A key motivation for studying the effect of membrane stretching and composition on the pressure profile is to understand the gating mechanism of the mechanosensitive channel MscL. MscL of Escherichia coli has served as a model for how cells sense and respond to osmotic shock (Blount et al., 1997; Batiza et al., 2002), due to its large conductance (3.6 nS (Sukharev et al., 1999)) as well as the availability of a crystal structure of the closed state of the channel (Chang et al., 1998). The measured conductance suggests a pore size of 36–42 Å (Sukharev et al., 1999; Cruickshank et al., 1997), but the diameter of the protein in the closed state revealed by the crystal structure is only 50 Å, indicating that gating requires a large conformational change. Structures of the open state obtained from steered MD simulations (Gullingsrud and Schulten, 2003), as well as models based in part on the known conductance of the open pore (Sukharev et al., 2001; Perozo et al., 2002a) consistently exhibit a change in average in-plane area of 1800–2200 Å2. This area change is considerably larger than the 350 Å2 (Chang et al., 1998; Sukharev et al., 1997) or 650 Å2 (Sukharev et al., 1999) area change deduced from patch-clamp experiments, although more recent studies taking into account the inhomogeneity in the gating state of the channels in a single patch suggest an area change of 2010 Å2, in agreement with structural models (S. Sukharev, private communication). In these experiments, the measured tension dependence of the probability Po for the channel to be open closely fits the Boltzmann weight of a two-state system,
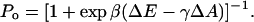 |
(5) |
Here, ΔE − γΔA is the total energy difference between the open and closed state, and γ is the applied tension. ΔE is the intrinsic (zero-tension) energy difference between the open and closed states of the channel, ΔA is the difference in area between the open and closed state, and β = 1/kBT is the temperature factor. A tension-dependent change in membrane properties that favored the closed state would cause ΔA to be underestimated.
Although hydrogen bonding and other interactions between the protein and the water and lipid environment may be important (Elmore and Dougherty, 2003), lateral pressures imparted by the membrane and solvent can have a decisive impact on protein conformations as well. As pointed out by Cantor (1997, 1999), the pressure profile can have a strong effect on protein conformation equilibria, if the conformations available to the protein have different cross-sectional area profiles. The dose-response curve in Eq. 5 does not take into account any change in shape of the channel, only its increase in average cross-sectional area, through the γΔA contribution. It is thus worth considering whether the lateral pressure distribution needs to be taken into account in determining the energy difference between the closed and open state of the channel.
To address this question, we adopt the simplest nontrivial model for the shape of the channel. We consider a bilayer centered in a coordinate system with z = 0 at the center of the bilayer, and the z axis normal to the bilayer (see Fig. 9 a). If the difference between lateral and normal pressure at a distance z from the center of the bilayer is denoted by p(z), then the mechanical work W required to create a protein-shaped cavity in the bilayer with cross-sectional area A(z) can be written (Cantor, 1999)
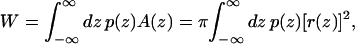 |
(6) |
where r(z) is the depth-dependent radius of the channel. Note that Eq. 6 does not describe the total free energy required for protein insertion, only the work done against the pressure profile. The bounds of integration are free to extend to ±∞ since p(z) drops to zero outside the bilayer.
FIGURE 9.

Analysis of pressure profile interaction with MscL conformational changes. (a) The protein is represented as a truncated cone with radius r(z) and slope s interacting with a pressure profile p(z). (b) Closed form of MscL from an E. coli homology model (Sukharev et al., 2001) at the start of the steered MD simulation of Gullingsrud and Schulten (2003). (c) Open form of MscL at the end of the steered MD simulation of Gullingsrud and Schulten (2003); the cross-sectional area of the channel varies with z.
