

Abstract
Solid-state NMR methods employing 2H NMR and geometric analysis of labeled alanines (GALA) were used to study the structure and orientation of the transmembrane α-helical peptide acetyl-GWW(LA)8LWWA-amide (WALP23) in phosphatidylcholine (PC) bilayers of varying thickness. In all lipids the peptide was found to adopt a transmembrane α-helical conformation. A small tilt angle of 4.5° was observed in di-18:1-PC, which has a hydrophobic bilayer thickness that approximately matches the hydrophobic length of the peptide. This tilt angle increased slightly but systematically with increasing positive mismatch to 8.2° in di-C12:0-PC, the shortest lipid used. This small increase in tilt angle is insufficient to significantly change the effective hydrophobic length of the peptide and thereby to compensate for the increasing hydrophobic mismatch, suggesting that tilt of these peptides in a lipid bilayer is energetically unfavorable. The tilt and also the orientation around the peptide axis were found to be very similar to the values previously reported for a shorter WALP19 peptide (GWW(LA)6LWWA). As also observed in this previous study, the peptide rotates rapidly around the bilayer normal, but not around its helix axis. Here we show that these properties allow application of the GALA method not only to macroscopically aligned samples but also to randomly oriented samples, which has important practical advantages. A minimum of four labeled alanine residues in the hydrophobic transmembrane sequence was found to be required to obtain accurate tilt values using the GALA method.
INTRODUCTION
Membrane proteins perform many important functions in cells, but relatively little is known about the mechanisms by which they work. To understand the mode of action of membrane proteins on a molecular level, it is important to have a detailed knowledge of the structural properties of these proteins, such as precise backbone structure and tilt angles of the transmembrane segments. In addition, since membrane proteins are embedded in a lipid bilayer, it is important to know how and to what extent these properties can be influenced by the lipid environment. An important factor could be, for example, the extent of hydrophobic matching (reviewed in Killian, 1998). If the hydrophobic part of a transmembrane segment is long with respect to the thickness of the bilayer formed by the surrounding lipids, the protein may adopt a tilted orientation to achieve hydrophobic matching. Alternatively, such a mismatch could lead to (local) changes in peptide backbone structure which might decrease the effective length of the protein. Clearly, both types of mismatch responses could have functional consequences because they could influence the structure, and hence the activity of the protein.
Since large proteins are difficult to study in detail, a number of groups have started to study single transmembrane helices that mimic the transmembrane segments of membrane proteins (Huschilt et al., 1989; Killian et al., 1996; Harzer and Bechinger, 2000; Liu et al., 2001; Mall et al., 2001; Sharpe et al., 2002; Caputo and London, 2003; Subczynski et al., 2003; reviewed in De Planque and Killian, 2003). One example of these model peptides is the family of α-helical transmembrane model peptides called WALP, with an amino acid sequence given in Table 1. These peptides have a hydrophobic transmembrane stretch of alanines and leucines of variable length and are flanked on both sides by tryptophan residues. These flanking residues were chosen because aromatic amino acid residues are frequently found at the interfacial region in membrane proteins (Landolt-Marticorena et al., 1993; Arkin and Brunger, 1998), where they are believed to have favorable interactions with the lipid-water interface (Yau et al., 1998; Persson et al., 1998). Also in the WALP peptides interfacial anchoring properties of the tryptophan residues have been shown to play an important role in the interaction of the peptides with surrounding lipids (De Planque et al., 2003).
TABLE 1.
Amino acid sequences of the peptides used
| Peptide | Sequence |
|---|---|
| WALP19 | Acetyl-GWW(LA)6LWWA-ethanolamine |
| WALP23 | Acetyl-GWW(LA)8LWWA-amide |
The use of peptide families, such as the WALP peptides, allows systematic analysis of the effects of the lipid environment on structural and dynamic properties of transmembrane segments of α-helical membrane proteins. WALP peptides have been studied extensively in lipid bilayers with a variety of biophysical methods (Killian et al., 1996; De Planque et al., 1998, 2003; Morein et al., 2000; Rinia et al., 2000; Demmers et al., 2000; Strandberg et al., 2002). Circular dichroism and Fourier transform infrared experiments have shown that WALP peptides are mostly transmembrane and α-helical (Killian et al., 1996; De Planque et al., 1999; 2001). Recently, a solid-state NMR method was developed, based on geometric analysis of labeled alanines (GALA) to study the orientation and backbone structure of transmembrane peptides (Van der Wel et al., 2002). The method uses CD3 labels on different alanine residues in the transmembrane part of peptides. The peptide-containing bilayers are macroscopically oriented between glass plates, and the quadrupolar splittings from 2H NMR spectra are measured for each labeled position. The results are analyzed based on a peptide model with α-helical geometry. Due to this geometry, GALA is a very sensitive method to study peptide tilt and backbone conformation, allowing a high resolution of the tilt angle. Detection of changes in the tilt for a specific peptide, assuming that the dynamics do not change significantly, can be made with a resolution of <1°. The method was applied to study the backbone and tilt angle of WALP19 in lipid bilayers of phosphatidylcholine with different lengths of the hydrocarbon chains (Van der Wel et al., 2002). The tilt was found to be essentially independent of the lipid length, and even in the relatively short di-C12:0-PC only a very small tilt and no backbone distortions were found (Van der Wel et al., 2002).
In the present study a longer peptide, WALP23, is used together with the same lipids as in the previous WALP19 study, resulting in peptide/lipid systems with a significantly larger positive mismatch. Therefore, if hydrophobic mismatch is important for determining the tilt angles, a significant tilt could be expected for this peptide, in particular in the shortest lipids. WALP23 also has more alanines in the hydrophobic region, giving more potential data points to use in the analysis. Since this feature would yield more data points than would be required to determine the tilt angle for a regular α-helix, application of this method to WALP23 allows a detailed analysis of possible deviations from an α-helical structure if small changes in the backbone structure would occur. In addition, we explore the possibility of analyzing tilt and precise backbone structure by using nonoriented bilayers. Such an approach would not only have practical advantages; such samples also better mimic the situation in biological membranes, in terms of water content and lipid packing.
Our results show that analysis of peptide tilt in unoriented bilayers indeed is possible. In all lipid systems investigated, only a small tilt of WALP23 was observed, which increased slightly with the extent of mismatch. The results will be compared with those previously obtained on WALP19 and discussed in light of other hydrophobic mismatch responses.
MATERIALS AND METHODS
Materials
WALP23 (amino acid sequence acetyl-GWW(LA)8LWWA-amide) was synthesized using Fmoc/tBu solid phase peptide synthesis and purified as described elsewhere for related KALP peptides (De Planque et al., 1999). Deuterated L-alanine-d4 was obtained from Sigma-Aldrich (St. Louis, MO) and its amino functionality was protected by an 9-fluorenylmethyloxycarbonyl (Fmoc) group as described by Ten Kortenaar et al. (1986) before being used in the synthesis. The WALP peptides were isotopically labeled with one of the alanine residues in the transmembrane domain deuterium-labeled. 1,2-dilauroyl-sn-glycero-3-phosphatidylcholine (di-C12:0-PC), 1,2-ditridecanoyl-sn-glycero-3-phosphatidylcholine (di-C13:0-PC), 1,2-dimyristoyl-sn-glycero-3-phosphatidylcholine (di-C14:0-PC), and 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (di-C18:1-PC) were purchased from Avanti Polar Lipids (Alabaster, AL) and used without further purification. Trifluoroacetic acid (TFA) and 2,2,2-trifluoroethanol (TFE) were obtained from Merck (Darmstadt, Germany). Deuterium-depleted water was obtained from Sigma Aldrich. All other chemicals were of analytical grade. Water was deionized and filtered with a Milli-Q Water purification system from Millipore (Bedford, MA).
