

Abstract
We present elastic and inelastic incoherent neutron scattering data from a series of trehalose glasses diluted with glycerol. A strong correlation with recently published protein stability data in the same series of glasses illustrates that the dynamics at Q ≥ 0.71 Å−1 and ω > 200 MHz are important to stabilization of horseradish peroxidase and yeast alcohol dehydrogenase in these glasses. To the best of our knowledge, this is the first direct evidence that enzyme stability in a room temperature glass depends upon suppressing these short-length scale, high-frequency dynamics within the glass. We briefly discuss the coupling of protein motions to the local dynamics of the glass. Also, we show that Tg alone is not a good indicator for the protein stability in this series of glasses; the glass that confers the maximum room-temperature stability does not have the highest Tg.
INTRODUCTION
Over a decade ago it was discovered that carbohydrate glass plays a central role in anhydrobiosis (Carpenter et al., 1987; Mouradian et al., 1984). Since then preservation of biological agents in nominally dry carbohydrate glasses has become a problem of both technological and scientific interest. Rapidly growing sectors of the pharmaceutical and tissue engineering industries rely increasingly on the ability to deliver functional proteins from a dry state. Despite the technological significance of this issue, the mechanisms by which viscous sugars impart dehydration stability are not fully understood. Rational design of bioprotective glasses for proteins will require a more detailed understanding of the underlying mechanisms of preservation.
Intrinsic characteristics of a hydrophilic glass that are important to protein stabilization appear to include its amorphous nature and retarded dynamics relative to physiological conditions. Hydrogen bonding sites of a bioprotective glass seem to provide a “substitute” for water in terms of the protein's local environment (Allison et al., 1999; Cleland et al., 2001; Costantino et al., 1998; Tanaka et al., 1991). The intimate contact necessary for effective hydrogen bonding to occur is made possible by the amorphous character of the glass. It seems likely that spatial constraints imposed by the crystalline phase do not allow this, as is evidenced by the observation that proteins are not stabilized in the crystalline phase of an otherwise effective preservant (Izutsu et al., 1994; Pikal and Rigsbee, 1997).
The inherently slow relaxation dynamics of the polyhydroxyl glass is expected to retard motions and reactions, both intrinsic and extrinsic to the protein, that lead to degraded protein function, such as denaturation, aggregation, deamidation, or oxidation of peptide side chains. Much progress has been made in understanding the relationship between protein reaction dynamics and host (solvent) dynamics at low to intermediate viscosities (Ansari et al., 1992; Beece et al., 1980; Doster, 1983; Gavish, 1980; Kleinert et al., 1998; Steinbach et al., 1991). However, the precise nature of the protein-solvent dynamic relationship is presently an open question for the very high viscosity regime, such as is encountered in the glass (Gottfried et al., 1996; Hagen et al., 1995; Lichtenegger et al., 1999; Sastry and Agmon, 1997; Schlichter et al., 2001).
While there is some uncertainty as to the role that solvent dynamics plays in protein dynamics at very high viscosities, the situation is even less clear when anhydrous biopreservation is considered. In the context of a pharmaceutical lyoprotective glass (i.e., when protein stability is of central interest), dynamics-related discussions are often couched in terms of the glass transition temperature (Tg) of the lyoprotective host. For example, it was suggested that some sugars are better preservants than others based on higher Tg values, and insensitivity of Tg to moisture (Green and Angell, 1989). Although it is often true that stability trends with Tg (Bell et al., 1995; Buitink et al., 2000; Duddu et al., 1997; Wang, 2000), counterexamples can be found (Buera et al., 1999; Davidson and Sun, 2001;).
Previously we demonstrated that adding small amounts of a low-Tg diluent to a bioprotective glass leads to a reduced Tg, but substantially increased stability for model proteins sequestered in the glass (Cicerone et al., 2003). Glasses in general exhibit rich dynamics over many decades in frequency, which cannot be described by simple parameters such as Tg, or even viscosity. In this article we affirm that Tg of the biopreservation glass alone does not predict the degree of stability that a glass can impart to a protein. Further, we show that, for the series of glasses studied here, it is the amplitude of the local, high frequency dynamics of the glass that correlates with the ability to impart protein stability. Specifically, we use incoherent neutron scattering to show that small amounts of diluent suppress local relaxations and “stiffen” collective vibrations that occur with timescales of a nanosecond and faster. The stiffening or suppression of these fast, local motions correlates precisely with the effectiveness of the glass at stabilizing labile proteins. This strong correlation suggests that dynamics of the protein and glass remain coupled.
MATERIALS AND METHODS
Samples
Glycerol, d8-glycerol, and α-α (d) trehalose were obtained from Sigma-Aldrich (St. Louis, MO) and used without further purification. (Certain commercial equipment and materials are identified in this article to specify adequately the experimental procedure. In no case does such identification imply recommendation by the National Institute of Standards and Technology nor does it imply the material or equipment identified is necessarily the best available for this purpose.) The neutron scattering samples were prepared by freeze drying aqueous solutions of trehalose and glycerol. The individual concentrations of trehalose and glycerol varied, but the combined concentration was maintained at a mass fraction of 0.20. Control samples were made from solutions also containing 0.100 mol/L CaCl2, 300 μg/mL Tween, and 0.050 mol/L histadine buffer, pH 6.0. These conditions are consistent with the previous enzyme activity measurements (Cicerone et al., 2003). All solutions were prepared using Milli-Q deionized water (18 MΩ cm). A typical freezedrying protocol is provided in Table 1. By measuring the final sample mass, we confirmed that diluent (glycerol) is not lost during the freezedrying process.
TABLE 1.
Freezedrying parameters
| Temperature | Duration | Pressure |
|---|---|---|
| −40°C | ≈1 h (or until frozen) | 101 kPa |
| −20°C | ≈6 h (or until primary drying is done) | 4.0 Pa |
| −8°C | ≈3 h | 4.0 Pa |
| 25°C | ≈24 h* | 4.0 Pa |
Final drying is probably complete before the 24 h; we have not attempted to determine the minimum time required for the final drying step.
The freeze dried glasses were handled in one of two ways. In an initial set of experiments, using hydrogenated glycerol, the glasses were briefly exposed to air while loading the neutron-scattering sample cells. Moisture uptake for these samples was not quantified, but this brief exposure to ambient air was consistent with the protocol for the enzyme stability studies published previously (Cicerone et al., 2003). In subsequent experiments using d8-glycerol, the vials containing the freeze dried glasses were backfilled with argon and sealed in the freeze drier. Those samples were stored and loaded into the hermetically sealed neutron scattering sample cells under an argon atmosphere, without any exposure to ambient air.
Incoherent neutron scattering
The elastic incoherent neutron scattering measurements were performed at the National Institute of Standards and Technology Center for Neutron Research on the High Flux Backscattering (HFBS) spectrometer (Gehring and Neumann, 1997), located on the NG2 beam line. The HFBS spectrometer operates with an incident neutron wavelength of 6.271 Å and a 0.85 μeV full width at half-maximum energy resolution. The accessible momentum transfer (Q) range is 0.25–1.75 Å−1. Here, the spectrometer operates in the fixed-window mode where the elastic scattering intensity is recorded as a function of Q while the sample is heated at 1 K/min from 40 K to above the calorimetric Tg. For organic hydrocarbons, like our samples, the scattering is dominated by hydrogen, which has an incoherent scattering cross section ≈20 times greater than carbon, oxygen, nitrogen, sulfur, or most other elements common to biological systems. The Q-dependence of the incoherent elastic scattering Iinc was analyzed in terms of the Debye-Waller factor (a harmonic oscillator model) where the hydrogen-weighted mean-square atomic displacement 〈u2〉 is given by
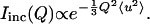 |
(1) |
In this simple model, ln(Iinc) vs. Q2 is linear with the slope proportional to 〈u2〉.
