

Abstract
Computational methods are employed to simulate interaction of scorpion toxin ScyTx in complex with the small conductance calcium-activated potassium channel rsk2. All of available 25 structures of ScyTx in the Protein Data Bank determined by NMR were considered for improving performance of rigid protein docking of ZDOCK. Four main binding modes were found among a large number of predicted complexes by using clustering analysis, screening with expert knowledge, energy minimization, and molecular dynamics simulations. The quality and validity of the resulting complexes were further evaluated by molecular dynamics simulations with the generalized Born solvation model and by calculation of relative binding free energies with the molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) in the AMBER 7 suit of programs. The complex formed by the 22nd structure of the ScyTx and rsk2 channel was identified as the most favorable complex by using a combination of computational methods, which contain further introduction of flexibility without restraining residue side chain. From the resulted spatial structure of the ScyTx and rsk2 channel, ScyTx associates the mouth of the rsk2 channel with α-helix rather than β-sheet. Structural analysis first revealed that Arg13 played a novel and vital role of blocking the pore of the rsk2 channel, whose role is remarkably different from that of highly homologous scorpion toxin P05. Between the interfaces in the ScyTx-rsk2 complex, strong electrostatic interaction and hydrogen bonds exist between Arg13 of ScyTx and Gly-Tyr-Gly-Asp sequential residues located in the four symmetrical chains of the pore region. Simultaneously, five hydrogen bonds between Arg6 of ScyTx and Asp341(C), Val366(C), and Pro367(C), and electrostatic interaction between Arg6 of ScyTx and Asp364(B) and Asp341(C) are also found by structural analysis. In addition, His31 located at the C-terminal of ScyTx is surrounded by Val342(A), Asp364(A), Met365(A), Pro367(B), and Asn366(B) within a contact distance of 4.0 Å. These simulation results are in good agreement with experimental data and can effectively explain the binding phenomena between ScyTx and the potassium channel at the level of molecular spatial structure. The consistency between results of molecular modeling and experimental data strongly suggests that our spatial structure model of the ScyTx-rsk2 complex is reasonable. Therefore, molecular docking combined with molecular dynamics simulations followed by molecular mechanics Poisson-Boltzmann surface area analysis is an attractive approach for modeling scorpion toxin-potassium channel complexes a priori for further biological studies.
INTRODUCTION
Potassium channels, as a diverse and ubiquitous family of membrane proteins, play critical roles in cellular signaling processes regulating neurotransmitter release, heart rate, neuronal excitability, etc. Increasing efforts are devoted to the structural and functional characterization of K+ channels (Doyle et al., 1998; Gandhi et al., 2003; Gu and Jan, 2003). In this regard, specific receptors of potassium channels are of particular interest. Scorpion toxins constitute the largest group of potassium channel blockers, which are useful pharmacological probes to identify the pore region of K+ channels and elucidate the structural topology of the extracellular surface of channels (Ranganathan et al., 1996; Srinivasan et al., 2002). Given the diversity of potassium channels, understanding the precise composition of channel-scorpion toxin complexes at the atomic level remains a huge challenge nowadays.
ScyTx, a scorpion toxin from the venom of the scorpion Leiurus quinquestriatus hebraeus, acts on the apamin-sensitive small conductance Ca2+-activated K+ channel (SKCa) (Vergara et al., 1998; Desai et al., 2000). It is a polypeptide containing 31 residues cross-linked by three disulfide bridges, and possesses binding and physiological properties similar to scorpion toxin P05 with 87% sequence similarity (Fig. 1). The structure-function relationship studies by chemical modifications indicated that Arg6 and Arg13 are essential both for binding to SKCa and for the functional effect of the toxin (Auguste et al., 1992). Subsequently, the three dimensional (3D) structure was determined by NMR in 1995 (Protein Data Bank (PDB) code: 1SCY), from which Arg6 and Arg13 are located in the α-helix (Martins et al., 1995). Similar to P05 (Cui et al., 2002), these data suggest strongly that the α-helix was mainly used to associate with SKCa instead of β-sheet, which is often used for scorpion toxins ChTX (Park and Miller, 1992), Lq2 (Cui et al., 2001), AgTx2 (Eriksson and Roux, 2002), and MTX (Fu et al., 2002).
FIGURE 1.

The sequence alignments of ScyTx and P05.
Prediction of protein-protein interactions by docking methods (Smith and Sternberg, 2002) has been the preference when having no high-resolution 3D structures of protein complex. It is because accurate predictive docking methods could provide substantial structural knowledge about complexes, from which functional information could be inferred or experiments designed to obtain it. To make progress in characterizing the interface for the ScyTx-SKCa complex at the atomic level, ZDOCK (Chen et al., 2003a) was used to determine the 3D structure of the ScyTx in complex with the rsk2 channel, which is the highly apamin-sensitive SKCa from rat (Vergara et al., 1998; Cui et al., 2002). After obtaining near-native complexes by ZDOCK, the quality of generated binding modes is further examined by molecular dynamics (MD) simulation and by calculating the relative binding free energy using molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) in the AMBER 7 suit of programs (Wang et al., 2001; Case et al., 2002). Then, the final equilibrated 3D structure of the ScyTx-rsk2 complex was produced for identifying residues involved in complex formation and elucidating the mechanism underlying the specificity of toxin-channel interaction.