Methods
Sample preparation
Stock solutions were prepared of the lipid in chloroform and the amount of lipid was checked by a phosphorus assay. To remove residual traces of TFA in the peptide powder after synthesis, peptides were dissolved in 1 ml TFE and dried to a film in a rotavapor and resolubilized in TFE. This procedure was repeated twice. The peptide solution was then added to an appropriate amount of the lipid solution. The mixture was vortexed and dried to a film in a rotavapor. The samples were further dried overnight under vacuum.
For oriented samples, the dry lipid-peptide film was redissolved in 2 ml methanol/chloroform (1:1 volume ratio) and the solution was spread on 50 glass plates of size 24 × 4.8 × 0.07 mm (Marienfeld Laboratory Glassware, Lauda-Koenigshofen, Germany). Dry peptide/lipid films were obtained by subsequent air-drying followed by drying under vacuum overnight. The dry plates were stacked in a 27 × 6 × 6 mm glass cuvette under a N2(g) flow and hydrated with deuterium-depleted water to get a hydration of 40% (w/w). The cuvette was sealed with an end-glass plate using fast-drying epoxy glue. Samples were allowed to equilibrate at 37°C for typically one week, until they became transparent, which is an indication for good alignment on the glass plates. Subsequently samples were stored at room temperature before measurements. Typically 2 μmol of peptide and 40 μmol of lipid was used for each sample. Lower P/L ratios were tried in a few samples, by keeping the amount of peptide constant while increasing the lipid content. However, this resulted in less-well-oriented samples, probably due to the high amount of sample material per glass plate.
For unoriented samples, the dry lipid-peptide film was hydrated in 200–300-μl deuterium-depleted water and the suspension was transferred to 7-mm diameter glass tubes. The pH of the suspension was checked to be at 6 or above. The tubes were sealed with a silicon stopper and epoxy glue. In unoriented samples, typically 1 μmol of peptide and 100 μmol of lipid was used for each sample, with a peptide/lipid molar ratio of 1:100. In a test series with P/L between 1:20 and 1:200 it was found that P/L = 1:100 or lower gave better resolved splittings, whereas the value of the splitting did not depend on the peptide concentration in this range.
NMR spectroscopy
NMR experiments were carried out on a Bruker Avance 500 MHz NMR spectrometer (Bruker Biospin, Karlsruhe, Germany). Unless stated otherwise, measurements were performed at 40°C. Samples were allowed to equilibrate at this temperature for at least 10 min before measurements.
31P NMR experiments were performed at 202.5 MHz using a one-pulse experiment with a 13.4-μs 90° pulse, 1.3-s relaxation delay time, 100-kHz spectral width, 1024 data points, and gated proton-noise decoupling. Between 200 and 2000 scans were collected. Spectra were processed on the spectrometer by DC offset correction, zero-filling to 2048 data points, and a 50-Hz exponential multiplication before Fourier transformation.
2H NMR experiments were performed at 76.78 MHz using a quadrupolar echo experiment with a 5.6-μs 90° pulse, an echo delay of 50 μs, a 100-ms relaxation delay time, 1 MHz spectral width, and 1024 data points. Between 200,000 and 1,000,000 scans were collected. Acquisition was started before the echo and the time domain data was left-shifted to get the FID starting at the echo maximum before further processing by zero-filling to 8192 data points and a 100-Hz exponential multiplication followed by Fourier transformation.
Calculations
Quadrupolar splittings from the labeled positions were measured from 2H NMR spectra. The data were fitted to a model of an α-helix in a manner that parallels the previous procedure (Van der Wel et al., 2002), with small modifications. In oriented spectra the quadrupolar splittings from samples oriented with the bilayer normal parallel to the magnetic field direction were used. For unoriented samples, the measured splittings were multiplied with 2 to obtain comparable splittings.
Data were fitted to the equation
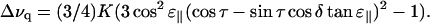 |
(1) |
Equation 1 is similar to the treatment of Jones et al. (1998) and Whiles et al. (2002), and is derived in the Appendix. Here, Δνq is the quadrupolar splitting, K is a constant with dimension frequency, and the angles τ, ɛ∥, and δ depend on the peptide geometry and orientation, and are defined in Fig. 1. The τ is the tilt angle, defined as the angle between the peptide helical axis and the bilayer normal (which is assumed to be along the magnetic field direction, Fig. 1 A), ɛ∥ is the angle between the peptide helix axis and the Cα-CD3 bond vector (Fig. 1 B), and δ is the rotation angle, defining the position of the bond vector around the helical axis (Fig. 1 C). In turn, δ depends on three angles as shown in Fig. 1 D,
 |
(2) |
where ρ is the rotation of the helix compared to a standard orientation with Cα of Gly1 in the xz plane, ɛ⊥ is the angle of the bond vector with respect to a vector from Cα to the peptide axis, and ϕ is the angle between the reference point Gly1 and the labeled residue in the peptide. For a regular helix, ϕ is given by −(n–1)ψ , where n is the residue number and ψ is the pitch angle. Using our notation, in an ideal α-helix, ψ = 100°, which value was used in the calculations unless otherwise stated; ɛ⊥ was estimated from molecular models using Insight II database to be −43.3°, which was used in all calculations; and τ, ρ, and ɛ∥ were used as fitting parameters in the calculations (see also Van der Wel et al., 2002).
FIGURE 1.

Definition of angles used in the calculations. (A) The tilt angle τ between the peptide axis and the bilayer normal (assumed to be along the magnetic field direction). (B) The bond angle ɛ∥ between the C-CD3 bond and the peptide axis. (C) The rotation angle δ giving the orientation of the C-CD3 bond. (D) The angle δ is determined by three contributions: ρ is the rotation of the whole peptide, defined as the anticlockwise rotation angle of Cα of Gly1, compared to the reference position; ϕ is the angle by which another amino acid residue is rotated around the peptide axis compared to Gly1; and ɛ⊥ is the angle of the Cα-CD3 bond projected onto a plane perpendicular to the helical axis compared to the direction from the peptide axis to the Cα. (Angles in the figure are chosen for clarity and do not represent real values.)
The constant K is defined as
 |
(3) |
where e2qQ/h is the quadrupolar coupling constant and S is an order parameter taking into account the molecular motion. For a deuterium bound to a carbon, the quadrupolar coupling constant is 167 kHz (Davis, 1983). In the case of methyl groups, this value is averaged by the fast rotation of the methyl groups giving a third of this value, or 56 kHz. Additional motions or a small deviation from tetrahedral geometry can cause further averaging, which is here included in the order parameter S. It was previously shown for WALP19 peptides that the fitting procedure was not very sensitive to changes of the value of K between 47 and 56 kHz (Van der Wel et al., 2002). In this study a K-value of 49 kHz was used, corresponding to S = 0.875, which was obtained from the splittings in a powder sample of dry WALP23-Ala-d4 peptide (data not shown).
Optimum values for τ, ρ, and ɛ∥ were calculated to minimize the error function
 |
(4) |
where the sum is over all labeled positions. All angles were varied in steps of 0.1°, τ was varied in the range 0–45°, and ρ in the range 0–360°, whereas ɛ∥ was only varied within a few degrees from 56.2° (the value for Ala in the Insight II database) until a minimum was found.
The error values for best-fits will be presented as root mean-square deviations (RMSD), defined as RMSD = (Error/number of data points)1/2.