Fig. 1 shows the Q-dependence of the elastic scattering intensity from a pure trehalose glass at a few different temperatures. In all of our measurements, Iinc is normalized by the scattering intensity at the lowest temperature available, which is 40 K. As we are primarily interested in dynamics and therefore the incoherent scattering, this normalization helps remove small artifacts of coherent scattering (static structure) that might affect the Q variations of the data. This normalization also means that the slope of ln(Iinc) vs. Q2 is zero at 40 K, i.e., 〈u2〉 = 0 at 40 K. We compensate this effect by fitting a straight line through the 〈u2〉 vs. T data between 40 K and 200 K and vertically offsetting the data so that the 〈u2〉 = 0 intercept of this line occurs at T = 0 K. With increased temperature there is a reduction of the elastic scattering intensities as thermal mobility is introduced to the sample. As seen in the linear fits of Fig. 1, this leads to an increase in the amplitude of the slopes, or an increase in 〈u2〉. It is important to realize that the 0.85 μeV resolution of the HFBS spectrometer means that only those motions faster than ≈200 MHz, or a few nanoseconds in the time domain, give rise to a increase of 〈u2〉; slower motions appear as static.
FIGURE 1.

Elastic scattering intensity scales with Q2 differently in a high Q and low Q regime at 347 K (▾) and 403 K (○). The reference data was obtained at 40 K (×). The sample is trehalose. Error bars represent standard uncertainties of mean ±1 SD.
The Debye-Waller formalism is a harmonic approximation, which is typically an oversimplification in soft matter at ambient conditions. This is evident by the fact that a single linear function does not fit the data in Fig. 1; the dependence is almost bilinear with break in the slope near Q2 = 0.5 Å−2. Nonlinear dependencies of ln(Iinc) on Q2 have been reported in both polymer systems (Frick and Fetters, 1994) and biological systems (Paciaroni et al., 2002; Settles and Doster, 1996), and there have been attempts to calculate 〈u2〉 using more complicated models. These models generally include both a Gaussian (harmonic) and a non-Gaussian component, with the latter being thermally activated and dominant at low Q. However, in our view one must fit the data over a very broad range of Q to reliably separate the local harmonic motions (high Q) from the long-range anharmonic contributions (low Q). The HFBS spectrometer is not well suited for this separation as it does not access a sufficiently large region of Q-space. Therefore we simplify the situation and only use the harmonic approximation for the high Q data, beyond Q2 = 0.5 Å−2, where the motions are smaller and the harmonic approximation is most appropriate. Despite its simplicity, the harmonic oscillator approximation has proven useful in qualitatively analyzing several biological systems, as demonstrated in a recent review (Zaccaï, 2000). The small set of lower Q data (below Q2 = 0.5 Å−2) that we neglect contains potentially interesting information about longer-range motions in the system, but attempts are not made to interpret this data because of the limited Q-range; the significance of the fits would be marginal.
Incoherent inelastic neutron scattering measurements were also performed at the National Institute of Standards and Technology Center for Neutron Research, using the Fermi-Chopper time-of-flight spectrometer (FCS) on the NG6 beam line. Fig. 2 displays a typical inelastic neutron scattering spectrum, in this case for pure trehalose at 100 K. These measurements utilized 4.8 Å neutrons with an energy resolution of 140 μeV full width at half-maximum. This means that the FCS sensitivity is limited to motions faster than the HFBS spectrometer, on the order of 30 GHz (≈30 ps) and faster. The FCS spectra were adjusted for the different macroscopic scattering cross sections, corrected for detector efficiency with a vanadium standard, and summed over the accessible Q-range of the instrument (approximately the same Q-range as the HFBS spectrometer), and Bose-scaled to account for trivial temperature variations in the density of states. The inset to Fig. 2 shows trehalose spectra at 100 K and 296 K; the Bose scaling results in overlap of the data at higher energies as expected.
FIGURE 2.

Inelastic scattering function for trehalose at 100 K. ○ is data; solid line is overall fit; dashed and dotted lines are background (bkg), boson peak (pb), inelastic scattering (is), and instrument resolution function (irf). (Inset) Inelastic scattering functions for trehalose at 100 K and 296 K, as marked. Axes are the same as for the main figure.
Boson peak energies are extracted from the inelastic spectra to compare peak positions between different glasses. To this end the spectra were fitted to several components, as illustrated in Fig. 2 for the 100 K spectrum. These components include a 140 μeV instrumental resolution function, as is indicated by the narrow Gaussian centered at 0 meV; a broad quasielastic scattering component (QES, or is in Fig. 2, a Lorentzian lineshape) due to fast relaxations centered at 0 meV; and a broad, asymmetric boson peak (BP or pb in Fig. 2) with a distinct maximum between −4 and −5 meV.
At 100 K the relaxations are strongly suppressed (i.e., the QES intensities are relatively low), meaning that the QES and boson peak are well separated. In this limit, fitting the boson peak position is essentially model-independent. Boson peaks have been fit with a number of physical models and or empirical functions. Here we parameterize the lineshape with a model-independent lognormal distribution function (Malinovsky et al., 1991) as
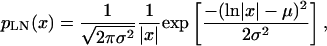 |
(2) |
where μ and σ are the center and width of the asymmetric distribution. These are related to the peak maximum (mode) through the relationship xpeak = exp (μ–σ2); this mode is reported as the boson peak position. In addition to the instrumental-resolution function Gaussian, QES Lorentzian, and BP log-normal, a flat background is needed to fit the data. Physically, this background represents the Debye-level density of states that reaches a plateau at E = 0 meV. The height of this plateau depends upon the velocity of sound, which has not been measured for these materials. In the absence of this knowledge, we assign the intensity of this background to equal the total signal intensity at −15 meV in the high frequency tail of the boson peak. The inset of Fig. 2 shows that the high frequency tails of the Bose-scaled spectra coincide in this region, suggesting that this assignment is reasonable. Furthermore, it should be apparent that background assignment has no effect on fitting of the boson peak position, and relatively little effect on the peak width. The background assignments were similar from sample to sample in all the glasses studied here.
RESULTS
Fig. 3 displays the 〈u2〉 values obtained by applying the Debye-Waller formalism to the elastic incoherent neutron scattering for five freezedried trehalose glasses diluted with d8-glycerol; the glycerol mass fractions (φglycerol) range from 0 to 0.20 in increments of 0.05. These glasses were prepared from H2O, so only the nonexchangeable sites remained deuterated (i.e., each glycerol contains 3 −OH groups, not −OD). The neutron scattering studies were performed to compare with previously published protein stability studies (Cicerone et al., 2003). We emphasize that none of these glasses contain protein. The protein stability studies were performed with glass/protein mass ratios of >106:1. Such small amounts of protein would be undetectable by neutron scattering, and including protein at a detectable level would completely change the character of the glass. The glasses studied here also do not contain buffer or surfactant, which are common components of protein-bearing glasses. We have performed the control experiments to demonstrate that the presence of salt and surfactant at levels consistent with our previous activity studies do not have a noticeable effect on 〈u2〉.