To a good approximation, MscL's transmembrane helices are assembled such that only cylindrical or conical cross sections are accessible; this is shown schematically in Fig. 9, b and c. The radius in either the closed or open state can thus be written r(z) = R + sz, where R is the average radius (25 Å in the closed state) and s is the slope. The closed form of the protein, as determined by the crystal structure, is nearly cylindrical; i.e., s = 0 in the closed state. Steered MD simulations of the gating process (Gullingsrud and Schulten, 2003) obtained a slope of 0.2 for the open state, corresponding to a tilt of 11.3°. In the models of Sukharev et al. (2001), MscL reaches a tilt of 0.2 in the open state, but in the opposite direction, i.e., the cross-sectional area of the channel is greater at the bottom than at the top. The difference is immaterial for the considerations in this study. The work as given in Eq. 6 can then be rewritten as
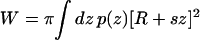 |
(7) |
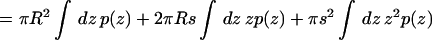 |
(8) |
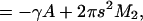 |
(9) |
where γ is the tension and M2 is the second moment of the pressure profile in each monolayer, proportional to the Gaussian curvature modulus (Safran, 1994). The contribution from the first moment vanishes since p(z) is symmetric in z. Equation 9 shows that this model of the gating motion of MscL includes the average change in area and tension through the γA term, as well as a contribution from the pressure profile through M2. Including these pressure profile contributions, and using s = 0 in the closed state, we can rewrite Eq. 5 as
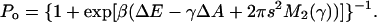 |
(10) |
Table 3 shows the pressure profile moments calculated from the simulations presented here. The moments for simulations D1–D4 in Table 3 exhibit no clear dependence on tension; there is, therefore, no shift in the measured change in cross-sectional area due to the pressure profile.
TABLE 3.
Surface tension γ and first and second moments of the pressure profile calculated from the simulations of Table 1
| Simulation | Description | γ (dyn/cm) | M1/kBT (Å−1) | M2/kBT |
|---|---|---|---|---|
| A1 | DLPE (60 bins) | 17.8 (8.5) | −0.7 (0.4) | −7.4 (8.0) |
| A2 | DLPE (120 bins) | 4.2 (5.0) | −0.5 (0.1) | −4.1 (1.2) |
| B | DLPC | 9.4 (2.9) | (−0.2) (0.1) | 4.2 (2.7) |
| C1 | POPE (48.5 Å2) | −51.9 (8.2) | 1.1 (0.3) | 17.6 (6.6) |
| C2 | POPE (53.3 Å2) | −15.2 (8.0) | −0.18 (0.3) | −5.1 (6.8) |
| C3 | POPE (59.1 Å2) | 5.6 (2.2) | −0.70 (0.1) | −11.2 (2.5) |
| C4 | POPE (64.9 Å2) | 8.9 (1.7) | −0.58 (0.08) | −6.5 (2.1) |
| D1 | POPC (64.0 Å2) | 11.4 (0.8) | −0.59 (0.05) | −3.7 (0.2) |
| D2 | POPC (67.24 Å2) | 18.6 (0.9) | −0.80 (0.05) | −7.7 (1.2) |
| D3 | POPC (70.56 Å2) | 21.7 (0.9) | −0.80 (0.06) | −6.7 (1.8) |
| D4 | POPC (73.96 Å2) | 22.9 (2.2) | −0.81 (0.07) | −6.6 (1.0) |
Values are averaged over the two monolayers as in Table 2.
It is also interesting to examine whether pressure profiles might be responsible for the difference in tension threshold of MscL in different lipid compositions. For example, MscL from Mycobacterium tuberculosis (Tb-MscL), from which the crystal structure of MscL was obtained, gates at twice the tension threshold as MscL from E. coli (Moe et al., 2000), suggesting that Tb-MscL's tension threshold might be lower in its native membrane environment. Our calculations show that POPC lowers the open state energy of MscL relative to the closed state by ≈1.7 kBT; moreover, the variation in M2 from which this stabilization is derived is less than a factor of 2. Hence, only 2–4 kBT of the 50 kBT free-energy difference between the open and closed states could be expected to vary with lipid type. Pressure profile moments computed by Cantor also varied by less than a factor of 2 for an even wider range of lipid types (Cantor, 2002), although the magnitude of M2 was found to be four times higher. Order of magnitude agreement seems reasonable considering the significant difference between the all-atom models with explicit headgroup and solvent used here and the coarse-grained lattice approach employed by Cantor, especially since the largest contribution to the moments comes from the headgroup region, where the two approaches differ the most.
DISCUSSION
Pressure profiles of lipid bilayers with different headgroups and lipid areas along the membrane normal had been previously characterized in a series of calculations based on mean-field theories (Harries and Ben-Shaul, 1997; Cantor, 1997), a coarse-grained lipid model (Goetz and Lipowsky, 1998), and all-atom simulations (Lindahl and Edholm, 2000). Knowledge of these profiles is fundamental for any analysis of membrane protein function and an accurate description is essential. To this end we have carried out the all-atom simulations described above. In general, our results are in agreement with those of Lindahl and Edholm (2000), both in terms of the magnitude of the calculated lateral pressures and the shape of the profile.