RESULTS
WALP23 in di-C14:0-PC
WALP23 was labeled at different positions with deuterated alanine. As shown in Fig. 2 in a helical wheel model, these deuterated sites are regularly distributed around the helical axis. Oriented samples were prepared of all labeled peptides in di-C14:0-PC. The bilayer organization of the lipids and their predominant orientation with their long axis perpendicular to the glass plates was confirmed by 31P NMR (data not shown). 2H NMR spectra of these samples, measured with the orientation of the bilayer normal parallel to the external magnetic field (Fig. 3, left column), show that the magnitude of the 2H splittings varies with the position of the labeled site around the helical axis, as was previously also observed for WALP19 (Van der Wel et al., 2002). The distribution of splittings is clearly nonrandom, with every other alanine showing a large splitting, and with small splittings for the intervening alanines. Since the angular distance between two consecutive alanines in a regular α-helix is 200° (Fig. 2), this pattern suggests that alanines on one face of the peptide exhibit larger splittings, whereas on the opposite face the splittings are smaller. Such a behavior would be consistent with that of a helix that is tilted away from the bilayer normal.
FIGURE 2.

Helical wheel projection of WALP23, with Gly1 at the top. The positions of the labeled alanine residues are marked with circles, and the tryptophan residues are marked with rectangles. The arrow marks the side of the peptide pointing away from the membrane normal in di-C14:0-PC bilayers.
FIGURE 3.

2H NMR spectra of WALP23/di-C14:0-PC for the different labeled alanine residues. The row with a deuterium label at position 5 is indicated by A5, etc. (Left column) Oriented samples with the bilayer normal parallel to the magnetic field. (Middle column) Oriented samples with the bilayer normal perpendicular to the magnetic field. (Right column) Unoriented samples. The isotropic peak in the middle of the spectra originates from residual deuterium in H2O.
When oriented samples are measured with the orientation of the bilayer normal perpendicular to the external magnetic field (Fig. 3, middle column), the quadrupolar splittings are reduced with a factor of two, within the margin of error (see below) of the measurements (Table 2). This observation indicates that the peptides are rotating fast with respect to the bilayer normal (Killian et al., 1992; Van der Wel et al., 2002). The results also indicate that the peptide does not rotate around its own helical axis, since this motion would average all the splittings to the same value, at least for a regular α-helix conformation. An implication of fast rotation about the bilayer normal, but not around the helical axis, is that it should be possible to obtain similar information from quadrupolar splittings in nonoriented samples as from oriented samples. In unoriented samples, the main splittings should correspond to those from oriented samples for which the bilayer normal is perpendicular to the magnetic field. The right column of Fig. 3 shows spectra from unoriented samples, and the splittings are indeed close to those of the middle column, as shown in Table 2.
TABLE 2.
Measured 2H NMR splittings of unoriented and oriented di-C14:0-PC samples in kHz
| Peptide | di-C14:0-PC oriented 0° | di-C14:0-PC oriented 90° | di-C14:0-PC unoriented |
|---|---|---|---|
| WALP23A5 | 9.3 | 4.3 | 4.7 |
| WALP23A7 | 1.0* | 0.5* | 0.5* |
| WALP23A9 | 11.3 | 5.8 | 6.6 |
| WALP23A11 | 2.0* | 1.0* | 2.3 |
| WALP23A13 | 12.8 | 5.65 | 7.75 |
| WALP23A15 | 1.0* | 0.5* | 0.5* |
| WALP23A17 | 12.3 | 6.0 | 6.5 |
| WALP23A19 | 2.0 | 1.5 | 0.6 |
Splittings that could not be resolved are marked with an asterisk and an estimated value is given.
WALP23 in different lipid systems
Next, samples were studied using unoriented di-C12:0-PC, di-C13:0-PC, and di-C18:1-PC bilayers. Control experiments using 31P NMR demonstrated that in all of these samples the lipids are organized in a bilayer (data not shown). Fig. 4 shows selected 2H NMR spectra. The splittings for all labeled positions in WALP23 in unoriented samples for di-C12:0-PC, di-C13:0-PC, di-C14:0-PC, and di-C18:1-PC are given in Table 3. It can be noted that splittings are rather similar in di-C14:0-PC and di-C18:1-PC samples, with slightly larger splittings for di-C14:0-PC. Along the peptide sequence, the splittings again vary systematically between large and small for every other splitting.
FIGURE 4.

2H NMR spectra for labeled alanines at positions 5, 7, and 11 in WALP23 incorporated in bilayers of di-C12:0-PC (top row), di-C13:0-PC (middle row), or di-C18:1-PC (bottom row). For details see text.
TABLE 3.
Measured 2H NMR splittings of unoriented samples in kHz
| di-C12:0-PC unoriented | di-C13:0-PC unoriented | di-C14:0-PC unoriented | di-C18:1-PC unoriented | |
|---|---|---|---|---|
| WALP23A5 | 4.75 | 5.3 | 4.7 | 4.7 |
| WALP23A7 | 5.5 | 3.5 | 0.5* | 0.35 |
| WALP23A9 | 7.6 | 7.5 | 6.6 | 5.9 |
| WALP23A11 | 5.45 | 4.4 | 2.3 | 0.5* |
| WALP23A13 | 8.65 | 8.7 | 7.75 | 6.7 |
| WALP23A15 | 2.05 | 1.65 | 0.5* | 0.5* |
| WALP23A17 | 7.1 | 6.7 | 6.5 | 5.6 |
| WALP23A19 | 0.75 | 0.75 | 0.6 | 1.65 |
Splittings that could not be resolved are marked with an asterisk and an estimated value is given.
In the series of saturated lipids of different length, di-C12:0-PC, di-C13:0-PC, and di-C14:0-PC, most splittings are similar but there are some systematic differences. In general, the splittings are larger in shorter lipids. In particular, WALP23A7 and WALP23A11 show a small splitting in di-C18:1-PC, but the splitting increases considerably as the lipids are shortened, as illustrated in Fig. 4. Interestingly, these two positions, 7 and 11, are approximately one helical turn from each other and on the same face of the helix (Fig. 2).
In the case of di-C14:0-PC, the full series of labeled positions have been examined in both oriented and unoriented samples. For each of the other lipids, three oriented samples of different labeled peptides were also made for a comparison with the unoriented samples. In all cases the observed quadrupolar splittings differed <0.5 kHz between oriented and unoriented samples (data not shown), indicating that the peptides behave very similarly in the two types of sample systems.
Error estimate
The error in the measurement of the quadrupolar splitting in each data point is estimated from duplicate samples to be ∼0.5 kHz. An error of this size must be considered rather small, because at the sensitive region around ɛ∥ = 56° a change in the peptide tilt of only 1° can already change the splitting up to 2 kHz, depending on the position around the peptide axis. The splitting is also very sensitive to the exact value of ɛ∥. Assuming that the peptide does not significantly change its structure and dynamics, the GALA method is able to detect very small changes in, for example, tilt angles.
Calculation of tilt angles
Next, the experimental values of the quadrupolar splittings were fitted to a model of an α-helical peptide in a tilted membrane-spanning orientation as described above. Experimental data from WALP23 in unoriented di-14:0-PC bilayers are presented in Fig. 5 together with the theoretical best-fit curve. In this figure, data are given for the different alanine positions according to their relative positions around the helical axis. In this case, the position of the residue Gly1 is defined as 0°, placing Trp3 at 200°, Ala5 at 400° (= 40°), and so on.
FIGURE 5.

Data of WALP23 in unoriented di-C14:0-PC bilayers given together with a theoretical best-fit curve corresponding to a tilt angle of 5.5°, a rotation angle of 158.3°, and a bond angle ɛ∥ of 58.2°. The error bar shows 1 kHz.
Best-fit values for WALP23 in all lipid systems investigated are given in Table 4. The results clearly show that there is a change to larger tilt angles of WALP23 when shorter lipids are used. The changes are small, as the tilt angle only changes from 4.4° to 8.2°. The rotation angle is similar in different lipids, indicating that the peptide is tilting in almost the same direction in all cases.