FIGURE 3.

Debye-Waller factors from trehalose glasses with φglycerol = 0 (+), 0.05 (♦), 0.10 (▾), 0.15(▴), and 0.20 (•). (Inset) Spring constants of low temperature glasses. Error bars are approximately the size of the symbols and represent standard uncertainties of mean ±1 SD. The lines are guides to the eye.
We see in Fig. 3 that 〈u2〉 evolves linearly with T below 200 K for all samples, as is characteristic for a harmonic solid. Within this framework, the equipartition theorem (kT/2 per degree of freedom, where k is Boltzmann's constant) is used to calculate an effective spring constant κ; stiffer harmonic vibrations have a larger κ or a smaller temperature dependence of 〈u2〉. The inset to Fig. 3 displays κ-values derived from the slope of linear fits to the 〈u2〉 data in the range 40–200 K. We see that the trehalose with φglycerol = 0.05 provides the “stiffest” glassy environment in the low temperature regime. Without applying the harmonic spring constant model this qualitatively reflects the observation that 〈u2〉 is the smallest for the sample with φglycerol = 0.05 not only in the low temperature linear regime, but also extending to nearly 350 K. The harmonic oscillator assumption implicit in the determination of κ is of course not appropriate above 200 K. However, as suggested by Zaccaï (2000), it is still useful to think of reduced 〈u2〉 at these higher temperatures qualitatively as an increased environmental spring constant of the soft medium.
Aside from the nonmonotonic behavior of κ, this series of glasses behaves more or less as expected. In Fig. 3 vertical arrows indicate the calorimetric Tg values, which monotonically shift to lower T with increasing glycerol content. Likewise, the upturn in 〈u2〉, expected near Tg (Angell et al., 2000; Frick and Richter, 1995), occurs at temperatures that decrease with increasing glycerol content. The amplitude of 〈u2〉 at Tg also follows a trend that might be expected, generally decreasing with decreasing Tg. For example, 〈u2〉|T = Tg = 0.25 Å2 for pure trehalose (Tg = 392 K) and decreases toward 〈u2〉|T = Tg = 0.11 Å2 for trehalose with φglycerol = 0.20 (Tg = 304 K). These monotonic variations with φglycerol in relation to Tg are in stark contrast with the environmental spring constant stiffening which reaches a maximum at φglycerol = 0.05. The nonmonotonic variation in κ precisely reflects the protein stability data (see below). This clearly indicates that a higher Tg alone is not sufficient for enhanced protein stability, and that suggests that local dynamics plays an important role in stabilization.
Elastic scattering experiments were also performed on trehalose glasses diluted with hydrogenated glycerol at the same φglycerol loadings. These were the samples briefly exposed to atmosphere while being transferred to the scattering cell so that the data could be directly compared with the protein stability data (Cicerone et al., 2003). These experiments reach the same conclusion, a pronounced maximum in the suppression of 〈u2〉 at φglycerol = 0.05. The magnitude of the maximum suppression at φglycerol = 0.05 is slightly less than seen in Fig. 3 (≈35 N/m vs. 59 N/m). In addition to the peak at φglycerol = 0.05 (which showed the strongest difference between the deuterated and hydrogenated analogs), there was also a slight suppression in all the κ-values for the fully hydrogenated glasses. We cannot say whether the difference is due primarily to the absorption of small amounts of atmospheric water in the hydrogenated samples, or to scattering from the nonexchangeable hydrogens of glycerol, which is masked in Fig. 3 as a result of the deuterium labeling. Consistent with the observed reduction in κ, the presence of moisture has been shown to soften similar glasses in the frequency window sensed by the backscattering experiments (Conrad and de Pablo, 1999).
Inelastic scattering was performed on the trehalose glasses diluted with deuterated glycerol at 100 K and 296 K, as illustrated in Fig. 2 and inset. Besides the instrumental resolution and background level, there are two primary components of interest in the spectra. The first is the QES component centered on 0 meV, which reflects relaxations or diffusive-type motions. This QES component increases with temperature, as would be expected, but we note that even at 100 K its intensity is measurable. The structural origin of this low temperature relaxation is not completely understood. The present data are insufficient for further understanding this motion because fitting of the peak heights and widths is highly convoluted at the present spectral resolution. High-resolution time-of-flight neutron scattering measurements are currently underway to remedy this problem.
The second feature of interest is the boson peak, a broad collective excitation found near −5 meV at 100 K, and at lower energies with increasing temperature (as shown in the inset to Fig. 2). The boson peak is a more-or-less ubiquitous feature for glassforming materials below Tg. They originate from a collective excitation/vibration (not a relaxation like the QES) that is lower frequency than the optic modes typically seen in infrared and Raman spectroscopy, but higher in frequency than the acoustic modes. Although the exact nature of the boson peak is still uncertain, estimates indicate that somewhere between 10 atoms and 100 atoms participate in this high frequency collective mode (Buchenau et al., 1991;Yamamuro et al., 2000).
Fig. 4 a shows the 100 K boson peak energies (EBP) of trehalose glasses as a function of the glycerol content. The boson peak maximum in pure trehalose lies at (4.59 ± 0.05) meV, and rises quickly for φglycerol = 0.05. For further additions of glycerol the boson peak maximum remains constant to within the uncertainty, if not showing a slight maximum near φglycerol = 0.10.
FIGURE 4.

Inelastic scattering from trehalose glasses diluted with d-glycerol in varying amounts. (a) Boson peak energies at 100 K. (b) Ratios of integrated quasielastic scattering intensity to integrated boson peak intensity at 100 K (•) and 296 K (□); see text for explanation. Error bars represent standard uncertainties of mean ±1 SD.
It is worth mentioning that the boson peak in the φglycerol = 0.20 sample stiffened dramatically to (5.20 ± 0.08) meV (not shown here). We are uncertain of the reason for this notable stiffening. However, we suspect that in this specific sample the glycerol and trehalose phase-separated, since 5.2 meV is nominally consistent with the boson peak of pure glycerol (Sokolov et al., 1994). We occasionally notice evidence for phase separation in samples with φglycerol ≥ 0.15, and have seen evidence for crystallization, through x-ray diffraction, in the samples with this level of glycerol loading when they were exposed to ambient moisture for ≈30 min. This possible phase-separation did not appear to be an issue in any of the HFBS experiments or in the protein stability studies.
The trend of EBP with glycerol content is qualitatively similar at room temperature (where the protein stability measurements were made) to that at 100 K, however there is an overall softening of the boson peak, or shift of EBP to lower energies. We do not present the EBP peak assignments here because the softening of the boson peak and the increasing of the QES slightly convolute the peak fitting. We present this higher temperature inelastic scattering data only as a ratio between the QES and BP intensity, which can be obtained in a model-independent manner.