METHODS AND THEORY
Atomic coordinates
The atomic coordinates of scorpion toxin ScyTx (PDB code: 1SCY) were downloaded from the Protein Data Bank (Berman et al., 2000). All 25 conformations from NMR were used for improving rigid docking performance of ZDOCK.
The potassium channel from Streptomyces lividans (KcsA) is an integral membrane protein with sequence similarity to most potassium channels, particularly in the pore region seen from sequence alignment of selected K+ channels with various types (Doyle et al., 1998) and demonstrated by experiments (Mackinnon et al., 1998). The homologous spatial structure of the pore region of rsk2 was modeled by using the crystallographic structure of the KcsA channel (PDB code: 1BL8) as a template through the SWISS-MODEL server (Guex and Peitsch, 1997). Sequence alignments of KcsA with the rsk2 channel can be found in literatures (Cui et al., 2002). The modeled 3D structure of rsk2 was subjected to refinement by 500-steps energy minimization by the SANDER module in the AMBER 7 suit of programs (Case et al., 2002).
ZDOCK
A rigid-body docking algorithm ZDOCK was recently developed. The latest ZDOCK 2.3 combines pairwise shape complementarity, desolvation, and electrostatic energies as the target function, which improved the performance of protein docking (Chen et al., 2003a). Good predictions were achieved during the Critical Assessment of Prediction of Interactions (CAPRI) Challenge (Chen et al., 2003b). The detailed algorithm and usage are described on their web site (http://zlab.bu.edu/rong/dock/index.shtml).
The rsk2 channel is embedded in the cell membrane. Toxin ScyTx has a rigid spatial structure stabilized by three disulfide bridges, and its rigid structure was used in protein engineering for designing the mini CD4 that reproduced the core of the CD4 site interacting with HIV-1 envelope glycoprotein (Vita et al., 1999). Thus, their complex structure is suitable to calculate by the rigid protein docking program of ZDOCK.
Energy minimization of ZDOCK structures
For each group of predicted complex structures, possible hits were obtained by clustering analysis and screening with expert knowledge among top 1000 predicted ligand orientations from ZDOCK (Chen et al., 2003b). Each possible toxin-channel complex is subjected to 500-steps energy minimization using the SANDER module in the AMBER 7 suit of programs. The ligand-receptor electrostatics energy (▵Eelec) was calculated with the ANAL program of AMBER 7 (Case et al., 2002) for further assessing 3D structures of the complex combined with previous structure-functional studies (Auguste et al., 1992).
Molecular dynamics simulations
Further 10 ps MD simulation was carried out using the distance-dependent dielectric with a time step of 2.0 fs. The nonbonded cutoff was set to 12.0 Å and heavy atoms were restrained with force constant 5.0 kcal/mol/ Å2. The most plausible binding mode of the ScyTx-rsk2 complex for 25 conformations of ScyTx was screened by interactive energy calculated by the ANAL program and structural analysis based on the structure-function experiments (Auguste et al., 1992).
In the comparison of binding free energies of different ScyTx orientations in complex with rsk2, it is important to ensure that each of these systems is equilibrated enough ahead of data collection and analysis (Kuhn and Kollman, 2000). In our work, the generalized Born (GB) solvation model in macromolecular simulations (Tsui and case, 2001) was used instead of explicit water during more sufficient MD simulation. GB-MD simulations of 50 ps (IGB = 2 in the AMBER 7) were carried out at 300 K with a time step of 2.0 fs. A cutoff distance of 12 Å was used for nonbonded interaction. We repeated the equilibration step three times starting from a larger force constant 5.0 kcal/mol/ Å 2 and then gradually reducing it for restraining all heavy atoms in the first 30 ps MD simulations. During the last 20 ps MD simulations, heavy atoms only in backbone were restrained with a force constant 0.25 kcal/mol/ Å 2 so that more flexibility was introduced into the side-chain conformations. Finally, 100 snapshots every 0.2 ps from the last 20 ps simulations were collected for postprocessing analysis. The ff99 force field (Parm99) (Wang et al., 2000) was used throughout the energy minimization and MD.
Calculation of binding free energy by MM-PBSA
In the MM-PBSA of AMBER 7.0, the binding free energy of A + B → AB is calculated using the following thermodynamic cycle:
SCHEME 1.

where T is the temperature, S is the solute entropy, ▵Ggas is the interaction energy between A and B in the gas phase, and 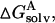
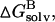 and
and 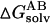 are the solvation free energies of A, B, and AB, which are estimated using a GB surface area (GBSA) method (Qiu et al., 1997; Tsui and Case, 2001; Case et al., 2002), i.e.,
are the solvation free energies of A, B, and AB, which are estimated using a GB surface area (GBSA) method (Qiu et al., 1997; Tsui and Case, 2001; Case et al., 2002), i.e., 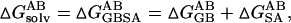 etc. ▵GGB and ▵GSA are the electrostatic and nonpolar term, respectively. ▵Ebond, ▵Eangle and ▵Etorsion are contributions to the intramolecular energy ▵Eintra of the complex. EvdW is van der Waals (vdW) interaction energy. Because of the constant contribution of −T▵S for each docked complex, we quote ▵G*binding, which is ▵Gbinding + T▵S, in the following discussion. By using MM-PBSA for postprocessing collected 100 snapshots, the relative binding free energy ▵G*binding can be calculated for assessing the quality and validity of resulting ScyTx-rsk2 complexes.
etc. ▵GGB and ▵GSA are the electrostatic and nonpolar term, respectively. ▵Ebond, ▵Eangle and ▵Etorsion are contributions to the intramolecular energy ▵Eintra of the complex. EvdW is van der Waals (vdW) interaction energy. Because of the constant contribution of −T▵S for each docked complex, we quote ▵G*binding, which is ▵Gbinding + T▵S, in the following discussion. By using MM-PBSA for postprocessing collected 100 snapshots, the relative binding free energy ▵G*binding can be calculated for assessing the quality and validity of resulting ScyTx-rsk2 complexes.