TABLE 4.
Best-fit results for WALP23 (unoriented) using data from all labeled positions
| Tilt angle τ/° | Rotation angle ρ/° | RMSD error (kHz) | Bond angle ɛ∥/° | |
|---|---|---|---|---|
| di-C12:0-PC | 8.2° | 142.7 | 2.19 | 56.9° |
| di-C13:0-PC | 7.4 | 145.4 | 1.41 | 57.4 |
| di-C14:0-PC | 5.5 | 158.3 | 0.90 | 58.2 |
| di-C18:1-PC | 4.4 | 153.5 | 0.48 | 58.1 |
| di-C14:0-PC oriented | 4.5 | 158.0 | 0.73 | 58.0 |
The angle between the Cα-CD3 bond and the helix axis (ɛ∥ in Eq. 1) is difficult to determine with a high accuracy from existing experimental data; for example, from crystal structures of membrane proteins. According to the Insight II database the angle is 56.2°, but this angle may fluctuate or deviate (the Insight II database gives values between 55° and 60° for different side chains in an α-helix). For di-C14:0-PC and di-C18:1-PC, a bond angle of ∼58° was found to give the best fit to the experimental data, in good agreement with our earlier findings of 58.8° and 59.2° for WALP19 in di-C14:0-PC and di-C18:1-PC, respectively (Van der Wel et al., 2002). For the shorter di-C13:0-PC and di-C12:0-PC lipid systems, slightly smaller ɛ∥-angles were suggested by the calculations. Also the RMSD values were found to increase with shorter lipids, suggesting that perhaps slight distortions of the backbone occur with increasing mismatch. However, in all lipids, the RMSD values can be reduced to zero if each of the backbone angles is allowed to fluctuate slightly from its average value. For di-C18:1-PC a fluctuation of only ∼0.4° in ɛ∥ would be required to obtain an RMSD value of zero; in di-C14:0-PC the errors are larger and a fluctuation of 0.9° is required, but even for the worst fit, in di-C12:0-PC, a maximum deviation of only ∼±1.8° would be sufficient. It is important to note that, although varying the ɛ∥-angle within a few degrees resulted in improved fits (lower RMSDs), it did not influence the optimal values of tilt angle and rotation angle. For example, if ɛ∥ was set to 55° (uniformly for all residues) in di-C14:0-PC, the best-fit values were τ = 5.4° and ρ = 149.8°, very close to the optimal values, even though the RMSD error was as large as 5.21 kHz.
The molecular model that we used per se does not assume a specific type of helix, but in all fits shown until this point, the angle between consecutive amino acid residues was fixed to 100°, the value for an α-helix. When this angle (ψ) was also allowed to vary, together with the backbone/side-chain angle (ɛ∥), it was found that the angle between consecutive residues giving the best-fit remains near 100° for di-C18:1-PC, di-C14:0-PC, and di-C13:0-PC, as illustrated in Fig. 6 for di-C14:0-PC. A large region of (ψ, ɛ∥) space was sampled where different helical structures are located. In the figure is shown the most relevant region where RMSD values below 4 kHz were found. Low RMSD values were also found in mirror regions (not shown) at the line ψ = 90°, which was due to the symmetry of the equations, and around the magic angle at ɛ∥ = 54.7°, where there is a pseudo-mirror symmetry due to the lack of sign of the quadrupolar splitting. The fit indicates that the structure is not changed to another type of helix. However, for di-C12:0-PC, there was not a good fit, and there was no clear minimum, which could indicate a distortion of the peptide backbone for large mismatch. The most likely position for such a distortion would be near Ala7 and Ala11, because at this position a prominent increase of the splittings was found upon decreasing lipid-chain length (Table 3).
FIGURE 6.

Figure of best fit for different pitch angle (ψ) and bond angles (ɛ∥) for WALP23 in unoriented di-C14:0-PC. The best fit is found for ψ = 99.5°, corresponding closely to an α-helical configuration of the peptide. The scale is RMSD error in kHz.
Combined analysis in different lipid systems
The results above used eight data points in each lipid system to get a fit, giving four separate fits where not only the tilt angle but also the bond and rotation angles were allowed to vary between lipid systems. Using the assumption that the peptide structure is the same and that only the tilt of the peptide should change depending on the lipid chain length, another fit was made, using the data from all lipid systems, keeping the bond and rotation angles the same and only allowing variation of the tilt angle in each lipid system. In this way, 32 data points are used to obtain six parameters in total. Interestingly, values of the rotation and bond angle were found that corresponded to an average of the values obtained in the individual lipid systems (ɛ∥ = 57.6° and ρ = 146.9°). The four tilt angles were found to be almost exactly the same as in the separate fits: 4.4° in di-C18:1-PC, 5.5° in di-C14:0-PC, 7.5° in di-C13:0-PC, and 8.3° in di-C12:0-PC. The RMSD error in this combined fit was quite large, 1.71 kHz. A more detailed analysis indicated that most of the error was due to the problem of fitting data from Ala7 and Ala11 in the shortest lipids. When these two residues were not included in the fit, using the other 24 data points a much smaller RMSD of 0.61 kHz was found, together with ɛ∥ = 58.3° and ρ = 157.8°, which is close to the values found in di-C14:0-PC or di-C18:1-PC. The best-fit tilt angles in this latter analysis were 4.3° in di-C18:1-PC, 5.3° in di-C14:0-PC, 6.4° in di-C13:0-PC, and 6.6° in di-C12:0-PC. This fit suggests that there indeed could be some local distortion of the peptide at positions 7 and 11 in shorter lipids, possibly affecting the ɛ∥-angles at these positions, whereas the rest of the peptide is well described by an ideal α-helix.
Number of data points required for accurate tilt angle determination
In WALP23 eight positions have been deuterium-labeled, giving eight data points to use in the fits for each lipid system. An analysis was made of how the results depend on the number of data points used. Since the best fit was found for di-C18:1-PC and the worst for di-C12:0-PC, a comparison was made for these two lipid systems. The results are shown in Fig. 7 as a plot of the maximum and minimum values of tilt, rotation angles, and RMSD error, obtained for different combinations of different numbers of the eight data points. For each parameter, the range of values is clearly decreasing using more data points.
FIGURE 7.

Results when a different number of data points were used in the fits, in di-C18:1-PC (top row) and di-C12:0-PC lipids (bottom row). For a certain number of data points, all different combinations of data points were used to get best-fit results. The squares indicate the maximum and minimum values, and the lines indicate the region where values are found, of each parameter for each number of data points used. In all cases the ɛ∥-angle was fixed to the optimal value from Table 4.
For di-C18:1-PC, using all eight data points, the fit yields a tilt angle of 4.4°. Using all possible combinations of only three data points yields tilt angles between 3.0° and 8.6°. However, even in this case, 54 of 56 choices of the set of the three data points gave tilt angles between 3.0° and 4.9° (not shown). Using four data points, the calculated tilt is between 3.7° and 4.8°, in reasonably good agreement with the best fit from all eight data points. In all cases the maximum RMSD value is small, near 0.6 kHz. Thus, it would seem that using four labels is sufficient to get a reasonably reliable determination of the tilt angle for WALP23 in di-C18:1-PC.
For di-C12:0-PC, the fit is much less good than for di-C18:1-PC, as seen from the much larger RMSD error. For this system, six data points seem to be required to narrow the range to only ±0.8° from the value of 8.2° that is obtained with all data points. Five data points gives a range of ±1.3° and four or fewer data points gives a very wide range of values.
A similar behavior is found for the rotation angle, where the use of more data points narrows the range of the best-fit values. For di-C18:1-PC already at four data points a narrow range of rotation angles close to 150° is observed, but for di-C12:1-PC six data points are required to obtain such a narrow range.
