

Abstract
The interchain 13C-19F dipolar coupling measured in a rotational-echo double-resonance (REDOR) experiment performed on mixtures of differently labeled KIAGKIA-KIAGKIA-KIAGKIA (K3) peptides (one specifically 13C labeled, and the other specifically 19F labeled) in multilamellar vesicles of dipalmitoylphosphatidylcholine and dipalmitoylphosphatidylglycerol (1:1) shows that K3 forms close-packed clusters, primarily dimers, in bilayers at a lipid/peptide molar ratio (L/P) of 20. Dipolar coupling to additional peptides is weaker than that within the dimers, consistent with aggregates of monomers and dimers. Analysis of the sideband dephasing rates indicates a preferred orientation between the peptide chains of the dimers. The combination of the distance and orientation information from REDOR is consistent with a parallel (N–N) dimer structure in which two K3 helices intersect at a cross-angle of ∼20°. Static 19F NMR experiments performed on K3 in oriented lipid bilayers show that between L/P = 200 and L/P = 20, K3 chains change their absolute orientation with respect to the membrane normal. This result suggests that the K3 dimers detected by REDOR at L/P = 20 are not on the surface of the bilayer but are in a membrane pore.
INTRODUCTION
Antimicrobial peptides are short, 10–40 residue polypeptides that serve as part of the innate defense system in both vertebrates and invertebrates (Andreu and Rivas, 1998; Epand and Vogel, 1999; Hancock and Diamond, 2000; van't Hof et al., 2001). Their mode of action involves an interaction with the pathogenic cell membrane where they are thought to form ion channels or pores, the exact mechanism of which is not clearly understood yet. Cytotoxicity studies and leakage experiments on artificial phospholipid membranes indicate that a minimal antimicrobial peptide concentration is required to observe lysis (van't Hof et al., 2001; Epand et al.; 1999, Shai, 1999). This observation gives rise to one of the fundamental questions concerning the peptide-bilayer interaction: do the peptide chains act individually while interacting with membrane systems, or do they associate with each other and form aggregates?
Electrophysiological and leakage studies of magainin, an antimicrobial peptide from the African clawed frog, Xenopus laevis (Zasloff, 1987), have shown cooperativity between the peptide chains (Cruciani et al., 1992; Matsuzaki et al., 1994, 1998a,b,c). The “unit” of cooperativity, however, is not well determined, and different studies sometimes seem to give conflicting results. Whereas fluorescent studies have indicated a pentameric magainin pore (Matsuzaki et al., 1995), fluorescence energy-transfer measurements have indicated a random distribution of magainin monomers on the surface of phospholipid vesicles (Schumann et al., 1997). Large pores with a diameter of ∼30 Å were detected by neutron diffraction in the bilayer in the presence of the peptide, which suggests that a relatively large number of magainin chains have to participate in the pore formation (Ludtke et al., 1996). There is no direct evidence, though, as to whether the peptide chains are in contact with each other or not (Huang, 2000).
In the previous companion article (Toke et al., 2004), rotational-echo double resonance (REDOR) (Gullion and Schaefer, 1989a,b) was used to determine accurately the local secondary structure of the magainin-like peptide antibiotic KIAGKIA-KIAGKIA-KIAGKIA (K3) (Maloy and Kari, 1995) in a lipid bilayer, and to establish qualitatively the overall positioning of K3 relative to the phospholipid headgroups and tails. In this article, REDOR is used to characterize the size and structure of K3 peptide aggregates. Two different sets of labels were inserted into K3 (cf. below) so that the heteronuclear dipolar couplings observed in mixtures of K3 chains now reveal aggregation and orientation directly.
A complication in the use of dipolar couplings to determine proximities of labels in bilayers is that the couplings are reduced by motional averaging. Thus, weak couplings associated with internuclear distances of 10 Å or more between lipids and peptides in membrane bilayers at 37°C simply cannot be determined by the available REDOR technology. The solution to this problem is to freeze the bilayer to stop all large-amplitude motions. Although dipolar couplings can now be determined by REDOR performed on frozen suspensions of multilamellar vesicles (MLVs), the sensitivity of the experiment is compromised by a poor filling factor. Most of the sample is still water. This means that accurate weak-coupling determinations of peptide-peptide separation and orientation are difficult. The solution to this problem is to lyophilize the sample to remove the nonstructural water. The lyophilization is done in the presence of sugar lyoprotectants to preserve important hydrogen bonds (Crowe and Crowe, 1984).