RESULTS AND DISCUSSION
Candidates of ScyTx-rsk2 complex screened by ZDOCK and MD
During ZDOCK, both ScyTx and rsk2 were treated as rigid bodies; thus, the effect of residue flexibility was neglected. To improve the docking performance, each available NMR conformation of ScyTx was considered. After energy minimization of ZDOCK structures and further 10 ps MD simulations using the distance-dependent dielectric combined with structure-functional studies (Auguste et al., 1992), four main binding modes were identified (Fig. 2). All α-helixes of toxin ScyTx from four modes were used to associate with the pore region of rsk2, which is in agreement with experimental results. Simultaneous neutralization of the positive charges of guanidinium groups of residues R6 and R13 resulted in total loss of binding affinity (Auguste et al., 1992). Similar to its highly homologous toxin P05 (Sabatier et al., 1993), a positive patch of electrostatic potential around the side chain of Arg6, Arg7, and Arg13 was used to recognize the rsk2 pore with strongly negative electrostatic potential (Cui et al., 2002). Table 1 lists the possible residue contacts and interactive energies after 10 ps MD between ScyTx and rsk2 and interactive energies.
FIGURE 2.

Relative positions of the Arg13 side chain and differential spatial orientations of ScyTx in complex with the rsk2 channel among four binding modes. (Binding mode I) Slope α-helix of ScyTx by diagonal way around the central axes of the rsk2 channel; (binding mode II) slope α-helix of ScyTx by middle way around the central axes of the rsk2 channel; (binding mode III) flat α-helix of ScyTx by diagonal way around the central axes of the rsk2 channel; and (binding mode IV) flat α-helix of ScyTx by middle way around the central axes of rsk2 channel.
TABLE 1.
Plausible binding modes and interactive residue pairwise between ScyTx and rsk2 within a contact distance of 4.0 Å
| ScyTx structure | Contacts between residue pairwise (ScyTx residue–rsk2 residue) | ▵Eelec | ▵EvdW | ▵Einter | Binding mode | ||
|---|---|---|---|---|---|---|---|
| 1 | R6-D341 | R13-G363 | H31-N368 | −289.88 | −79.64 | −369.52 | I |
| (12, 13) | R6-D364 | R13-Y362 | H31-W351 | ||||
| 22 | R6-D341 | R13-Y362 | H31-N368 | −357.21 | −74.07 | −431.28 | II |
| (3,4,8,16,17,19,23,24) | R6-N368 | R13-G361 | H31-V342 | ||||
| 9 | R6-Y362 | R13-N368 | H31-Q339 | −257.91 | −64.33 | −322.24 | III |
| (2,6,14,15,20,21) | R6-G363 | R13-D341 | H31-N368 | ||||
| 10 | R6-V366 | R13-G363 | H31-D341 | −371.64
|
−68.75
|
−440.39
|
IV
|
| R6-N368 | R13-Y362 | H31-M365 | |||||
All energies are in kcal/mol. Interactive energies were estimated by the ANAL module in AMBER 7.
There are some differences among the four binding modes in Fig. 2. The α-helix of ScyTx presents slope or flat orientations by diagonal or just middle around the central axes of the rsk2 channel. Unlike P05, an interesting finding is the Arg13 residue, whose side chain just hangs around the mouth of the rsk2 channel in binding mode I including the 1st, 4th, 12th, and 16th structures of ScyTx, or directly plugs into the pore region in the binding mode II containing the 3rd, 8th, 13th, 17th, 19th, 22nd, 23rd, and 24th conformations of ScyTx. Such phenomenon of blocking the channel pore often takes place when β-sheets of scorpion toxins are used to contact potassium channels with a considerably conserved Lys residue (Park and Miller, 1992; Cui et al., 2001; Eriksson and Roux, 2002; Fu et al., 2002). Other conformations of ScyTx with similar binding modes are also listed in Table 1. Binding modes III and IV are different from the above spatial orientations of ligands. Their α-helixes simply span the mouth of rsk2 channel, which is similar to the P05 complex (Cui et al., 2002), and other conformations of ScyTx using similar spatial orientation are also listed in Table 1.
To better identify the resulting ScyTx-rsk2 complexes, the interactive electrostatic energy is not enough (Cui et al., 2001, 2002; Fu et al., 2002). The binding free energy was often necessary for further discrimination between the different modes (Kuhn and Kollman, 2000; Wang et al., 2001; Eriksson and Roux, 2002).
MD simulations with GB solvation model and binding free energy
The solvent environment plays an important role on molecular structures, dynamics, and energetics. Calculation of MD simulations of biological macromolecules in explicitly water molecules is extremely costly on the nanosecond timescale. However, an analytical GB model efficiently describes electrostatics of molecules in a water environment. It represents the solvent implicitly as a continuum with the dielectric properties of water, and includes the charge screening effects of salt. Thus, long (100 ps or more) solvent equilibration is not required when the GB is used. The theory and applications of the GB model are recently reviewed (Tsui and Case, 2001). The GB model (IGB = 2) of AMBER 7 was adopted to introduce more flexibility of residue side chain for further refining ScyTx-rsk2 complex obtained by ZDOCK in this work than previous studies (Cui et al., 2001, 2002; Fu et al., 2002).