The QES/BP intensity ratios, shown in Fig. 4 b, reflect the ratio of relaxations to collective vibrations on the picosecond timescale. The BP vibrations can be thought of as attempts at relaxational motion (like diffusion) whereas the QES reflect successful attempts, so the ratio is an indicator of the efficiency with which relaxation occurs in a glass; a high ratio denoting efficient relaxation. We estimate the ratio of the relaxational (QES) to the vibrational (BP) motions of these glasses in a model-independent way by using the minimum in S(Q,E) as a dividing line between the inelastic and quasielastic components. This minimum occurs at −1.70 and −1.85 meV in the low-temperature and high-temperature data, respectively (see Fig. 2 for example). We simply integrate the total scattering on the high-energy side of the minimum, out to −15 meV, to estimate the BP intensity, and likewise integrate the total scattering between the minimum and −0.35 meV to estimate the total QES contribution. Before determining this ratio the Debye-like background was subtracted. However, neglecting this subtraction makes no difference in the trend of the ratios shown in Fig. 4 b.
Fig. 4 b shows that the relaxation processes are much more efficient at higher temperatures, as expected. This figure also shows that there is a distinct minimum in the success rate for relaxation near φglycerol = 0.05 at low temperature. Although relaxations at room temperature are equally efficient in the trehalose glass and the φglycerol = 0.05 glass, the overall amplitude of scattering is much lower in the φglycerol = 0.05 sample, as seen in Fig. 3. Thus, this glass exhibits less relaxation even at room temperature.
The QES/BP ratio has been shown to correlate with fragility of glassy systems (i.e., with the temperature dependence of dynamics), the stronger temperature-dependence being correlated with a larger QES/BP ratio (Sokolov et al., 1993). Fragility is typically evaluated from α-relaxation data at Tg. We have not measured α-relaxation in these systems; however, if we assume equivalence between a relaxation time τfast and 1/〈u2〉 (see below), we note a weak positive correlation between increased temperature-dependence of τfast and larger ratios of QES/PB.
DISCUSSION
We have shown that the addition of small amounts of glycerol can suppress the amplitude of fast (200 MHz and faster) dynamics in glassy trehalose. It is important to realize that this dynamic suppression occurs even though the diluent plasticizes the glass (reduces its Tg). There is a large body of literature that reports a plasticization of Tg with a concomitant antiplasticization of the sub-Tg dynamics in the kHz frequency range. This phenomenon has been seen in sugars (Lourdin et al., 1997; Noel et al., 1996) and synthetic polymers (Bergquist et al., 1999; Casalini et al., 2000). The dynamic signatures of this antiplasticization are qualitatively similar to the observations here, although they occur on very different timescales.
In the previous article (Cicerone et al., 2003) we showed that stability of the enzymes horseradish peroxidase (HRP) and yeast alcohol dehydrogenase (YADH) sequestered in sugar glasses could be significantly improved by adding low-Tg diluents to the glass. This effect seems to be fairly general; we demonstrated it for several disaccharides and polymeric sugars diluted with any of several diluents, including glycerol, dimethylsulfoxide, propylene glycol, ethylene glycol, or oligomeric polyethylene glycol. However, we did not observe an improvement in stability when raffinose (a trisaccharide) was diluted. We do not know the reason for this. Fig. 5 a shows the temperature-dependent stability lifetimes (τdeact) for HRP and YADH in pure trehalose glass from that work. The data were acquired by heating the enzyme-bearing glass to a specified temperature for varying lengths of time, dissolving the glass in a buffer solution, and then quantifying the residual enzyme activity. The exposure temperatures were below the Tg of the sequestering glass. The deactivation time constants (τdeact) displayed an Arrhenius-like temperature dependence, consistent with previous reports for dynamic properties (Nozaki and Mashimo, 1987; Shamblin et al., 1999) and stability of biological systems (Mazzobre et al., 1997; Yoshioka et al., 1994) below Tg.
FIGURE 5.

(a) Enzyme deactivation times in trehalose glass, plotted in Arrhenius format: HRP (▪) and YADH (▴). (b) Enzyme deactivation times extrapolated to room temperature for bioprotective glasses plasticized with varying amounts of glycerol. Symbols are the same as in a; lines are a guide to the eye. Error bars represent standard uncertainties of mean ±1 SD.
Fig. 5 b shows resulting values of τdeact at 296 K for HRP and YADH in trehalose glasses with differing glycerol content. All data in Fig. 5 b are extrapolated in temperature from Arrhenius plots, except HRP at φglycerol = 0, which was obtained directly. In both enzymes a maximum in the stability is conferred in the vicinity of φglycerol = 0.05. This is the same glycerol content at which a minimum in 〈u2〉, or peak in κ, occurs in Fig. 3.
The primary finding here is that small additions of a low-Tg diluent to a carbohydrate glass can suppress local, fast dynamics, and that those glassy mixtures which show suppressed local dynamics also show enhanced protein stabilization. This coincidence suggests a relationship between the preservation of the enzymes and suppression of high frequency (200 MHz and faster) local dynamics measured by the HFBS and FCS spectrometers.
It is remarkable that the enzyme deactivation lifetimes correlate with the dynamics measured by incoherent neutron scattering; the process of deactivation can occur slowly over thousands of hours, whereas the neutron scattering is sensitive to processes on the order of a nanosecond or faster, a separation of 15 decades in time. However, these are not the first correlations between the amplitude of 〈u2〉 and biological activity. Parak et al. (1982) illustrated that proteins undergo a dynamic transition, Td in the vicinity of 200 K, from a regime of purely harmonic to anharmonic motions. Brooks et al. (1988) suggested that these anharmonic atomic fluctuations act as the lubricant that enables conformational fluctuations on a physiological timescale, and this idea was supported by the discovery that Td coincides with the temperature for the onset of biological activity (Doster et al., 1989). In further support of this relationship, studies of bacteriorhodopsin have revealed a positive correlation between regions of biological activity in Subramaniam et al. (1993), and regions of large amplitude fluctuations, reflected in Reat et al. (1998).
Cordone et al. (1999) found that trehalose suppressed the 〈u2〉 in myoglobin significantly in comparison to an aqueous environment. This led them and others (Branca et al., 2001; Caliskan et al., 2003; Tsai et al., 2000; Zaccaï, 2000) to speculate that an effective lyophilization medium will suppress fast dynamics of the protein reflected in 〈u2〉. The data presented here strongly supports this idea. We see a marked positive correlation between improved protein preservation and suppression in 〈u2〉 of the preservation glass itself. The neutron scattering measurements were made in absence of protein.
Coupling of protein and glass dynamics
The implication that a suppression of local motions (〈u2〉) of a glassy host also indicates a suppression of similar motions of a protein located in that host assumes that the fast, local dynamics of the protein and the host are coupled. Because the degree of protein coupling to solvent seems to be an open question in the literature, we briefly address this question. Kramers' theory (Kramers, 1940), in its usual interpretation, predicts that the rate for the dynamics (k) should have an inverse relationship with viscosity (k ∝ η−1). This model is frequently the starting point for considering a coupling between solvent and solute dynamics. However, this theory was formulated in the high friction limit for a dynamically homogeneous system. These approximations may not hold in many solute-liquid systems, and they do not seem appropriate for considering solute motions in a glass. We present evidence and arguments below to the effect that, although much of the important dynamics of the protein may not be coupled with viscosity of the host fluid, there seems to be a firm link between the local dynamics of the protein with that of the host.