However, compared to Lindahl and Edholm (2000), our calculations represent an important methodological advance, in that the length scale of inherent features has been identified through extensive sampling to be the single angstrom level, whereas Lindahl and Edholm (2000) had smoothed their results with a filter of ∼5 Å. Calculations of correlation times and error bars (Fig. 2) show that the most challenging region for calculations of the profile is at the aqueous interface, where a low density of water molecules leads to large fluctuations in the pressure, and a tight packing of lipid acyl chains leads to long autocorrelation times. In fact, all pressure profile calculations that treat the membrane as a self-assembled bilayer in equilibrium with water (Goetz and Lipowsky, 1998; Lindahl and Edholm, 2000) found that the pressure profile is quite complicated in the headgroup region. Although statistical error is a bigger problem with smaller bin sizes, oversmoothing will result in inaccurate calculation of the pressure profile moments, which have a direct bearing on protein conformational equilibria (Cantor, 1999).
The moments calculated in our simulations are a factor of 2–3 smaller than those calculated by Cantor; this stems primarily from differences in the treatment of the headgroups since, by virtue of their distance from the center of the bilayer, they contribute most to the moments (M1 and M2 in Table 3). The pressure profile in the headgroup region is also the most difficult to calculate, as shown in Fig. 2. The extent to which the pressure profile can be calculated accurately in the headgroup region has important implications for the modeling of protein-lipid interactions. For example, the effect of pressure profile redistribution on the gating of mechanosensitive channels such as MscL may be analyzed in terms of simple geometrical models of the closed and open states of the channel. In such an analysis, our results discount an effect of higher pressure profile moments on gating.
Mechanical forces mediated by the pressure profile provide a general framework for understanding the nonuniform distribution of compounds within lipid bilayers. For example, the pressure trough may play a role in positioning anaesthetics within the bilayer, providing a zone where molecules can be readily absorbed. In this zone, anaesthetics may exert an influence on mechanosensitive channels as observed in Martinac et al. (1990) and Patel et al. (1998). As demonstrated through steered MD simulations (Gullingsrud and Schulten, 2003), this zone dominates the mechanosensitivity of MscL. Extending this property of MscL to other channels may suggest a mechanism for anaesthetics. The pressure trough may also play a role in accommodating bulky, aromatic side chains that membrane proteins often expose to the bilayer aqueous interface to position themselves along the membrane normal (Killian and von Heijne, 2000). These side groups are indeed found near the pressure trough (cf. Fig. 7); the pressure profile may thus mechanically position membrane proteins.
The accuracy and predictive power of lipid membrane simulations have progressed dramatically in just the past few years (Scott, 2002; Feller, 2000), drawing on improvements in force fields and methodology. A decade ago, 30,000 atom simulations of lipid bilayers for 250 ps became possible only with a self-built parallel (60 processor) machine requiring years of run time (Heller et al., 1993). Today, 100,000 atom simulations lasting over 100 ns have become feasible, in part through public investment into massively parallel machines with thousands of processors as well as software able to take advantage of such hardware (Kalé et al., 1999). Molecular dynamics is now capable of calculating pressure profiles using a completely self-consistent model that treats all elements of the bilayer and solvent with equal accuracy. The simulations presented here, based on pioneering earlier studies (Lindahl and Edholm, 2000; Ben-Shaul, 1995), point the way to a deeper understanding of biological membranes and membrane proteins.
Acknowledgments
We thank M. Jensen and J. Sonne for helpful discussions, as well as our reviewers for their thoughtful suggestions. This work was supported by the National Institutes of Health (PHS5 P41RR05969-04) and the National Science Foundation (MCB-9982629). The authors also acknowledge computer time provided by National Resource Allocations Committee grant MCA93S028. The molecular images in this article were created with the molecular graphics program VMD (Humphrey et al., 1996).