Introduction of residue side-chain flexibility can reduce the number of candidates of ScyTx-rsk2 complexes for MD simulations at certain degrees. To get a balance between calculation cost and amount of near-native ScyTx-rsk2 complexes, two ScyTx complexes were selected for each of binding modes I, II, and III. In this work, two criteria of choosing candidates of ScyTx complexes were used: one is complex with the middle and the highest interactive energy, the other is a different conformation of residue Arg13 hanging or blocking the rsk2 pore in the binding modes I and II. Thus, a total of seven candidates from Table 1 were used for further discrimination.
MD simulations of 50 ps were then performed for all seven candidates of ScyTx-rsk2 complexes; 100 snapshots for further processing analysis were selected from the last 20 ps of the trajectories with only slightly restraining heavy atoms of backbone for exploring more significant rotameric states of the side chains. After 30 ps MD simulations for the 22nd conformation of ScyTx bound with rsk2 channel, root mean-square deviation (RMSD) of all heavy atoms and only Cα atoms compared with the starting complex are shown in Fig. 3, whose little variance indicated that the system was equilibrated during snapshot collection. RMSD of other six possible complex structures are also much similar. The MM-PBSA analysis allows us to separate the total free energy of binding into electrostatic and vdW solute-solute and solute-solvent interactions, thereby gaining additional insights into the physics of the ScyTx-rsk2 complex association process.
FIGURE 3.

Root mean-square deviations (Å) of all heavy atoms (upper line) and the α-carbons (lower line) of the ScyTx-rsk2 complex from 30 ps to 50 ps MD simulations compared with starting docked structures.
Table 2 lists the components of molecular mechanics and solvation energies. Overall, binding mode II is most favorable among the four binding modes, which is in agreement with initial analysis by ZDOCK and MD listed in Table 1. The complex of the 22nd conformation of ScyTx and rsk2 has the lowest binding free energy, which is −58.92 kcal/mol, ∼10 kcal/mol more negative than the second most stable complex of the 16th conformation of the ScyTx and rsk2 channel. For all binding modes, the electrostatic energies are the dominant contributor to interactive energies, which can explain why local patches with large positive electrostatic potential in scorpion toxins are used to associate with mouths of potassium channels bearing a large negative electrostatic potential (Cui et al., 2001, 2002; Fu et al., 2002). However, there is no predominant factor for leading to stable complex formation because no obvious difference is found between electrostatic energies (▵Eelec + ▵▵GGB) and van der Waals energies (▵EvdW + ▵▵GSA) among all binding modes in the implicit solvent environment.
TABLE 2.
Relative binding free energies of four binding modes of ScyTx in complex with the rsk2 channel
| Binding mode | ScyTx structure | ▵Eelec | ▵EvdW | ▵Einter | ▵▵GGB | ▵▵GSA | ▵G*binding |
|---|---|---|---|---|---|---|---|
| I | 1 | −287.74 | −86.09 | −373.82 | 349.35 | −11.54 | −36.02 |
| 13 | −296.57 | −85.02 | −381.59 | 350.30 | −12.21 | −43.50 | |
| II | 16 | −285.52 | −94.56 | −380.08 | 344.09 | −12.68 | −48.67 |
| 22 | −338.95 | −102.65 | −441.60 | 396.28 | −13.61 | −58.92 | |
| III | 9 | −312.00 | −71.18 | −383.18 | 370.97 | −9.31 | −21.52 |
| 20 | −297.90 | −90.64 | −388.55 | 366.73 | −11.53 | −33.35 | |
| IV | 10 | −359.03 | −87.94 | −446.97 | 418.70 | −11.22 | −39.49 |
All energies are in kcal/mol.
Discrimination between the binding modes assisted by the computational alanine scanning
To further discriminate among the binding modes, the difference in the binding free energies between mutated and wild-type complexes (▵▵Gbinding) was calculated by the computational alanine scanning in MM-PBSA (Masova and Kollman, 1999). Table 3 lists the calculated ▵▵Gbinding for complex conformations with lower binding free energies among each binding mode. Excellent agreement was found between the calculated and experimental data for the complex of the 22nd conformation of ScyTx and rsk2. ScyTx modified on Arg6 and Arg13 was completely unable to inhibit the binding of both 125I-apamin and 125I-[Tyr2]ScyTx to their receptors (Auguste et al., 1992), which corresponds to the enormous value of 31.97 kcal/mol in the ▵▵Gbinding when simultaneously mutating Arg6 and Arg13. His31-modified ScyTx was 40-fold less potent than ScyTx in inhibiting the binding of the two 125I-labeled toxins to their receptors, which agrees well with the bigger value of 9.56 kcal/mol in the ▵▵Gbinding. Less differences were found in ▵▵Gbinding values for mutants of Glu27 and three Lys residues because such modification did not alter ScyTx properties (Auguste et al., 1992). Though most computational data of ▵▵Gbinding are in good agreement with experiment for the complex of the 13th conformation of ScyTx and rsk2 channel, the only 1.48 kcal/mol of ▵▵Gbinding cannot imply that His31 is important both for the binding activity. In addition, it is easy to find that computational data do not agree with experimental data for binding modes III and IV. Therefore the most stable spatial orientation of the 22nd structure in complex with rsk2 channel was used to elucidate structure-function relation of ScyTx.