A large body of work on protein dynamics in various solvents indicates that Kramers' theory does not always predict protein dynamics properly, i.e., the product of k and η is not always constant as η is changed. A general trend is observed in that coupling between solvent viscosity and diffusive motions within a protein become weaker at elevated viscosity, and as increasingly local or interior protein motions are considered. While large amplitude (global) protein motions, and particularly those involving exterior portions of the protein, appear to follow Kramers' relation at visco-sities above ≈1 cP (Ansari et al., 1992; Hagen et al., 1995; Iben et al., 1989), diffusive motions involving smaller portions of the protein are less likely to follow Kramers' relation at elevated viscosity, and a power-law relationship between viscosity and these dynamics emerges: k ∝ η−ν, with ν in the range 0.35–1 (Beece et al., 1980; Gavish and Werber, 1979; Kleinert et al., 1998; Lavalette and Tetreau, 1988; Ng and Rosenberg, 1991; Rosenberg et al., 1989; Tian et al., 1996; Yedgar et al., 1995). Furthermore, diffusive motions that primarily involve the interior of the protein seem to follow a similar non-Kramers behavior in any viscosity regime (Beece et al., 1980; Kleinert et al., 1998). In stark contrast to this trend, fast, local atomic displacements (0.1 Å2 ≤ 〈u2〉 ≤ 1 Å2) of solvent and protein track one another very well throughout the volume of the protein, even at extremely high viscosity in the glass (Lichtenegger et al., 1999; Reat et al., 2000; Vitkup et al., 2000).
While the strong coupling of local atomic displacements between solvent and protein may seem peculiar in light of the observed non-Kramers' behavior, we suggest that these observations are quite compatible. Deviations from Kramers' relation for exterior protein motions in mixed solvent sys-tems have been attributed to differences of solvent composition and viscosity in the immediate vicinity of the protein compared to that of the bulk (Kleinert et al., 1998; Lavalette et al., 1999; Lichtenegger et al., 1999). Such deviations have been attributed to position-dependent, intrinsic viscosity for motions interior to the protein (Ansari et al., 1992; Gavish, 1980). We point out that a linear coupling between viscosity and kinetics would likely hold in the presence of either of these effects if the reaction rates were compared to the local viscosity rather than the bulk viscosity. Another mechanism becomes important at sufficiently high viscosity which could also lead to a breakdown of Kramers' relation despite strong local coupling of solute and solvent dynamics. Nanoscopic domains of heterogeneous dynamics appear to become long-lived as viscosity is increased in the supercooled and glassy regime for fragile glassformers. Solvent and solute dynamics remain coupled locally, within a domain, but pronounced decoupling of solute diffusion coefficients from bulk viscosity occurs due to a difference in sampling the distribution of dynamics by diffusion and viscosity measurements (Cicerone and Ediger, 1996; Cicerone et al., 1997).
Even though the relationship ημk−1 may hold locally for each of the mechanisms discussed above, in none of these cases could the dependence of solute dynamics on bulk viscosity be determined without specific knowledge of how the host and solute interact dynamically. On the other hand, the primary experimental result of the present work seems to suggest that we can know something of the dynamics of the solute by measuring the solvent dynamics alone.
Another mechanism leading to a breakdown in the Kramers' relation has been proposed by Grote and Hynes (1980), who modified Kramers' approach by including the concept of time-dependent friction, allowing for the case that the solvent motions may not be infinitely fast compared to those of the solute. Doster used the idea of time-dependent friction, along with intrinsic protein viscosity, to make a quantitative accounting for the non-Kramers behavior of ligand-binding rates involving both interior and exterior portions of myoglobin (Doster, 1983). Time-dependent friction was also used to explain observed violations of Kramers' relation for isomerization of small molecules (Courtney and Fleming, 1985; Flom et al., 1986; Rothenberger et al., 1983) and synthetic polymers (Glowinkowski et al., 1990; Zhu and Ediger, 1997) in single-component solvent systems of low and intermediate viscosity. It is worth noting that a power-law relation was found between viscosity and solute dynamics in the polymer systems, specifically k ∝ η−ν with ν in the range 0.3–0.95, and with ν approached unity as the moving moiety became large.
Within the Grote-Hynes framework, the barrier-crossing rate for a particle will depend on the short-time, nonadiabatic friction experienced during the crossing when the solvent dynamics are comparable to or slower than the intrinsic solute dynamics (Grote and Hynes, 1980). Thus, at high solvent viscosity it will be particularly true that the high-frequency portion of the solvent dynamics, and not the zero-shear viscosity, will most impact the reaction rate constant k. Although the Grote-Hynes approach implicitly assumes a dynamically homogeneous medium, and thus does not capture all of the important physics, the notion of a persistent relation between fast dynamics of solute and solvent is consistent with our data, with the simulations of Vitkup et al. (2000) and with recent findings of Caliskan et al. (2003) that picosecond dynamics of lysozyme and the host (glycerol or trehalose) track precisely, even though the protein dynamics clearly do not track the bulk viscosity of the solvents (Gottfried et al., 1996; Sastry and Agmon, 1997; Schlichter et al., 2001).
In Fig. 6 we show further evidence for coupling between the fast dynamics of the glassy host and reactivity of the protein. In this case the coupling is between the fast dynamics of the trehalose/glycerol glasses and the deactivation dynamics of enzymes in identical systems. In Fig. 6, top panel, we plot 1/〈u2〉 vs. 1000/T for the glasses (without protein). Fig. 6, second and third panels, show Arrhenius plots of the stability data for HRP and YADH in these glasses. The motivation for comparing 1/〈u2〉 with Arrhenius plots of stability data comes from the expectation, based on theoretical arguments, that the timescale of local relaxations of the glass should scale exponentially with 1/〈u2〉 (Hall and Wolynes, 1987).
FIGURE 6.

Enzyme preservation and dynamics in plasticized trehalose glasses with φglycerol = 0 (▿), 0.05 (□), and 0.10 (○). Top panel shows 1/〈u2〉, which is proportional to local dynamics (see text). Second panel shows HRP activity lifetimes, and third panel shows YADH activity lifetimes. Fourth panel shows apparent activation energies as a function of glycerol content measured by HRP lifetimes (H), YADH stability (Y), and 1/〈u2〉 (n°). Error bars represent standard uncertainties of mean ±1 SD.
The similarities between the protein stability data and the neutron scattering data are striking. The processes are universally slowest for φglycerol = 0.05 at low temperatures and for the undiluted trehalose at higher temperatures. Also, the dynamical processes are universally fastest for φglycerol = 0.10. These similarities are further emphasized in Fig. 6, fourth panel, which compares apparent activation energies for the 1/〈u2〉 data with that of the enzyme stability data; both the 1/〈u2〉 and τdeact activation energies show the same trend with increasing glycerol content. The clear correlation between the temperature dependencies of host dynamics and degradation kinetics of the protein embody the statement by Grote and Hynes (1980) that fast solute dynamics will most influence k in highly viscous fluids. By extension, this result implies a coupling between the fast dynamics of the solvent and the protein.