The RMSD error for a smaller data set can be either larger or smaller than for the full set of eight data points, which is expected if there is a random error in each data point. This means that for some small sets of data, a very small RMSD error may be found fortuitously, even if the calculated tilt and rotation angles are far away from the “real” values. For example, the combination of three data points (residues 5, 17, and 19) giving the most deviating values for di-C18:1-PC, gave a tilt of 8.6° and ρ = 274.5° while showing a very small RMSD value of 0.06 kHz.
As a conclusion, it would seem that, depending on the experimental errors (or on how close the peptide conformation resembles an ideal α-helix), six data points are sufficient to obtain a reliable value of the tilt angle, and four labeled positions is probably a minimum. Results from three or fewer labeled positions should be used only with great caution.
Calculation for the N- and C-terminal parts of the peptide
The result above that four labeled positions can be enough for the analysis, allowed us to test the possibility of a bent or kinked helix. This possibility was investigated by independent calculations of the tilt and rotation angles for the four N-terminal labeled residues (5, 7, 9, 11) and the four C-terminal labels (13, 15, 17, 19). In these calculations the same ɛ∥-angles were used for the different lipid systems as the best-fit values in Table 4. For WALP23 in di-C18:1-PC, di-C14:0-PC, and di-C13:0-PC bilayers the tilt differed <1° between the two parts of the peptide, and the rotation angle differed <10°. This is within the error of the method and indicates that the peptide is very straight and regular in all these lipid systems. In contrast, for WALP23 in di-C12:0-PC, the best-fit value of the tilt was 19.4° for the N-terminal part of the peptide and 7.7° for the C-terminal part. The rotation angles changes from 201° in the N-terminal part to 139° in the C-terminal part. This could be an indication that the peptide forms a kink, or bends when the hydrophobic mismatch is too large. It should be noted, however, that this model gives only a slight decrease in the overall RMSD for WALP23 in di-C12:0-PC, from 2.19 to 1.65 kHz, which is still higher than in the other lipid systems (see Table 4).
DISCUSSION
In this study, we use solid-state NMR on isotopically labeled WALP23 peptides to study the structure and dynamics of transmembrane peptides in a lipid bilayer. First, we will discuss the results from oriented and unoriented samples in di-C14:0-PC. Then we will discuss the effect of hydrophobic mismatch, as investigated by using lipids with different chain length. The results will be compared to those obtained from the shorter WALP19 peptide (Van der Wel et al., 2002). Finally we will discuss some advantages and disadvantages of different NMR methods to study tilt of transmembrane peptides.
Behavior of WALP23 in di-C14:0-PC
The 2H NMR spectra of the WALP23 peptide in all investigated lipid systems shows a variation of quadrupolar splittings from different labeled alanine positions. For a peptide with a regular α-helical structure this indicates that the peptide is tilted and not rotating fast around the peptide axis. The tilt is quite small (4.4–8.2°) and comparable to that previously obtained for the shorter but otherwise similar WALP19 (3.6–4.0°, Van der Wel et al., 2002). Oriented samples give rise to twice-as-large quadrupolar splittings when the bilayer normal is oriented parallel to the magnetic field than when oriented perpendicular, indicating that the peptide is rotating fast around the bilayer normal. A similar peptide motion was found previously for WALP19 (Van der Wel et al., 2002) as well as for other synthetic transmembrane peptides (Jones et al., 1998; Kovacs et al., 2000; Morrow and Grant, 2000; Sharpe et al., 2001; 2002; Whiles et al., 2002). These are nontrivial results; for if the peptide axis is not tilted with respect to the bilayer normal, then the two motions, rotation about the bilayer normal and rotation about the helix axis, would be identical.
The rotational motion could be explained by a random diffusion of the peptide in the bilayer, while tilted in a specific preferred direction. The reason for this preferred rotational angle could be a favorable conformation of the large and bulky flanking tryptophan residues, in combination with a favorable interaction with the lipid/water interface, where tryptophans have been proposed to anchor. The best-fit value of the rotation angle in each lipid (Table 4) can be visualized as the direction of tilt of the peptide, telling which residue is pointing up against the bilayer normal in the tilted peptide. In Fig. 2, the rotation angle for WALP23 in unoriented di-C14:0-PC bilayers is indicated with an arrow. In other words, Cα of Ala17 is in the plane formed by the bilayer normal and the peptide axis, pointing away from the membrane normal, where the maximum splittings are found. It is likely that some (limited) rotational motion around the peptide axis is present, and that the observed value is an average orientation. The fit is much less sensitive to the parameter ρ than to the tilt angle τ, and a small change does not much influence the fit. It is therefore interesting that the rotation angle is very similar in all lipid systems for WALP23, ∼150° ±8°. This is somewhat lower than the rotation angles found for WALP19 in the same lipid systems, ∼192° ±20°.
Comparison of oriented and unoriented systems
It is shown here that the GALA experiments can be performed on both oriented and unoriented samples. The same information is obtained, but the use of unoriented samples has a number of practical advantages: 1), it allows a wider application of the method, e.g., to investigate the effects of lipids that are notoriously difficult to orient or to analyze the effects of environmental factors such as salt concentration or pH; 2), it is less time-consuming to prepare samples; 3) using the same sample volume, much more material may be used, allowing us to obtain a better signal/noise ratio or save spectrometer time; and 4), unoriented samples better mimic biological membranes in terms of hydration and lipid packing. In the literature there are several examples where such unoriented systems have been successfully used to investigate structure and dynamics of single-span membrane peptides by solid-state NMR methods (Jones et al., 1998; Sharpe et al., 2002).
The splittings are rather similar in oriented and unoriented samples, especially considering the fact that they are very sensitive to small changes in tilt or ɛ∥. Indeed, there is only a small change of best-fit tilt value for WALP23 in di-C14:0-PC from 4.5° in oriented samples to 5.5° in nonoriented samples. A possible explanation for the difference is that in the oriented samples the bilayer thickness is larger because the lipids are more ordered. It is also possible that the macroscopic orientation of lipid bilayers on glass plates can make the bilayers less flexible, which could make it more difficult for peptides to tilt. The lower water content in the oriented samples may have a similar effect. It may also be noted that the peptide concentration is lower in the unoriented samples where P/L = 1:100 whereas in the oriented samples P/L = 1:20. In total, the changes of splittings are small and the peptide tilt seems not to be much affected by the different conditions between oriented and unoriented samples.
Effects of hydrophobic mismatch
Hydrophobic mismatch is expected to be energetically unfavorable, and therefore there should be some compensating mechanism to minimize it. A too-long peptide could tilt, which would reduce the effective hydrophobic length. The nonzero tilt of WALP19 was found to be close to 4° in oriented samples of di-C12:0-PC, di-C14:0-PC, and di-C18:1-PC (Van der Wel et al., 2002). In the case of WALP19 it could be that the mismatch is small enough to be compensated by other mechanisms. WALP23 has four more amino acids in the α-helix, increasing its length with 6 Å, so for this peptide a much larger mismatch is expected. As shown in Table 4, the tilt in di-C18:1-PC is still low, ∼4.5°, but it increases in the shorter lipids. However, much larger tilt angles would be needed to provide a considerable relief of the mismatch. In a simple model, the effective hydrophobic length is the projection of the peptide length in the membrane, Leff = Lpcosτ, where Lp is the peptide length, τ is the tilt angle, and Leff should be equal to the lipid bilayer hydrophobic thickness. Let us assume that WALP23 in di-C18:1-PC is matching when the tilt is the 4.4° found in this study. The hydrophobic thickness of di-C18:1-PC is 27 Å (Nagle and Tristram-Nagle, 2000). This implies that to get matching conditions for WALP23 in di-C12:0-PC, with a hydrophobic thickness of 20 Å (De Planque et al., 1998), the peptide needs to tilt by 42°. The much smaller change from 4.4° to 8.2° observed here would only change Leff from 27.0 Å to 26.8 Å.