TABLE 3.
Computational alanine scanning mutagenesis results for complex of ScyTx with the rsk2 channel (▵▵G = ▵Gmutant − ▵Gwild-type)
| Binding mode | Asn4Ala | Leu5Ala | Arg6Ala | Met7Ala | Gln9Ala | Leu10Ala | Ser11Ala | Arg13Ala | Ser14Ala | Leu15Ala |
|---|---|---|---|---|---|---|---|---|---|---|
| I(13th structure) | 1.91 | 0.75 | 16.6 | −0.14 | −0.19 | 2.45 | −0.27 | 13.45 | −0.77 | 0.33 |
| II(22nd structure) | 1.27 | 0.85 | 14.33 | 1.24 | −0.15 | 4.31 | −0.33 | 17.67 | −0.34 | 0.42 |
| III(20th structure) | −0.28 | 1.43 | 2.24 | −0.19 | 0.43 | 2.42 | −0.22 | −39.69 | −0.65 | 0.45 |
| IV(10th structure) | 8.45 | 9.10 | 16.05 | 9.14 | 8.64 | 13.49 | 8.43 | 17.48 | 10.73 | 9.29 |
| Arg6Ala | Lys20Ala | |||||||||
| Lys25Ala | ||||||||||
| Leu17Ala | Leu18Ala | Lys20Ala | Lys25Ala | Glu27Ala | Val29Ala | Lys30Ala | His31Ala | Arg13Ala | Lys30Ala | |
| I(13th structure) | 0.05 | 2.16 | −0.11 | 0.12 | −0.94 | 0.10 | 0.48 | 1.48 | 30.11 | 0.64 |
| II(22nd structure) | 0.36 | 2.85 | 1.42 | 0.15 | −1.00 | 0.01 | 1.34 | 9.56 | 31.97 | 2.92 |
| III(20th structure) | 0.79 | 2.71 | 0.00 | 0.18 | −1.05 | 0.65 | −0.05 | 4.66 | 13.51 | 0.33 |
| IV(10th structure) | 10.41 | 10.38 | 8.72 | 8.98 | 8.11 | 8.80 | 11.39 | 9.74 | 24.67 | 11.41 |
*All energies are in kcal/mol.
Here, our work shows that MM-PBSA is very useful to rank complexes that are closely related. However, there are still things to be done, such as force-field, MD sampling, and entropy-estimated, to make MM-PBSA more efficient and reliable (Wang et al., 2001).
Differential binding modes between highly homologous ScyTx and P05
From the most plausible binding mode of the 22nd conformation of ScyTx and rsk2 in our work (shown in Figs. 4 and 5), a considerably interesting thing is differential binding modes between slope orientation of ScyTx and flat orientation of P05 docked into the rsk2 channel (Cui et al., 2002) while having much highly homologous sequence (shown in Fig. 1) and much similar spatial structure (RMSD of Cα atoms: ∼1.68 Å). Such difference is caused by the essential discrepancy of their molecular macroscopical properties shown by their dipole moments (Darbon et al., 1999). Actually, difference of binding modes owing to the dipole moments was early noticed among scorpion toxins, and dipole moments of nine scorpion toxins were calculated (Darbon et al., 1999). All subsequent modeled complexes of scorpion toxins in complex with potassium channels show the consistency between binding modes and directions of dipole moments for Lq2 (Cui et al., 2001), AgTx2 (Eriksson and Roux, 2002), MTX (Fu et al., 2002), and P05 (Cui et al., 2002). Similarly, the big difference of dipole moments for ScyTx and P05 further leads to their differential binding modes. In this work, we first present dominant contribution of electrostatic energies among interactive energies during the molecular recognition process between scorpion toxin and potassium channel (shown in Table 2).
FIGURE 4.

Plausible interaction between function residues of ScyTx and residues of the rsk2 channel within a contact distance of 4.0 Å. Upper two figures are Arg6 of ScyTx surrounded by residues from the rsk2 channel. Lower two figures are Arg13 of ScyTx plugging into the pore of the rsk2 channel. Dotted line represents hydrogen bond between two atoms.
FIGURE 5.

Plausible interaction between residue pairwise between His31 of ScyTx and residues of the rsk2 channel (left figure) and relative positions of ScyTx residues unbinding the rsk2 channel (right figure).
Different from Arg13 in the α-helix of P05 interacting with Asp341(A) and Asp364(B) in the outer region of the selective filter of the rsk2 channel (Cui et al., 2002), a more interesting and significant phenomenon is that Arg13 of ScyTx plugged its side chain into the pore for exerting its function, which is first predicted among scorpion toxins. Within a contact distance of 4.0 Å, the side chain of Arg13 was surrounded by a considerably conservative Gly-Tyr-Gly signature motif of potassium channels (Doyle et al., 1998). The RMSDs for backbone and side-chain atoms are 0.27 and 0.17 Å for the Gly-Tyr-Gly signature motif between unbound and equilibrated rsk2 channels, respectively. This shows much less distortion of the selectivity filter for further explaining structural conservation of the potassium channel (Mackinnon et al., 1998). However, much more conformational change happens between unbound and bound Arg13 residue in ScyTx by further introduction of side-chain flexibility during the MD with the GB solvation model. When backbone atoms of Arg13 are well supposed (RMSD, 0.07 Å), the distances of corresponding CB, CG, CD, NE, CZ, NH1, and NH2 atoms (same atomic name as its PDB file) are 0.26, 0.76, 0.82, 1.67, 2.52, 2.70, and 3.27 Å, respectively, between unbound and bound atoms of Arg13. Thus, the side chain of Arg13 extends as long as possible for matching the limited space after binding to the rsk2 channel (figure not shown). Different from Lys residue blocking the pore of the potassium channel from the β-sheet of scorpion toxin (Cui et al., 2001; Fu et al., 2002), Arg13 of ScyTx also can contact the first Gly of the conservative Gly-Tyr-Gly signature motif, which is farther from the mouth of the rsk2 channel, because of a longer side chain of Arg than Lys. Taken together, such phenomena of different binding modes are much helpful to understand bioactive diversity of numerous scorpion toxins blocking potassium channels.