We note that 1/〈u2〉 scales linearly with log(η) for at least two glassforming systems over a tremendous viscosity range when the data are treated properly (Buchenau and Zorn, 1992; Kanaya et al., 1999). In their original correlation with viscosity, Buchenau and Zorn (1992) subtracted the 〈u2〉 values of crystalline Se from the amorphous Se data to remove the hard, or crystalline-like vibrations that do not directly lead to viscous flow. Kanaya et al. (1999) applied a comparable correction. It is not certain that this relationship will be universal, even with the prescribed data treatment.
We did not make an attempt to remove the effect of hard vibrations from our data for two reasons: It is not immediately clear to us that the crystalline-like vibrations should be entirely eliminated from consideration for understanding solvent-solute coupling, and our data is influenced much less by the higher frequency crystalline-like dynamics than was that of Buchenau and Zorn, or Kanaya et al. (The HFBS spectrometer used in these studies has energy resolution of 0.85 μeV, as compared to 0.2 meV and 0.02 meV of the spectrometers used by Buchenau et al. and Kanaya et al., respectively.) Although we did not make an effort to subtract the scattering component due to the crystalline-like vibrations, it appears that the trends shown in Fig. 6, top panel, and thus the similarities between the different samples, would be unaffected by such a correction.
Finally, we note that we have not considered possible thermodynamic factors related to the presence of the diluent glycerol influencing protein stabilization. Miller and de Pablo (2000) showed a correlation between the effectiveness of lyoprotective glasses and heat of solution; connections between hydrogen bonding and effective stabilization have also been made (Allison et al., 1999). We cannot with certainty address whether heat of solution or protein-glass hydrogen-bonding strengths would correlate with effectiveness of stabilization for the series of glasses studied herein, as we have not performed such measurements on these glasses. Such studies would be useful as they might further elucidate the important factors in the biopreservation efficacy of these glasses.
SUMMARY
We have shown for the first time that the MHz-GHz dynamics, as probed by inelastic and elastic neutron scattering on length scales of 1 nm and smaller, are of direct importance to the preservation of proteins in a glassy host. The connection between the preservation efficacy and the high frequency, local dynamics of the glass provides valuable insight as to how the dynamics of the glass couple to the protein. The observed relationship suggests that a time-dependent friction approach is valuable in understanding the effect of solvent dynamics on protein motions.
We show that an effective preservation medium will strongly suppress the local, fast motions that appear to be precursors for protein denaturation or other deactivation pathways. From a practical perspective, these observations could serve as a basis for metrology methods that could be applied during the design of future bioprotective glasses. The use of such metrologies could significantly reduce the trial-and-error aspect of lengthy and tedious long-term stability studies.
Acknowledgments
The authors are indebted to Jack Douglas for many stimulating discussions, and especially the suggestion to plot the 〈u2〉 data in an Arrhenius format to extract apparent activation energies. Additionally, we thank Alexei Sokolov, Amos Tsai, and Dan Neumann for their valued suggestions.
References
- Allison, S. D., B. Chang, T. W. Randolph, and J. F. Carpenter. 1999. Hydrogen bonding between sugar and protein is responsible for inhibition of dehydration-induced protein unfolding. Arch. Biochem. Biophys. 365:289–298. [DOI] [PubMed] [Google Scholar]
- Angell, C. A., K. L. Ngai, G. B. McKenna, P. F. McMillan, and S. W. Martin. 2000. Relaxation in glassforming liquids and amorphous solids. J. Appl. Phys. 88:3113–3157. [Google Scholar]
- Ansari, A., C. M. Jones, E. R. Henry, J. Hofrichter, and W. A. Eaton. 1992. The role of solvent viscosity in the dynamics of protein conformational changes. Science. 256:1796–1798. [DOI] [PubMed] [Google Scholar]
- Beece, D., L. Eisenstein, H. Frauenfelder, D. Good, M. C. Marden, L. Reinisch, A. H. Reynolds, L. B. Sorensen, and K. T. Yue. 1980. Solvent viscosity and protein dynamics. Biochemistry. 19:5147–5157. [DOI] [PubMed] [Google Scholar]
- Bell, L. N., M. J. Hageman, and L. M. Muraoka. 1995. Thermally induced denaturation of lyophilized bovine somatotropin and lysozyme as impacted by moisture and excipients. J. Pharm. Sci. 84:707–712. [DOI] [PubMed] [Google Scholar]
- Bergquist, P., Y. Zhu, A. A. Jones, and P. T. Inglefield. 1999. Plasticization and antiplasticization in polycarbonates: the role of diluent motion. Macromolecules. 32:7925–7931. [Google Scholar]
- Branca, C., S. Magazu, G. Maisano, and F. Migliardo. 2001. Vibrational and relaxational contributions in disaccharide/H2O glass formers. Phys. Rev. B. 6422:art-224204.
- Brooks, C. L., M. Karplus, and B. M. Pettitt. 1988. Proteins: A Theoretical Perspective Of Dynamics, Structure, and Thermodynamics. Advances in Chemical Physics, Vol. LXXI. John Wiley & Sons. 1–200.
- Buchenau, U., Y. M. Galperin, V. L. Gurevich, and H. R. Schober. 1991. Anharmonic potentials and vibrational localization in glasses. Phys. Rev. B. 43:5039–5045. [DOI] [PubMed] [Google Scholar]
- Buchenau, U., and R. Zorn. 1992. A relation between fast and slow motions in glassy and liquid selenium. Europhys. Lett. 18:523–528. [Google Scholar]
- Buera, M. P., S. Rossi, S. Moreno, and J. Chirife. 1999. DSC confirmation that vitrification is not necessary for stabilization of the restriction enzyme EcoRI dried with saccharides. Biotechnol. Prog. 15:577–579. [DOI] [PubMed] [Google Scholar]
- Buitink, J., I. J. van den Dries, F. A. Hoekstra, M. Alberda, and M. A. Hemminga. 2000. High critical temperature above Tg may contribute to the stability of biological systems. Biophys. J. 79:1119–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caliskan, G., A. Kisliuk, A. M. Tsai, C. L. Soles, and A. P. Sokolov. 2003. Protein dynamics in viscous solvents. J. Chem. Phys. 118:4230–4236. [Google Scholar]
- Carpenter, J. F., L. M. Crowe, and J. H. Crowe. 1987. Stabilization of phosphofructokinase with sugars during freeze-drying: characterization of enhanced protection in the presence of divalent cations. Biochim. Biophys. Acta. 923:109–115. [DOI] [PubMed] [Google Scholar]