The lyophilized MLVs are not completely dehydrated (we have detected 15% w/w water) and many features of the structure and location of K3 in a bilayer are preserved, but a lyophilized MLV sample is nevertheless not suitable for a rigorous determination of lipid structure. This situation was examined in detail in I. In addition, it is possible that lyophilization has affected peptide aggregation. However, tightly packed large aggregates are not present in the lyophilized MLVs (see below), so they certainly are not present in fully hydrated MLVs. Moreover, the small tightly packed peptide aggregates that are found (at high resolution) in lyophilized MLVs are also found (at low resolution) in fully hydrated vesicles. Thus, it seems reasonable to accept the high-resolution characterization of the aggregates (in particular, the determination of size and internal structure) as biophysically relevant.
The REDOR experiments described in II were performed on K3 incorporated into MLVs of synthetic phospholipid bilayers. In the first REDOR experiment, one version of K3 was labeled by [1-13C]Ala10-[15N]Gly11 and the other by [2-2H, 3-19F]Ala10. (In these experiments the deuterium label was not used.) The 13C-19F dipolar coupling between fluorine and the labeled carbonyl carbon of Ala10 (the latter selected by one-bond 13C-15N dipolar coupling to the 15N of Gly11) necessarily measures interchain distances. In a second REDOR experiment, a different labeling scheme was used to determine whether K3 chains are parallel (N–N) or antiparallel (N–C). To characterize whether there is a unique orientation between the chains of a K3 dimer, slow-spinning 13C{19F} REDOR experiments were performed to determine relative dephasing rates of spinning sidebands, which are dependent on interchain orientation (O'Connor and Schaefer, 2002). Finally, to obtain information on the absolute orientation of K3 chains within the lipid bilayer, static 19F NMR experiments were performed on [2-2H, 3-19F]Ala14-(KIAGKIA)3-NH2 in lipid bilayers oriented on glass plates.
MATERIALS AND METHODS
Peptide synthesis and vesicle preparation
Labeled peptides were synthesized by Genaera Pharmaceuticals (Plymouth Meeting, PA) on an Applied Biosystems model 431A peptide synthesizer as described in I (Toke et al., 2004). Details of vesicle preparation are also described in I.
Preparation of macroscopically aligned samples
A peptide-lipid mixture was prepared in 70% methanol and deposited on thin glass slides (0.08 × 7.5 × 18 mm) (Salgado et al., 2001; Grage et al., 2002). After initial evaporation of the solvent, the films were dried under vacuum overnight. Depending on the amount of material used, 5–15 coated slides were stacked (0.8 mg peptide for lipid/peptide molar ratio (L/P) = 20 and L/P = 100, and 0.4 mg for L/P = 200). The stack was hydrated for 2 days at 48°C in an atmosphere of 98% relative humidity, resulting in spontaneous orientation of bilayers. The block was wrapped with parafilm and polyethylene foil to maintain the hydration of the sample during the NMR measurement. The degree of orientation of the lipid membranes (typically 80–90%) was determined with 31P NMR at 0° tilt as described before (Ulrich et al., 1992).
NMR spectroscopy
REDOR experiments were performed as described in I. Static 19F NMR measurements were performed on an 11.7-T wide-bore Unity Inova spectrometer (Varian, Palo Alto, CA), equipped with a 19F/1H double-tuned flat coil probe head with a 9 × 20 × 2.4 mm rectangular sample space in a susceptibility-matched ceramic housing (Doty Scientific, Columbia, SC). A standard echo-pulse sequence was used as described before (Salgado et al., 2001, Grage et al., 2002; Afonin et al., 2004), with 20 kHz 1H decoupling. Spectra were acquired above (35°C) and below (15°C) the lipid phase transition temperature, and referenced against CFCl3.
Calculation of REDOR dephasing
Dephasing was calculated using Bessel function expressions for a spin-½ pair (Mueller, 1995). This expression was summed over a Gaussian distribution of dipolar couplings corresponding to a distribution of isolated 13C-19F spin pairs. A single static 19F with an average position on the methyl C3 axis was assumed (cf. below). The effects of orientation and averaging resulting from the motion of the 19F about the methyl C3 axis were ignored. This is a reasonable approximation for the long-range 13C distances from the labeled carbonyl carbon of one K3 chain to the CH2F- group of another chain (O'Connor et al., 2002). Variation of the parameters of the distribution (mean and width) and the overall scaling was used to minimize the root mean-square deviation between the experimental and calculated total dephasing (O'Connor and Schaefer, 2002; O'Connor et al., 2002). Experimental and calculated values of sideband dephasing were compared using the ratio of the sideband dephasing relative to the total dephasing (O'Connor and Schaefer, 2002). Differences in centerband and sideband dephasing rates are indications of a preferred relative orientation between the chemical shift anisotropy (CSA) and dipolar tensors and the existence of a local molecular order (O'Connor and Schaefer, 2002). The principal values of the alanine carbonyl-carbon CSA-tensor used in the analysis were σ11 = 251.8 ppm, σ22 = 185.2 ppm, and σ33 = 92.8 ppm (Wei et al., 2001).