The system should proceed to a state of lowest free energy, and apparently a tilt of the peptide will not give the lowest energy, even if it results in a decrease of the mismatch. Either there are other mechanisms to relieve mismatch, or peptide tilt is more unfavorable than mismatch. Indeed, it is quite feasible that there is some energy term that makes tilting unfavorable, such as problems with lipid packing around a tilted peptide. Such packing problems may be less for peptides that form oligomeric structures, such as the M2 transmembrane peptide from Influenza A virus, for which rather large tilt angles were observed (Kovacs et al., 2000). Packing problems of course could be lipid-specific, and therefore it would be interesting to also investigate tilting of peptides in other lipid systems. The use of unoriented samples will make it possible to study a wide range of different lipids and different environmental conditions.
In principle, besides tilting, the peptides could also change their length by a change of conformation, which would mean to distort the backbone configuration away from an ideal α-helix. Our results show that the peptides do not change their helical pitch: they remain in a very stable α-helical conformation. However, there are some variations in the bond angle (ɛ∥). The results suggest a tendency of the angle to increase with decreasing mismatch. This was also observed for WALP19 (Van der Wel et al., 2002) and suggests that there may be very small, but systematic changes in backbone structure with increasing mismatch. Such changes would be extremely small because they could not be observed by Fourier transform infrared spectroscopy (De Planque et al., 2001).
Overall, the results suggest that the WALP peptides form surprisingly regular α-helices without distortions or kinks when there is a moderate extent of mismatch. However, for the shorter lipids the observed increase of the RMSD value may indicate slight local distortions of the backbone at increasing mismatch. The largest variations in quadrupolar splitting in different lipid systems are found at positions 7 and 11. When these positions are excluded from the fitting procedure, almost no difference in best-fit parameters is found in di-18:1-PC lipids, and the whole peptide fits very well with an ideal α-helical structure. In shorter lipids, these two residues yield increasingly larger quadrupolar splittings that are also increasingly away from the best-fit curve using the other six residues. One explanation would be that the bond angles ɛ∥ at positions 7 and 11 become distorted in shorter lipids, although leaving the backbone structure mostly intact. Since Ala7 and Ala11 are at the same face of the peptide (see Fig. 2), we may speculate that they could be part of a site of peptide-peptide interactions, and that the occurrence of such interactions may become more likely as the hydrophobic mismatch increases. Such peptide-peptide interactions may reduce the sensitivity of the peptide to mismatch, as was previously proposed for the Influenza M2 peptide (Kovacs et al., 2000).
Another possible explanation for the large RMSD values in the shortest lipids would be that the peptide changes from a straight to a bent helix. The calculations of tilt for the first and last part of the helix indicated that the peptide is not bent in the longer lipid systems, where the tilt of the first and second part of the helix was within the error of the method. Only in di-C12:0-PC was there a large difference between the N- and C-terminal parts, which could indicate that the peptide is bent or kinked. However, the fit is not very good even when a kink is included in the model, indicating some more distortions are present. A kinked α-helix has recently been observed by solid-state NMR methods in the channel-forming transmembrane domain of virus protein u (Vpu) of HIV-1 (Park et al., 2003).
Our experiments indicate that mismatch does not result in significant tilting or in clearly observable backbone adaptation, at least in the systems that we used here. Other mechanisms to compensate for hydrophobic mismatch could include stretching or compressing of the lipid acyl chains. However, results from 2H NMR studies on effects of different length WALP peptides on acyl chain order of perdeuterated lipids in unoriented peptide/lipid systems indicated systematic, but only very small mismatch-dependent, changes in bilayer thickness (De Planque et al., 1998). Moreover, in a recent x-ray diffraction study (Weiss et al., 2003), using oriented peptide/lipid bilayers and a lower water content, no change at all was found in bilayer thickness upon incorporation of different length WALP peptides.
Still another possible explanation for the lack of large mismatch-dependent effects is related to the observation that Trp-interfacial anchoring effects are more dominant than hydrophobic mismatch effects for peptides with a hydrophobic Leu-Ala core (De Planque et al., 2003). We speculate that, although Trp may prefer a defined localization at the interface, it can be located within a rather broad interfacial region >10 Å wide (Yau et al., 1998), at only a relatively small energetic cost, which may be further minimized by allowing reorientation of the Trp χ1 and χ2 side-chain angles. If the peptide is too long for the outer limit of this permitted range, mismatch effects like lipid stretching, peptide tilt, or other structural changes could become more pronounced. Thus, larger tilt values might be found in systems with an even larger mismatch than in this study.
Tilt determination of α-helices using different NMR methods
The GALA method to study tilt, rotation, and backbone conformation of transmembrane peptides uses CD3-labeled peptides studied by 2H NMR. So far, 15N in the peptide backbone amide has been the most commonly used isotopic label for NMR studies of peptides in lipid bilayers. The GALA method can complement 15N NMR experiments.
A practical advantage of the GALA method is that the labeled methyl groups, due to their intrinsic mobility and the presence of three chemically equivalent deuterons in each side chain, give rise to high intensity signals, which can easily be monitored in a simple one-dimensional NMR experiment. The low sensitivity of 2H is partly compensated by a short relaxation time. On the other hand, 15N is a label that can be incorporated in any peptide, irrespective of the content of alanine residues, also biosynthetically.
The two labels are independent, with the relevant C-CD3 bond forming an angle to the peptide helix axis of ∼58° (this article), whereas the corresponding angle for an NH bond is only ∼15° (Mesleh et al., 2003). To illustrate the importance of this, consider a peptide in oriented bilayers. When the peptide is tilted, for example, 10°, the angle θ between the N-H or C-CD3 bond vector and the magnetic field will vary around the helix axis. The N-H θ angle will vary between 5° and 25°, and the C–CD3 θ-angle will vary between 48° and 68°. There is a (3 cos2 θ−1) variation of the measured value (quadrupolar splitting for 2H and chemical shift for 15N), and from the graph of this function (Fig. 8) it is clear that the slope is higher close to 58°, whereas at 15° the function changes more slowly. In this respect, CD3 is a more sensitive probe.
FIGURE 8.

A plot of the function 3 cos2 θ–1, illustrating why Ala-CD3 labels are more sensitive to small tilt values than 15N labels. The boxes represent ±10° in θ and the corresponding changes in the value of 3 cos2 θ–1. The slope at 56° is approximately twice as large as at 15°.
A curve as in Fig. 8 can also be used to explain why the results for the GALA method in oriented bilayers will not be extremely sensitive to the orientation of the glass plates in the magnetic field. If the angle between the bilayer normal and the magnetic field is denoted β, there is a 3 cos2 β−1 dependence of the splittings. Thus, for glass plates oriented with their normal vector at an angle of 0° or 90° to the magnetic field direction, a deviation of the glass plates' orientation with 5° will only lead to a decrease of Δνq of 1%.
Although a larger value of ɛ∥ increases the sensitivity to tilt angles, small values of ɛ∥, as for NH labels, have the advantage that there is not a large change in quadrupolar splittings or chemical shift between amide labels at different positions around the helix. This means that a single 15N label allows a rough estimate of the tilt of an α-helical peptide (shown for several peptides in Bechinger et al., 1996), which is not the case for alanine-d3 labels.
It must also be noted that two-dimensional methods, where 15N-1H dipolar couplings are measured together with 15N CSA, are able to provide atomic resolution structures of peptides and proteins in lipid bilayers (Ketchem et al., 1996; Zeri et al., 2003). The tilt and rotation of peptides and proteins can be accurately determined using PISEMA/PISA wheel, or dipolar wave experiments (Wu et al., 1994; Tian et al., 1998; Marassi and Opella, 2000; Wang et al., 2000; Mesleh et al., 2003).