- Casalini, R., K. L. Ngai, C. G. Robertson, and C. M. Roland. 2000. α- and β-relaxations in neat and antiplasticized polybutadiene. J. Polym. Sci. B Polym. Phys. 38:1841–1847. [Google Scholar]
- Cicerone, M. T., and M. D. Ediger. 1996. Enhanced translation of probe molecules in supercooled o-terphenyl: signature of spatially heterogeneous dynamics? J. Chem. Phys. 104:7210–7218. [Google Scholar]
- Cicerone, M. T., A. Tellington, L. Trost, and A. Sokolov. 2003. Substantially improved stability of biological agents in dried form. BioProc. Int. 1:36–47. [Google Scholar]
- Cicerone, M. T., P. A. Wagner, and M. D. Ediger. 1997. Translational diffusion on heterogeneous lattices: a model for dynamics in glass forming materials. J. Phys. Chem. B. 101:8727–8734. [Google Scholar]
- Cleland, J. L., X. Lam, B. Kendrick, J. Yang, T. H. Yang, D. Overcashier, D. Brooks, C. Hsu, and J. F. Carpenter. 2001. A specific molar ratio of stabilizer to protein is required for storage stability of a lyophilized monoclonal antibody. J. Pharm. Sci. 90:310–321. [DOI] [PubMed] [Google Scholar]
- Conrad, P. B. and J. J. de Pablo. 1999. Computer simulation of the cryoprotectant disaccharide α,α-trehalose in aqueous solution. J. Phys. Chem. A. 103:4049–4055. [Google Scholar]
- Cordone, L., M. Ferrand, E. Vitrano, and G. Zaccai. 1999. Harmonic behavior of trehalose-coated carbon-monoxy-myoglobin at high temperature. Biophys. J. 76:1043–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino, H. R., K. G. Carrasquillo, R. A. Cordero, M. Mumenthaler, C. C. Hsu, and K. Griebenow. 1998. Effect of excipients on the stability and structure of lyophilized recombinant human growth hormone. J. Pharm. Sci. 87:1412–1420. [DOI] [PubMed] [Google Scholar]
- Courtney, S. H., and G. R. Fleming. 1985. Photoisomerization of stilbene in low viscosity solvents—comparison of isolated and solvated molecules. J. Chem. Phys. 83:215–222. [Google Scholar]
- Davidson, P., and W. Q. Sun. 2001. Effect of sucrose/raffinose mass ratios on the stability of co-lyophilized protein during storage above the Tg. Pharm. Res. 18:474–479. [DOI] [PubMed] [Google Scholar]
- Doster, W. 1983. Viscosity scaling and protein dynamics. Biophys. Chem. 17:97–103. [DOI] [PubMed] [Google Scholar]
- Doster, W., S. Cusack, and W. Petry. 1989. Dynamical transition of myoglobin revealed by inelastic neutron scattering. Nature. 337:754–756. [DOI] [PubMed] [Google Scholar]
- Duddu, S. P., G. Zhang, and P. R. Dal Monte. 1997. The relationship between protein aggregation and molecular mobility below the glass transition temperature of lyophilized formulations containing a monoclonal antibody. Pharm. Res. 14:596–600. [DOI] [PubMed] [Google Scholar]
- Flom, S. R., V. Nagarajan, and P. F. Barbara. 1986. Dynamic solvent effects on large-amplitude isomerization rates. I. 2-Vinylanthracene. J. Phys. Chem. 90:2085–2092. [Google Scholar]
- Frick, B., and L. J. Fetters. 1994. Methyl group dynamics in glassy polyisoprene—a neutron backscattering investigation. Macromolecules. 27:974–980. [Google Scholar]
- Frick, B., and D. Richter. 1995. The microscopic basis of the glass transition in polymers from neutron-scattering studies. Science. 267:1939–1945. [DOI] [PubMed] [Google Scholar]
- Gavish, B. 1980. Position-dependent viscosity effects on rate coefficients. Phys. Rev. Lett. 44:1160–1163. [Google Scholar]
- Gavish, B., and M. M. Werber. 1979. Viscosity-dependent structural fluctuations in enzyme catalysis. Biochemistry. 18:1269–1275. [DOI] [PubMed] [Google Scholar]
- Gehring, P. M., and D. A. Neumann. 1997. Backscattering spectroscopy at the NIST Center for Neutron Research. Physica B. 241:64–70. [Google Scholar]
- Glowinkowski, S., D. J. Gisser, and M. D. Ediger. 1990. C-13 Nuclear magnetic resonance measurements of local segmental dynamics of polyisoprene in dilute solution—nonlinear viscosity dependence. Macromolecules. 23:3520–3530. [Google Scholar]
- Gottfried, D. S., E. S. Peterson, A. G. Sheikh, J. Q. Wang, M. Yang, and J. M. Friedman. 1996. Evidence for damped hemoglobin dynamics in a room temperature trehalose glass. J. Phys. Chem. 100:12034–12042. [Google Scholar]
- Green, J. L., and C. A. Angell. 1989. Phase relations and vitrification in saccharide-water solutions and the trehalose anomaly. J. Phys. Chem. 93:2880–2882. [Google Scholar]
- Grote, R. F., and J. T. Hynes. 1980. The stable states picture of chemical reactions. II. Rate constants for condensed and gas-phase reaction models. J. Chem. Phys. 73:2715–2732. [Google Scholar]
- Hagen, S. J., J. Hofrichter, and W. A. Eaton. 1995. Protein reaction kinetics in a room-temperature glass. Science. 269:959–962. [DOI] [PubMed] [Google Scholar]
- Hall, R. W., and P. G. Wolynes. 1987. The aperiodic crystal picture and free-energy barriers in glasses. J. Chem. Phys. 86:2943–2948. [Google Scholar]
- Iben, I. E., D. Braunstein, W. Doster, H. Frauenfelder, M. K. Hong, J. B. Johnson, S. Luck, P. Ormos, A. Schulte, P. J. Steinbach, A. H. Xie, and R. D. Young. 1989. Glassy behavior of a protein. Phys. Rev. Lett. 62:1916–1919. [DOI] [PubMed] [Google Scholar]
- Izutsu, K., S. Yoshioka, and T. Terao. 1994. Effect of mannitol crystallinity on the stabilization of enzymes during freeze-drying. Chem. Pharm. Bull. (Tokyo). 42:5–8. [DOI] [PubMed] [Google Scholar]
- Kanaya, T., T. Tsukushi, K. Kaji, J. Bartos, and J. Kristiak. 1999. Microscopic basis of free-volume concept as studied by quasielastic neutron scattering and positron annihilation lifetime spectroscopy. Phys. Rev. E. 60:1906–1912. [DOI] [PubMed] [Google Scholar]
- Kleinert, T., W. Doster, H. Leyser, W. Petry, V. Schwarz, and M. Settles. 1998. Solvent composition and viscosity effects on the kinetics of CO binding to horse myoglobin. Biochemistry. 37:717–733. [DOI] [PubMed] [Google Scholar]
- Kramers, H. A. 1940. Brownian motion in a field of force and the diffusion model of chemical reactions. Physica. 7:284–304. [Google Scholar]
- Lavalette, D., and C. Tetreau. 1988. Viscosity-dependent energy barriers and equilibrium conformational fluctuations in oxygen recombination with hemerythrin. Eur. J. Biochem. 177:97–108. [DOI] [PubMed] [Google Scholar]
- Lavalette, D., C. Tetreau, M. Tourbez, and Y. Blouquit. 1999. Microscopic viscosity and rotational diffusion of proteins in a macromolecular environment. Biophys. J. 76:2744–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenegger, H., W. Doster, T. Kleinert, A. Birk, B. Sepiol, and G. Vogl. 1999. Heme-solvent coupling: a Mossbauer study of myoglobin in sucrose. Biophys. J. 76:414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourdin, D., H. Bizot, and P. Colonna. 1997. Correlation between static mechanical properties of starch-glycerol materials and low-temperature relaxation. Macromol. Symp. 114:179–185. [Google Scholar]