References
- Batiza, A. F., M. M.-C. Kuo, K. Yoshimura, and C. Kung. 2002. Gating the bacterial mechanosensitive channel MscL in vivo. Proc. Natl. Acad. Sci. USA. 99:5643–5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker, H., H. J. C. Berendsen, E. J. Dijkstra, S. Achterop, and R. van Drunen. 1993. GROMACS: a parallel computer for molecular dynamics simulations. Proc. 4th Intl. Conference Physics Computing '92. 252–256.
- Ben-Shaul, A. 1995. Molecular theory of chain packing, elasticity and lipid-protein interaction in lipid bilayers. In Handbook of Biological Physics, Vol. 1. R. Lipowsky and E. Sackmann, editors. Elsevier Science, Amsterdam. 359–401.
- Bezrukov, S. M. 2000. Functional consequences of lipid packing stress. Curr. Opin. Colloid Interface Sci. 5:237–243. [Google Scholar]
- Blount, P., M. J. Schroeder, and C. Kung. 1997. Mutations in a bacterial mechanosensitive channel change the cellular response to osmotic stress. J. Biol. Chem. 272:32150–32157. [DOI] [PubMed] [Google Scholar]
- Botelho, A. V., N. J. Gibson, R. L. Thurmond, Y. Wang, and M. F. Brown. 2002. Conformational energetics of rhodopsin modulated by nonlamellar-forming lipids. Biochemistry. 41: 6354–6368. [DOI] [PubMed] [Google Scholar]
- Cantor, R. S. 1997. The lateral pressure profile in membranes: a physical mechanism of general anesthesia. Biochemistry. 36:2339–2344. [DOI] [PubMed] [Google Scholar]
- Cantor, R. S. 1999. The influence of membrane lateral pressures on simple geometric models of protein conformational equilibria. Chem. Phys. Lipids. 101:45–56. [DOI] [PubMed] [Google Scholar]
- Cantor, R. S. 2002. Size distribution of barrel-stave aggregates of membrane peptides: influence of the bilayer lateral pressure profile. Biophys. J. 82:2520–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casado, M., and P. Ascher. 1998. Opposite modulation of NMDA receptors by lysophospholipids and arachidonic acid: common features with mechanosensitivity. J. Physiol. 513:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, G., R. H. Spencer, A. T. Lee, M. T. Barclay, and D. C. Rees. 1998. Structure of the MscL homolog from Mycobacterium tuberculosis: a gated mechanosensitive ion channel. Science. 282:2220–2226. [DOI] [PubMed] [Google Scholar]
- Cruickshank, C. C., R. F. Minchin, A. C. L. Dain, and B. Martinac. 1997. Estimation of the pore size of the large-conductance mechanosensitive ion channel of Escherichia coli. Biophys. J. 73:1925–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan, N., P. Pincus, and S. A. Safran. 1993. Membrane-induced interactions between inclusions. Langmuir. 9:2768–2771. [Google Scholar]
- Dan, N., and S. A. Safran. 1998. Effect of lipid characteristics on the structure of transmembrane proteins. Biophys. J. 75:1410–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliot, J., D. Needham, J. Dilger, and D. Haydon. 1983. The effects of bilayer thickness and tension on gramicidin single-channel life time. Biochim. Biophys. Acta. 735:95–103. [DOI] [PubMed] [Google Scholar]
- Elmore, D. E., and D. A. Dougherty. 2003. Investigating lipid composition effects on the mechanosensitive channel of large conductance (MscL) using molecular dynamics simulations. Biophys. J. 85:1512–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann, U., L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- Feller, S. E. 2000. Molecular dynamics simulations of lipid bilayers. Curr. Opin. Colloid Interface Sci. 5:217–223. [Google Scholar]
- Feller, S. E., and R. W. Pastor. 1999. Constant surface tension simulations of lipid bilayers: the sensitivity of surface areas and compressibilities. J. Chem. Phys. 111:1281–1287. [Google Scholar]
- Feller, S. E., Y. H. Zhang, R. W. Pastor, and B. R. Brooks. 1995. Constant pressure molecular dynamics simulation—the Langevin piston method. J. Chem. Phys. 103:4613–4621. [Google Scholar]
- Goetz, R., and R. Lipowsky. 1998. Computer simulations of bilayer membranes: self-assembly and interfacial tension. J. Chem. Phys. 108:7397–7409. [Google Scholar]
- Grubmüller, H. 1996. SOLVATE 1.0. http://www.mpibpc.gwdg.de/abteilungen/071/solvate/docu.html.