Structure-function relationship of ScyTx
From the most favorable 3D structure of ScyTx, it is considerably helpful to understand the structure-function relationship of ScyTx in complex with the rsk2 channel. Combined with computational data of the alanine scanning listed in Table 3 and essential Arg6 and Arg13 for bioactivity identified by experiment (Auguste et al., 1992), it is easy to understand both tight associations with the rsk2 channel shown in Fig. 4. Besides five hydrogen bonds between Arg13 of ScyTx and Gly361(B), Tyr362(B), Gly361(C), and Gly361(D) from the rsk2 channel, there is favorable electrostatic interaction between Arg13 of ScyTx and four Asp364 residues enclosing Arg13 within a contact distance of 5.0 Å. The importance of Arg13 in ScyTx was also observed from its mutant data, which is the biggest in Table 3. Thus, the considerably significant function of Arg13 of ScyTx can give a structurally novel clue for further biological studies in this work, though it was supposed to be essential for binding (Martins et al., 1995).
Fig. 4 also shows that another key residue Arg6 of ScyTx mainly interacts with residues of the C chain of the rsk2 channel and forms five hydrogen bonds with the carbonyl groups of the side chain of Asp341(C) and backbones of Val366(C) and Pro367(C). Strong electrostatic interaction exists between Arg6 of ScyTx and negatively charged residues Asp341(C) and Asp364(B) of the rsk2 channel. In addition, there are also some contributions for binding affinity of ScyTx between Arg6 and Gln342(C) and Asn368(C) of the rsk2 channel within a contact distance of 4.0 Å. The important effect of Arg6 on binding activity of ScyTx also corresponds to the second biggest value 14.33 kcal/mol in ▵▵Gbinding listed in Table 3. Structure-activity relationship studies of apamin and P05 showed that positively charged Arg residues from α-helixes are necessary for SKCa binding activity (Labbe-Jullie et al., 1991; Cui et al., 2002). This suggests that Arg6 and Arg13 in the α-helix of ScyTx control the productive recognition with the rsk2 channel, indicating that the 3D structure of the ScyTx-rsk2 complex in this work is reasonable.
Additionally, His31 is located at the C-terminal of ScyTx, and in vicinity residues of Val342(A), Asp364(A), Met365(A), Pro367(B), and Asn366(B) within a contact distance of 4.0 Å (Fig. 5). Their interaction was supported by the 40-fold decrease in the binding activity upon iodination of the His31 side chain for the integrity loss of the His imidazole ring in ScyTx (Auguste et al., 1992). When His31 is mutated into Ala31, the third biggest value 9.56 kcal/mol of ▵▵Gbinding is in very good agreement with experiment. There is also enough structural evidence that several residues, Lys20, Lys25, Glu27, and Lys30 are not involved in the binding process shown in Fig. 5. They are all located on the surface of antiparallel β-sheets, and just opposite to the bioactive surface of the α-helix. Esterification of Glu27 or unselective modification of Lys20, Lys27, and Lys30 leaves the binding affinity undisturbed (Auguste et al., 1992) because these charged residues are spatially far away from surface residues of the rsk2 channel. From their computational data of the alanine scanning listed in Table 3, little change of ▵▵Gbinding values can further explain experimental phenomena.
More recently, binding activity of ScyTx only decreases ∼75-fold for human SKCa2, which has the same residue sequence and spatial structure as rsk2 within the whole pore region of the potassium channel, after charge-neutralization mutation (Arg6 mutated into Leu6) (Shakkottai et al., 2001), and bioactivity of ScyTx completely loses after simultaneous neutralization of Arg6 and Arg13 (Auguste et al., 1992). However the neutralization of conserved residue Lys27Gln removed the scorpion toxin ChTX's characteristic voltage dependence (Park and Miller, 1992) and resulted in a 15,000-fold decrease of binding affinity for Shaker channel (Goldstein et al., 1994) because of a functional loss of the blocking channel pore. Such comparison of binding phenomena implies a remarkably different role of Arg6 and Arg13 during exerting their biological function for scorpion toxin ScyTx. Thus far the simulation of interaction of ScyTx and the rsk2 channel, confirming studies of structure-function relationship, strongly suggested that our 3D structure of ScyTx-rsk2 complex is reasonable. In addition, the function of Leu10 and Leu18 can be further identified by experiment because there is some contribution to binding free energies from mutant data in Table 3. In the spatial structure of the complex, Leu10 of ScyTx is adjacent to Asp341(C), Gly363(C), Asp364(C), and Val366(C), and Leu18 is close to Asp364(A) and Val366(B) within a contact distance of 4.0 Å. Other residues with less effect on binding activity of ScyTx are also listed in Table 3 through prediction with computational alanine scanning.