- Malinovsky, V. K., V. N. Novikov, and A. P. Sokolov. 1991. Log-normal spectrum of low-energy vibrational excitations in glasses. Phys. Lett. A. 153:63–66. [Google Scholar]
- Mazzobre, M. F., M. D. Buera, and J. Chirife. 1997. Glass transition and thermal stability of lactase in low-moisture amorphous polymeric matrices. Biotechnol. Prog. 13:195–199. [Google Scholar]
- Miller, D. P., and J. J. de Pablo. 2000. Calorimetric solution properties of simple saccharides and their significance for the stabilization of biological structure and function. J. Phys. Chem. B. 104:8876–8883. [Google Scholar]
- Mouradian, R., C. Womersley, L. M. Crowe, and J. H. Crowe. 1984. Preservation of functional integrity during long-term storage of a biological membrane. Biochim. Biophys. Acta. 778:615–617. [DOI] [PubMed] [Google Scholar]
- Ng, K., and A. Rosenberg. 1991. Possible coupling of chemical to structural dynamics in subtilisin BPN′ catalyzed hydrolysis. Biophys. Chem. 39:57–68. [DOI] [PubMed] [Google Scholar]
- Noel, T. R., R. Parker, and S. G. Ring. 1996. A comparative study of the dielectric relaxation behaviour of glucose, maltose, and their mixtures with water in the liquid and glassy states. Carbohydr. Res. 282:193–206. [Google Scholar]
- Nozaki, R., and S. Mashimo. 1987. Dielectric-relaxation measurements of polyvinyl acetate in glassy state in the frequency-range 10−6–106 Hz. J. Chem. Phys. 87:2271–2277. [Google Scholar]
- Paciaroni, A., S. Cinelli, and G. Onori. 2002. Effect of the environment on the protein dynamical transition: a neutron scattering study. Biophys. J. 83:1157–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parak, F., E. W. Knapp, and D. Kucheida. 1982. Protein dynamics—Mossbauer spectroscopy on deoxymyoglobin crystals. J. Mol. Biol. 161:177–194. [DOI] [PubMed] [Google Scholar]
- Pikal, M. J., and D. R. Rigsbee. 1997. The stability of insulin in crystalline and amorphous solids: observation of greater stability for the amorphous form. Pharm. Res. 14:1379–1387. [DOI] [PubMed] [Google Scholar]
- Reat, V., R. Dunn, M. Ferrand, J. L. Finney, R. M. Daniel, and J. C. Smith. 2000. Solvent dependence of dynamic transitions in protein solutions. Proc. Natl. Acad. Sci. USA. 97:9961–9966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reat, V., H. Patzelt, M. Ferrand, C. Pfister, D. Oesterhelt, and G. Zaccai. 1998. Dynamics of different functional parts of bacteriorhodopsin: H-H-2 labeling and neutron scattering. Proc. Natl. Acad. Sci. USA. 95:4970–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg, A., K. Ng, and M. Punyiczki. 1989. Activity and viscosity effects on the structural dynamics of globular proteins in mixed-solvent systems. J. Mol. Liq. 42:31–43. [Google Scholar]
- Rothenberger, G., D. K. Negus, and R. M. Hochstrasser. 1983. Solvent influence on photo-isomerization dynamics. J. Chem. Phys. 79:5360–5367. [Google Scholar]
- Sastry, G. M., and N. Agmon. 1997. Trehalose prevents myoglobin collapse and preserves its internal mobility. Biochemistry. 36:7097–7108. [DOI] [PubMed] [Google Scholar]
- Schlichter, J., J. Friedrich, L. Herenyi, and J. Fidy. 2001. Trehalose effect on low temperature protein dynamics: fluctuation and relaxation phenomena. Biophys. J. 80:2011–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settles, M., and W. Doster. 1996. Anomalous diffusion of adsorbed water: a neutron scattering study of hydrated myoglobin. Faraday Discuss. 103:269–279. [Google Scholar]
- Shamblin, S. L., X. L. Tang, L. Q. Chang, B. C. Hancock, and M. J. Pikal. 1999. Characterization of the time scales of molecular motion in pharmaceutically important glasses. J. Phys. Chem. B. 103:4113–4121. [Google Scholar]
- Sokolov, A. P., A. Kisliuk, D. Quitmann, A. Kudlik, and E. Rossler. 1994. The dynamics of strong and fragile glass formers—vibrational and relaxation contributions. J. Non Crystall. Sol. 172:138–153. [Google Scholar]
- Sokolov, A. P., E. Rossler, A. Kisliuk, and D. Quitmann. 1993. Dynamics of strong and fragile glass formers—differences and correlation with low-temperature properties. Phys. Rev. Lett. 71:2062–2065. [DOI] [PubMed] [Google Scholar]
- Steinbach, P. J., A. Ansari, J. Berendzen, D. Braunstein, K. Chu, B. R. Cowen, D. Ehrenstein, H. Frauenfelder, J. B. Johnson, and D. C. Lamb. 1991. Ligand binding to heme proteins: connection between dynamics and function. Biochemistry. 30:3988–4001. [DOI] [PubMed] [Google Scholar]
- Subramaniam, S., M. Gerstein, D. Oesterhelt, and R. Henderson. 1993. Electron diffraction analysis of structural changes in the photocycle of bacteriorhodopsin. EMBO J. 12:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, K., T. Takeda, and K. Miyajima. 1991. Cryoprotective effect of saccharides on denaturation of catalase by freeze-drying. Chem. Pharm. Bull. (Tokyo). 39:1091–1094. [Google Scholar]
- Tian, W. D., J. T. Sage, P. M. Champion, E. Chien, and S. G. Sligar. 1996. Probing heme protein conformational equilibration rates with kinetic selection. Biochemistry. 35:3487–3502. [DOI] [PubMed] [Google Scholar]
- Tsai, A. M., D. A. Neumann, and L. N. Bell. 2000. Molecular dynamics of solid-state lysozyme as affected by glycerol and water: a neutron scattering study. Biophys. J. 79:2728–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitkup, D., D. Ringe, G. A. Petsko, and M. Karplus. 2000. Solvent mobility and the protein “glass” transition. Nat. Struct. Biol. 7:34–38. [DOI] [PubMed] [Google Scholar]
- Wang, W. 2000. Lyophilization and development of solid protein pharmaceuticals. Int. J. Pharm. 203:1–60. [DOI] [PubMed] [Google Scholar]
- Yamamuro, O., K. Harabe, T. Matsuo, K. Takeda, I. Tsukushi, and T. Kanaya. 2000. Boson peaks of glassy mono- and polyalcohols studied by inelastic neutron scattering. J. Phys. Cond. Matter. 12:5143–5154. [Google Scholar]
- Yedgar, S., C. Tetreau, B. Gavish, and D. Lavalette. 1995. Viscosity dependence of O2 escape from respiratory proteins as a function of cosolvent molecular weight. Biophys. J. 68:665–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka, S., Y. Aso, K. Izutsu, and T. Terao. 1994. Application of accelerated testing to shelf-life prediction of commercial protein preparations. J. Pharm. Sci. 83:454–456. [DOI] [PubMed] [Google Scholar]
- Zaccaï, G. 2000. How soft is a protein? A protein dynamics force constant measured by neutron scattering. Science. 288:1604–1607. [DOI] [PubMed] [Google Scholar]
- Zhu, W., and M. D. Ediger. 1997. Viscosity dependence of polystyrene local dynamics in dilute solution. Macromolecules. 30:1205–1210. [Google Scholar]