- Grubmüller, H., H. Heller, A. Windemuth, and K. Schulten. 1991. Generalized Verlet algorithm for efficient molecular dynamics simulations with long-range interactions. Mol. Simulat. 6:121–142. [Google Scholar]
- Gullingsrud, J., and K. Schulten. 2003. Gating of MscL studied by steered molecular dynamics. Biophys. J. 85:2087–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill, O. P., and B. Martinac. 2001. Molecular basis of mechanotransduction in living cells. Physiol. Rev. 81:685–740. [DOI] [PubMed] [Google Scholar]
- Harries, D., and A. Ben-Shaul. 1997. Conformational chain statistics in a model lipid bilayer: comparison between mean field and Monte Carlo calculations. J. Chem. Phys. 106:1609–1619. [Google Scholar]
- Heller, H., M. Schaefer, and K. Schulten. 1993. Molecular dynamics simulation of a bilayer of 200 lipids in the gel and in the liquid crystal-phases. J. Phys. Chem. 97:8343–8360. [Google Scholar]
- Huang, H. 1986. Deformation free energy of bilayer membrane and its effect on gramicidin channel lifetime. Biophys. J. 50:1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- Kalé, L., R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan, and K. Schulten. 1999. NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 151:283–312. [Google Scholar]
- Killian, J. A., and G. von Heijne. 2000. How proteins adapt to a membrane-water interface. Trends Biochem. Sci. 25:429–434. [DOI] [PubMed] [Google Scholar]
- Leimkuhler, B. J., S. Reich, and R. D. Skeel. 1996. Integration methods for molecular dynamics. In Mathematical Approaches to Biomolecular Structure and Dynamics (IMA Volumes in Mathematics and Its Applications, Vol. 82). J. P. Mesirov, K. Schulten, and D. W. Sumners, editors. Springer-Verlag, New York. 161–185.
- Lindahl, E., and O. Edholm. 2000. Spatial and energetic-entropic decomposition of surface tension in lipid bilayers from molecular dynamics simulations. J. Chem. Phys. 113:3882–3893. [Google Scholar]
- Lindahl, E., B. Hess, and D. van der Spoel. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 7:306–317. [Google Scholar]
- Lundbaek, J. A., and O. S. Andersen. 1994. Lysophospholipids modulate channel function by altering the mechanical properties of lipid bilayers. J. Gen. Physiol. 104:645–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell, A. D., Jr., D. Bashford, M. Bellott, R. L. Dunbrack, Jr., J. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, I. W. E. Reiher, B. Roux, M. Schlenkrich, J. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus. 1998. All-hydrogen empirical potential for molecular modeling and dynamics studies of proteins using the CHARMM22 force field. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- MacKerell, A. D., Jr., D. Bashford, M. Bellott, R. L. Dunbrack, Jr., J. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, B. Roux, M. Schlenkrich, J. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus. 1992. Self-consistent parameterization of biomolecules for molecular modeling and condensed phase simulations. FASEB J. 6:A143. [Google Scholar]
- Marrink, S. J., and A. E. Mark. 2001. Effect of undulations on surface tension in simulated bilayers. J. Phys. Chem. B. 105:6122–6127. [Google Scholar]
- Marrink, S. J., and A. E. Mark. 2003. Molecular dynamics simulation of the formation, structure, and dynamics of small phospholipid vesicles. J. Am. Chem. Soc. 125:15233–15242. [DOI] [PubMed] [Google Scholar]
- Martinac, B., J. Adler, and C. Kung. 1990. Mechanosensitive ion channels of E. coli activated by amphipaths. Nature. 348:261–263. [DOI] [PubMed] [Google Scholar]
- Moe, P. C., G. Levin, and P. Blount. 2000. Correlating a protein structure with function of a bacterial mechanosensitive channel. J. Biol. Chem. 275:31121–31127. [DOI] [PubMed] [Google Scholar]
- Nagle, J. F., R. Zhang, S. Tristram-Nagle, W. Sun, H. I. Petrarche, and R. M. Suter. 1996. X-ray structure determination of fully hydrated lα phase dipalmitoylphosphatidylcholine bilayers. Biophys. J. 70:1419–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, C., M. Goulian, and O. S. Andersen. 1998. Energetics of inclusion-induced bilayer deformations. Biophys. J. 74:1966–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, C., and O. S. Andersen. 2000. Inclusion-induced bilayer deformations: effects of monolayer equilibrium curvature. Biophys. J. 79:2583–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, A. J., E. Honoré, F. Maingret, F. Lesage, M. Fink, F. Duprat, and M. Lazdunski. 1998. A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J. 17:4283–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozo, E., D. M. Cortes, P. Sompornpisut, A. Kloda, and B. Martinac. 2002a. Open channel structure of MscL and the gating mechanism of mechanosensitive channels. Nature. 418:942–948. [DOI] [PubMed] [Google Scholar]
- Perozo, E., A. Kloda, D. M. Cortes, and B. Martinac. 2002b. Physical principles underlying the transduction of bilayer deformation forces during mechanosensitive channel gating. Nat. Struct. Biol. 9:696–703. [DOI] [PubMed] [Google Scholar]
- Safran, S. A. 1994. Statistical Thermodynamics of Surfaces, Interfaces, and Membranes. Addison-Wesley, Reading, MA.