CONCLUSION
The most optimized 3D structure of scorpion toxin ScyTx in complex with the rsk2 channel was determined by ZDOCK and MD simulations followed by MM-PBSA analysis. Better performance of ZDOCK can be achieved by using different NMR conformations of ScyTx. Introduction of more flexibility for residue side chains by MD and relative binding free energy analysis by MM-PBSA are more helpful to discriminate more reasonable spatial structures of ScyTx and the rsk2 channel. The final refined 3D structure of ScyTx and the rsk2 channel agrees well with the primary clues from structure-function relationship studies. Though α-helixes are used to recognize pore entryway of the rsk2 channel for both ScyTx and P05 with high homology, the role of Arg13 exerting its biological function is vitally different between ScyTx and P05. Residue Arg13 of ScyTx was predicted to plug into the pore and interact with the considerably conservative Gly-Tyr-Gly signature motif. Thus, ScyTx can be used as pharmacological probes in studying the structural difference of SK channel pores after function of Arg13 is validated by further experiment in the future. Simultaneously, identified and unidentified residue pairwises between ScyTx and rsk2 channel, deduced from their 3D structure of complex, can be used in further biological studies of both ScyTx and the rsk2 channel. All these works can accelerate research of SKCa structure-function relationship, molecular design of more specific and biological inhibitors of SKCa, and understanding of the potential role of SKCa in epilepsy, sleep apnea, and neurodegenerative and smooth muscle disorders.
Acknowledgments
We highly appreciate Prof. James W. Caldwell from the University of California for offering us AMBER 7, and also greatly thank Mr. Rong Chen from Boston University for offering us the ZDOCK program and much help.
We are also grateful for financial support from the Key Project of the Ministry of Science and Technology of China (grant Nos. 2002BA49C and 2003CB514120).
References
- Auguste, P., M. Hugues, C. Mourre, D. Moinier, A. Tartar, and M. Lazdunski. 1992. Scyllatoxin, a blocker of Ca2+ activated K+ channels: structure-function relationships and brain localization of the binding sites. Biochemistry. 31:648–654. [DOI] [PubMed] [Google Scholar]
- Berman, H. M., J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov, and P. E. Bourne. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case, D. A., D. A. Pearlman, J. W. Caldwell, T. E. Cheatham 3rd, J. M. Wang, W. S. Ross, C. Simmerling, T. Darden, K. M. Merz, R. V. Stanton, A. Cheng, J. J. Vincent, M. Crowley, V. Tsui, H. Gohlke, R. Radmer, Y. Duan, J. Pitera, I. Massova, G. L. Seibel, U. C. Singh, P. Weiner, and P. A. Kollman. 2002. AMBER 7. University of California, San Francisco.
- Chen, R., L. Li, and Z. P. Weng. 2003a. ZDOCK: An initial-stage protein-docking algorithm. Proteins. 52:80–87. [DOI] [PubMed] [Google Scholar]
- Chen, R., W. W. Tong, J. Mintseris, L. Li, and Z. P. Weng. 2003b. ZDOCK predictions for the CAPRI Challenge. Proteins. 52:68–73. [DOI] [PubMed] [Google Scholar]
- Cui, M., J. H. Shen, J. M. Briggs, X. M. Luo, X. J. Tan, H. L. Jiang, K. X. Chen, and R. Y. Ji. 2001. Brownian dynamics simulations of interaction between scorpion toxin Lq2 and potassium ion channel. Biophys. J. 80:1659–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, M., J. H. Shen, J. M. Briggs, W. Fu, J. J. Wu, Y. M. Zhang, X. M. Luo, Z. W. Qi, R. Y. Ji, H. L. Jiang, and K. X. Chen. 2002. Brownian dynamics simulations of the recognition of the scorpion toxin P05 with the small-conductance calcium-activated potassium channels. J. Mol. Biol. 318:417–428. [DOI] [PubMed] [Google Scholar]
- Darbon, H., E. Blanc, and J. M. Sabatier. 1999. Three-dimensional structure of scorpion toxins: towards a new model of interaction with potassium channels. Perspect. Drug Discov. 15/16:41–60. [Google Scholar]
- Desai, R., A. Peretz, H. Idelson, P. Lazarovici, and B. Attali. 2000. Ca2+-activated K+ channels in human leukemic Jurkat T cells. Molecular cloning, biochemical and functional characterization. J. Biol. Chem. 275:39954–39963. [DOI] [PubMed] [Google Scholar]
- Doyle, D. A., J. M. Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Eriksson, M. A. L., and B. Roux. 2002. Modeling the structure of Agitoxin in complex with the Shaker K+ channel: a computational approach based on experimental distance restraints extracted from thermodynamic mutant cycles. Biophys. J. 83:2595–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, W., M. Cui, J. M. Briggs, X. Q. Huang, B. Xiong, Y. M. Zhang, X. M. Luo, J. H. Shen, R. Y. Ji, H. L. Jiang, and K. X. Chen. 2002. Brownian dynamics simulations of the recognition of the scorpion toxin maurotoxin with the voltage-gated potassium ion channels. Biophys. J. 83:2370–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi, C. S., E. Clark, E. Loots, A. Pralle, and E. Y. Isacoff. 2003. The orientation and molecular movement of a K+ channel voltage-sensing domain. Neuron. 40:515–525. [DOI] [PubMed] [Google Scholar]