In conclusion, our results show that GALA is a useful method to obtain high resolution information on the structure and orientation of peptides in lipid bilayers, with high precision and sensitivity. Hence, the method may allow monitoring of very subtle changes in tilt angle or backbone conformation of transmembrane segments that can be important for functional activity of membrane proteins. The method is complementary to previously used 15N NMR methods.
Acknowledgments
This work was supported by the Dutch Organization for Scientific Research and by grant MCB9816063 from the U.S. National Science Foundation.
APPENDIX
Derivation of an expression for the quadrupolar splitting in terms of tilt, rotation, and bond angles
In the derivation a methyl (CD3) group will be assumed, but it is equally valid for a carbon-deuterium bond (CD). The same angles defining the peptide orientation and structure as illustrated in Fig. 1 will be used throughout this derivation.
The quadrupolar splitting Δνq is given by
 |
(A1) |
where K is a strength constant as defined in Methods and θ is the angle between the C-CD3 bond and the magnetic field direction.
The bond vector of length r can now be located around the cone shown in Fig. 9 A. The z direction is defined to be along the magnetic field and the x axis is defined so that the center of the cone lies in the xz plane. The center of the cone is defined by the angle τ, the width of the cone by the angle ɛ∥, and the position of the bond around the cone by the angle δ. (Note that the values of the angles in the figures are chosen for clarity and may not be realistic.) From Fig. 9 A it can be seen that the angle θ is given by
 |
(A2) |
where z is the projection of the bond vector to the z axis. The next step is to find an expression for z.
FIGURE 9.

(A) The cone formed by the possible orientations of the C-CD3 bond vector. (B) The circle where the CD3 group can be located. (C) The circle in B shown from the side. The angles and distances are described in the text.
First, the CD3 group can be located on a circle of radius d shown in Fig. 9 B. As seen in Fig. 9 B, δ is defined as the anticlockwise angle between the bond and the a axis, with the a axis lying in the xz plane. Fig. 9 C gives the projection of the circle in Fig. 9 B to the xz plane. It is clear that
 |
(A3) |
where z0 is the position of the center of the circle. From Fig. 9 C we get
 |
(A4) |
 |
(A5) |
From Fig. 9 B we get
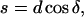 |
(A6) |
and from Fig. 9 A we see
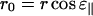 |
(A7) |
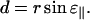 |
(A8) |
We can now combine Eqs. A4 and A7 to get
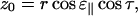 |
(A9) |
and from Eqs. A5, A6, and A8 we get
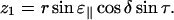 |
(A10) |
Combining Eqs. A2, A3, A9, and A10 we get the expression
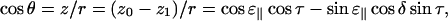 |
(A11) |
which gives
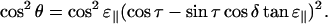 |
(A12) |
The final expression, Eq. 1, is now obtained by combining Eqs. A1 and A12:
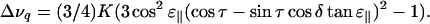 |
(A13) |
Erik Strandberg's present address is IFIA, Forschungszentrum Karlsruhe, POB 3640, 76012 Karlsruhe, Germany.
References
- Arkin, I. T., and A. T. Brunger. 1998. Statistical analysis of predicted transmembrane α-helices. Biochim. Biophys. Acta. 1429:113–128. [DOI] [PubMed] [Google Scholar]
- Bechinger, B., L. M. Gierasch, M. Montal, M. Zasloff, and S. J. Opella. 1996. Orientations of helical peptides in membrane bilayers by solid-state NMR spectroscopy. Solid State Nucl. Magn. Reson. 7:185–191. [DOI] [PubMed] [Google Scholar]
- Caputo, G. A., and E. London. 2003. Cumulative effects of amino acid substitutions and hydrophobic mismatch upon the transmembrane stability and conformation of hydrophobic α-helices. Biochemistry. 42:3275–3285. [DOI] [PubMed] [Google Scholar]
- Davis, J. H. 1983. The description of membrane lipid conformation, order and dynamics by 2H-NMR. Biochim. Biophys. Acta. 737:117–171. [DOI] [PubMed] [Google Scholar]
- De Planque, M. R. R., B. B. Bonev, J. A. A. Demmers, D. V. Greathouse, R. E. Koeppe II, F. Separovic, A. Watts, and J. A. Killian. 2003. Interfacial anchor properties of tryptophan residues in transmembrane peptides can dominate over hydrophobic matching effects in peptide-lipid interactions. Biochemistry. 42:5341–5348. [DOI] [PubMed] [Google Scholar]
- De Planque, M. R. R., E. Goormaghtigh, D. V. Greathouse, R. E. Koeppe II, J. A. W. Kruijtzer, R. J. M. Liskamp, B. de Kruijff, and J. A. Killian. 2001. Sensitivity of single membrane-spanning α-helical peptides to hydrophobic mismatch with a lipid bilayer: effects on backbone structure, orientation, and extent of membrane incorporation. Biochemistry. 40:5000–5010. [DOI] [PubMed] [Google Scholar]
- De Planque, M. R. R., D. V. Greathouse, R. E. Koeppe II, H. Schäfer, D. Marsh, and J. A. Killian. 1998. Influence of lipid/peptide hydrophobic mismatch on the thickness of diacylphosphatidylcholine bilayers. A 2H NMR and ESR study using designed transmembrane α-helical peptides and gramicidin A. Biochemistry. 37:9333–9345. [DOI] [PubMed] [Google Scholar]
- De Planque, M. R. R., and J. A. Killian. 2003. Protein-lipid interactions studied with designed transmembrane peptides: role of hydrophobic matching and interfacial anchoring. Mol. Membr. Biol. 20:271–284. [DOI] [PubMed] [Google Scholar]
- De Planque, M. R. R., J. A. W. Kruijtzer, R. M. J. Liskamp, D. Marsh, D. V. Greathouse, R. E. Koeppe II, B. de Kruijff, and J. A. Killian. 1999. Different membrane anchoring positions of tryptophan and lysine in synthetic transmembrane α-helical peptides. J. Biol. Chem. 274:20839–20846. [DOI] [PubMed] [Google Scholar]
- Demmers, J. A. A., J. Haverkamp, A. J. R. Heck, R. E. Koeppe II, and J. A. Killian. 2000. Electrospray ionization mass spectrometry as a tool to analyze hydrogen/deuterium exchange kinetics of transmembrane peptides in lipid bilayers. Proc. Natl. Acad. Sci. USA. 97:3189–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harzer, U., and B. Bechinger. 2000. Alignment of lysine-anchored membrane peptides under conditions of hydrophobic mismatch: A CD, 15N and 31P solid-state NMR spectroscopy investigation. Biochemistry. 39:13106–13114. [DOI] [PubMed] [Google Scholar]
- Huschilt, J. C., B. M. Millman, and J. H. Davis. 1989. Orientation of alpha-helical peptides in a lipid bilayer. Biochim. Biophys. Acta. 979:139–141. [DOI] [PubMed] [Google Scholar]
- Jones, D. H., K. R. Barber, E. W. Van der Loo, and C. W. M. Grant. 1998. Epidermal growth factor receptor transmembrane domain: 2H NMR implications for orientation and motion in a bilayer environment. Biochemistry. 37:16780–16787. [DOI] [PubMed] [Google Scholar]
- Ketchem, R. R., K. C. Lee, S. Huo, and T. A. Cross. 1996. Macromolecular structural elucidation with solid-state NMR-derived orientational constraints. J. Biomol. NMR. 8:1–14. [DOI] [PubMed] [Google Scholar]