- Schlenkrich, M., J. Brickmann, A. D. MacKerell Jr., and M. Karplus. 1996. Empirical potential energy function for phospholipids: criteria for parameter optimization and applications. In Biological Membranes: A Molecular Perspective from Computation and Experiment. K. M. Merz and B. Roux, editors. Birkhauser, Boston. 31–81.
- Schlick, T., R. Skeel, A. Brünger, L. Kalé, J. A. Board Jr., J. Hermans, and K. Schulten. 1999. Algorithmic challenges in computational molecular biophysics. J. Comput. Phys. 151:9–48. [Google Scholar]
- Schneider, M. J., and S. E. Feller. 2001. Molecular dynamics simulations of a phospholipid-detergent mixture. J. Phys. Chem. B. 105:1331–1337. [Google Scholar]
- Scott, H. L. 2002. Modeling the lipid component of membranes. Curr. Opin. Struct. Biol. 12:495–502. [DOI] [PubMed] [Google Scholar]
- Seelig, A., and J. Seelig. 1977. Effect of a single cis double bond on the structure of a phospholipid bilayer. Biochemistry. 16:45–50. [DOI] [PubMed] [Google Scholar]
- Seelig, J. 1977. Deuterium magnetic resonance: theory and application to lipid membranes. Q. Rev. Biophys. 10:353–418. [DOI] [PubMed] [Google Scholar]
- Shelley, J., M. Shelley, R. Reeder, S. Bandyopadhyay, P. Moore, and M. Klein. 2001. Simulations of phospholipids using a coarse grain model. J. Phys. Chem. B. 105:9785–9792. [Google Scholar]
- Sukharev, S., S. R. Durell, and H. R. Guy. 2001. Structural models of the MscL gating mechanism. Biophys. J. 81:917–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukharev, S. I., P. Blount, B. Martinac, F. R. Blattner, and C. Kung. 1994. A large-conductance mechanosensitive channel in E. coli encoded by MscL alone. Nature. 368:265–268. [DOI] [PubMed] [Google Scholar]
- Sukharev, S. I., P. Blount, B. Martinac, and C. Kung. 1997. Mechanosensitive channels of Escherichia coli—the MscL gene, protein, and activities. Annu. Rev. Physiol. 69:633–657. [DOI] [PubMed] [Google Scholar]
- Sukharev, S. I., W. J. Sigurdson, C. Kung, and F. Sachs. 1999. Energetics and spatial parameters for gating of the bacterial large conductance mechanosensitive channel, MscL. J. Gen. Physiol. 113:525–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templer, R. H., S. J. Castle, A. R. Curran, G. Rumbles, and D. R. Klug. 1998. Sensing isothermal changes in the lateral pressure in model membranes using di-pyrenyl phosphatidylcholine. Faraday Discuss. 111:41–53. [DOI] [PubMed] [Google Scholar]
- Venturoli, M., and B. Smit. 1999. Simulating the self-assembly of model membranes. Phys. Chem. Comm. 10:45–49. [Google Scholar]
- Xiang, T. X., and B. D. Anderson. 1994. Molecular distributions in interphases: statistical mechanical theory combined with molecular dynamics simulation of a model lipid bilayer. Biophys. J. 66:561–573. [DOI] [PMC free article] [PubMed] [Google Scholar]