- Goldstein, S. A., D. J. Pheasant, and C. Miller. 1994. The charybdotoxin receptor of a Shaker K+ channel: peptide and channel residues mediating molecular recognition. Neuron. 12:1377–1388. [DOI] [PubMed] [Google Scholar]
- Gu, C., and Y. N. Jan. 2003. A conserved domain in axonal targeting of Kv1 (Shaker) voltage-gated potassium channels. Science. 301:646–649. [DOI] [PubMed] [Google Scholar]
- Guex, N., and M. C. Peitsch. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 18:2714−2723. [DOI] [PubMed] [Google Scholar]
- Kuhn, B., and P. A. Kollman. 2000. A ligand that is predicted to bind better to avidin than biotin: insights from computational fluorine scanning. J. Am. Chem. Soc. 122:3909–3916. [Google Scholar]
- Labbe-Jullie, C., C. Granier, F. Albericio, M. L. Defendini, B. Ceard, H. Rochat, and J. Van Rietschoten. 1991. Binding and toxicity of apamin. Characterization of the active site. Eur. J. Biochem. 196:639–645. [DOI] [PubMed] [Google Scholar]
- Mackinnon, R., S. L. Cohen, A. Kuo, A. Lee, and B. T. Chait. 1998. Structural conservation in prokaryotic and eukaryotic potassium channels. Science. 280:106–109. [DOI] [PubMed] [Google Scholar]
- Martins, J. C., F. J. M. Van de Ven, and F. A. M. Borremans. 1995. Determination of the three-dimensional solution structure of scyllatoxin by 1H nuclear magnetic resonance. J. Mol. Biol. 253:590–603. [DOI] [PubMed] [Google Scholar]
- Massova, I., and P. A. Kollman. 1999. Computational alanine scanning to probe protein-protein interactions: a novel approach to evaluate binding free energies. J. Am. Chem. Soc. 121:8133–8143. [Google Scholar]
- Park, C. S., and C. Miller. 1992. Mapping function to structure in a channel-blocking peptide: electrostatic mutants of charybdotoxin. Biochemistry. 31:7749−7755. [DOI] [PubMed] [Google Scholar]
- Qiu, D., P. S. Shenkin, F. P. Hollinger, and W. C. Still. 1997. The GB/SA continuum model for solvation. A fast analytical method for the calculation approximate Born radii. J. Phys. Chem. 101:3005–3014. [Google Scholar]
- Ranganathan, R., J. H. Lewis, and R. MacKinnon. 1996. Spatial localization of the K+ channel selectivity filter by cycle-based structure analysis. Neuron. 16:131–139. [DOI] [PubMed] [Google Scholar]
- Sabatier, J. M., H. Zerrouk, H. Darbon, K. Mabrouk, A. Benslimane, H. Rochat, M. F. Martin-Eauclaire, and J. Van Rietschoten. 1993. P05, a new leiurotoxin I-like scorpion toxin: synthesis and structure-activity relationships of the α-amidated analog, a ligand of Ca2+-activated K+ channels with increased affinity. Biochemistry. 32:2763–2770. [DOI] [PubMed] [Google Scholar]
- Shakkottai, V. G., I. Regaya, H. Wulff, Z. Fajloun, H. Tomita, M. Fathallah, M. D. Cahalan, J. J. Gargus, J. M. Sabatier, and K. G. Chandy. 2001. Design and characterization of a highly selective peptide inhibitor of the small conductance calcium-activated K+ channel, SkCa2. J. Biol. Chem. 276:43145–43151. [DOI] [PubMed] [Google Scholar]
- Smith, G. R., and M. J. E. Sternberg. 2002. Prediction of protein-protein interactions by docking methods. Curr. Opin. Struct. Biol. 12:28–35. [DOI] [PubMed] [Google Scholar]
- Srinivasan, K. N., P. Gopalakrishnakone, P. T. Tan, K. C. Chew, B. Cheng, R. M. Kini, J. L. Y. Koh, S. H. Seah, and V. Brusic. 2002. SCORPION, a molecular database of scorpion toxins. Toxicon. 40:23–31. [DOI] [PubMed] [Google Scholar]
- Tsui, V., and D. A. Case. 2001. Theory and application of the generalized Born solvation model in macromolecular simulations. Biopolymers. 56:275–291. [DOI] [PubMed] [Google Scholar]
- Vergara, C., R. Latorre, and N. V. Marrion. 1998. Calcium-activated potassium channels. Curr. Opin. Neurobiol. 8:321–329. [DOI] [PubMed] [Google Scholar]
- Vita, C., E. Drakopoulou, J. Vizzavona, S. Rochette, L. Martin, A. Menez, C. Roumestand, Y. S. Yang, L. Ylisastigui, A. Benjouad, and J. G. Gluckman. 1999. Rational engineering of a miniprotein that reproduces the core of the CD4 site interacting with HIV-1 envelope glycoprotein. Proc. Natl. Acad. Sci. USA. 96:13091–13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., P. Cieplak, and P. A. Kollman. 2000. How well does a RESP (restrained electrostatic potential) model do in calculating the conformational energies of organic and biological molecules? J. Comput. Chem. 21:1049–1074. [Google Scholar]
- Wang, J. M., P. Morin, W. Wang, and P. K. Kollman. 2001. Use of MM-PBSA in reproducing the binding free energies to HIV-1 of TIBO derivatives and predicting the binding mode to HIV-1 RT of efavirenz by docking and MM-PBSA. J. Am. Chem. Soc. 123:5221–5230. [DOI] [PubMed] [Google Scholar]