- Killian, J. A. 1998. Hydrophobic mismatch between proteins and lipids in membranes. Biochim. Biophys. Acta. 1376:401–416. [DOI] [PubMed] [Google Scholar]
- Killian, J. A., I. Salemink, M. R. R. De Planque, G. Lindblom, R. E. Koeppe II, and D. V. Greathouse. 1996. Induction of nonbilayer structures in diacylphosphatidylcholine model membranes by transmembrane alpha-helical peptides: importance of hydrophobic mismatch and proposed role of tryptophans. Biochemistry. 35:1037–1045. [DOI] [PubMed] [Google Scholar]
- Killian, J. A., M. J. Taylor, and R. E. Koeppe, Ii. 1992. Orientation of the valine-1 side chain of the gramicidin transmembrane channel and implications for channel functioning. A 2H NMR study. Biochemistry. 31:11283–11290. [DOI] [PubMed] [Google Scholar]
- Kovacs, F. A., J. K. Denny, Z. Song, J. R. Quine, and T. A. Cross. 2000. Helix tilt of the M2 transmembrane peptide from Influenza A virus: an intrinsic property. J. Mol. Biol. 295:117–125. [DOI] [PubMed] [Google Scholar]
- Landolt-Marticorena, C., K. A. Williams, C. M. Deber, and R. A. Reithmeier. 1993. Nonrandom distribution of amino acids in the transmembrane segments of human type I single span membrane proteins. J. Mol. Biol. 229:602–608. [DOI] [PubMed] [Google Scholar]
- Liu, F., R. N. A. H. Lewis, R. S. Hodges, and R. N. McElhaney. 2001. A differential scanning calorimetric and 31P NMR spectroscopic study of the effect of transmembrane α-helical peptides on the lamellar-reversed hexagonal phase transition of phosphatidylethanolamine model membranes. Biochemistry. 40:760–768. [DOI] [PubMed] [Google Scholar]
- Mall, S., R. Broadbridge, R. P. Sharma, J. M. East, and A. G. Lee. 2001. Self-association of model transmembrane α-helices is modulated by lipid structure. Biochemistry. 40:12379–12386. [DOI] [PubMed] [Google Scholar]
- Marassi, F. M., and S. J. Opella. 2000. A solid-state NMR index of helical membrane protein structure and topology. J. Magn. Reson. 144:150–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesleh, M. F., S. Lee, G. Veglia, D. S. Thiriot, F. M. Maressi, and S. J. Opella. 2003. Dipolar waves map the structure and topology of helices in membrane proteins. J. Am. Chem. Soc. 125:8928–8935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morein, S., R. E. Koeppe II, G. Lindblom, B. de Kruijff, and J. A. Killian. 2000. The effect of peptide/lipid hydrophobic mismatch on the phase behavior of model membranes mimicking the lipid composition in Escherichia coli membranes. Biophys. J. 78:2475–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow, M. R., and C. W. Grant. 2000. The EGF receptor transmembrane domain: peptide-peptide interactions in fluid bilayer membranes. Biophys. J. 79:2024–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle, J. F., and S. Tristram-Nagle. 2000. Lipid bilayer structure. Curr. Opin. Struct. Biol. 10:474–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S. H., A. A. Morse, A. A. Nevzorov, M. F. Mesleh, M. Oblatt-Montal, M. Montal, and S. J. Opella. 2003. Three-dimensional structure of the channel-forming trans-membrane domain of virus protein u (Vpu) from HIV-1. J. Mol. Biol. 333:409–424. [DOI] [PubMed] [Google Scholar]
- Persson, S., J. A. Killian, and G. Lindblom. 1998. Molecular ordering of interfacially localized tryptophan analogs in ester- and ether-lipid bilayers studied by 2H-NMR. Biophys. J. 75:1365–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinia, H. A., R. A. Kirk, R. A. Demel, M. M. E. Snel, J. A. Killian, J. P. J. M. van der Eerden, and B. de Kruijff. 2000. Visualization of highly ordered striated domains induced by transmembrane peptides in supported phosphatidylcholine bilayers. Biochemistry. 39:5852–5858. [DOI] [PubMed] [Google Scholar]
- Sharpe, S., K. R. Barber, C. W. M. Grant, D. Goodyear, and M. R. Morrow. 2002. Organization of model helical peptides in lipid bilayers: insight into the behavior of single-span protein transmembrane domains. Biophys. J. 83:345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe, S., C. W. Grant, K. R. Barber, J. Giusti, and M. R. Morrow. 2001. Structural implications of a Val → Glu mutation in transmembrane peptides from the EGF receptor. Biophys. J. 81:3231–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strandberg, E., S. Morein, D. T. S. Rijkers, R. M. J. Liskamp, P. C. A. Van der Wel, and J. A. Killian. 2002. Lipid dependence of membrane anchoring properties and snorkeling behavior of aromatic and charged residues in transmembrane peptides. Biochemistry. 41:7190–7198. [DOI] [PubMed] [Google Scholar]
- Subczynski, W. K., M. Pasenkiewicz-Gierula, R. N. McElhaney, J. S. Hyde, and A. Kusumi. 2003. Molecular dynamics of 1-palmitoyl-2-oleoylphosphatidylcholine membranes containing transmembrane α-helical peptides with alternating leucine and alanine residues. Biochemistry. 42:3939–3948. [DOI] [PubMed] [Google Scholar]
- Tian, F., Z. Song, and T. A. Cross. 1998. Orientational constraints derived from hydrated powder samples by two-dimensional PISEMA. J. Magn. Reson. 135:227–231. [DOI] [PubMed] [Google Scholar]
- Ten Kortenaar, P. B. W., B. G. van Dijk, J. M. Peters, B. J. Raaben, P. J. H. M. Adams, and G. I. Tesser. 1986. Rapid and efficient method for the preparation of Fmoc-amino acids starting from 9-fluorenylmethanol. Int. J. Pept. Protein Res. 27:398–400. [Google Scholar]
- Van der Wel, P. C. A., E. Strandberg, J. A. Killian, and R. E. Koeppe, II. 2002. Geometry and intrinsic tilt of a tryptophan-anchored transmembrane α-helix determined by 2H NMR. Biophys. J. 83:1479–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., J. Denny, C. Tian, S. Kim, Y. Mo, F. Kovacs, Z. Song, K. Nishimura, Z. Gan, R. Fu, J. R. Quine, and T. A. Cross. 2000. Imaging membrane protein helical wheels. J. Magn. Reson. 144:162–167. [DOI] [PubMed] [Google Scholar]
- Weiss, T. M., P. C. A. Van der Wel, J. A. Killian, R. E. Koeppe II, and H. W. Huang. 2003. Hydrophobic mismatch between helices and lipid bilayers. Biophys. J. 84:379–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiles, J. A., J. K. Glover, R. R. Vold, and E. A. Komives. 2002. Methods for studying transmembrane peptides in bicelles: consequences of hydrophobic mismatch and peptide sequence. J. Magn. Reson. 158, 149–156. [DOI] [PubMed] [Google Scholar]
- Wu, C. H., A. Ramamoorthy, and S. J. Opella. 1994. High-resolution heteronuclear dipolar solid-state NMR spectroscopy. J. Magn. Reson. A. 109:270–272. [Google Scholar]
- Yau, W. M., W. C. Wimley, K. Gawrisch, and S. H. White. 1998. The preference of tryptophan for membrane interfaces. Biochemistry. 37:14713–14718. [DOI] [PubMed] [Google Scholar]
- Zeri, A. C., M. F. Mesleh, A. A. Nevzorov, and S. J. Opella. 2003. Structure of the coat protein in Fd filamentous bacteriophage particles determined by solid-state NMR spectroscopy. Proc. Natl. Acad. Sci. USA. 100:6458–6463. [DOI] [PMC free article] [PubMed] [Google Scholar]