MODELING
K3 helices were generated using Insight II (Molecular Simulations, San Diego, CA). After the appropriate orientation of the helices with respect to each other suggested by the REDOR data, the dimer structure was energy-minimized using the Discover module of Insight II with interhelical distance restraints of r([1-13C]Ala10-[3-19F]Ala10) = 4.5 ± 0.3 Å and r([1-13C]Ala17-[3-19F]Ala14) = 10.4 ± 0.5 Å. The relative orientation of the carbonyl CSA tensor of Ala10 and the [1-13C]Ala10-[3-19F]Ala10 interhelical dipolar vector was held fixed at azimuthal and polar angles of α ± 2° and β ± 2° suggested by the REDOR sideband analysis.
Small unilamellar vesicle preparation
A 1:1 molar mixture of dipalmitoylphosphatidylcholine (DPPC) and dipalmitoylphosphatidylglycerol (DPPG) was dissolved in CHCl3/MeOH (2:1) to ensure thorough mixing. The solvent was removed under dry N2 at 37°C. The lipid film was dried overnight under vacuum and then resuspended by vortex mixing in buffer (10 mM TRIS, 154 mM NaCl, 0.1 mM EDTA, pH 7.4) at 65°C to give an approximate lipid concentration between 20 and 40 mM. The suspension was sonicated in an ice/water bath for 50 min using a Virsonic 100 (VirTis Company, Gardiner, NY) ultrasonic homogenizer. Debris and vesicle aggregates were removed by centrifugation at 14,000 × g for 10 min. The supernatant was pressed through a 0.45 μm microfilter. Gel filtration on a Sephacryl S1000 column confirmed the existence of a main population of vesicles with a mean diameter of 60 nm. For preparation of calcein-containing small unilamellar vesicles (SUVs), 70 mM calcein buffer solution was added to the dried lipid. Untrapped calcein was removed from the vesicles by gel filtration on a Sephadex G75 column (eluent: buffer containing 10 mM Tris, 154 mM NaCl, 0.1 mM EDTA, pH 7.4). The final lipid concentration was determined by phosphorous analysis modified after Bartlett (Kates, 1986).
Calcein release assay
Aliquots of the peptide solution were injected into a cuvette containing a stirred lipid suspension at room temperature to give a final volume of 3.0 mL. Calcein release from the SUVs was determined fluorometrically by measuring the decrease in self-quenching (excitation at 490 nm, emission at 520 nm) on a Spex Tau2 spectrofluorimeter with right-angle detection and 5 nm bandpass in the excitation and detection legs. The fluorescence intensity corresponding to 100% release was determined by addition of 200 μL 10% (v/v) Triton X-100.
RESULTS
Fast motion of the 19FCH2 group
The position of the fluorine of the 19FCH2 moiety of labeled K3 is averaged by fast rotation about the Cα-Cβ bond. This was confirmed by comparison of the magic-angle spinning 19F NMR spectra (not shown) of the 19FCH2-labeled K3 and a 19FCH2-C(=O)-labeled fragment of emerimicin (Holl et al., 1992). The spinning sideband intensities in the two spectra were identical. Fast rotation was established for the 19FCH2-C(=O) group of the emerimicin fragment by a two-dimensional 13C{19F} REDOR experiment (Goetz et al., 1998). REDOR was performed on the emerimicin fragment rather than on K3 because a larger sample guaranteed greater sensitivity. The spacing between the dephasing pulses was varied from 30 μs to 100 μs (half the rotor period), and the dephasing of the carbonyl-carbon signal observed. If the 19FCH2 is static, 100% dephasing is expected for a pulse spacing of 40 μs; the expected dephasing in the presence of fast rotation about the Cα-Cβ bond is 70% (O'Connor and Schaefer, 2002). The observed dephasing was 75%, consistent with fast rotation. Full dephasing for the emerimicin fragment was not observed until the pulse spacing was increased to 60 μs. The fast motion for the emerimicin fragment means that there must be fast motion for the CH2F group of K3 because of the equal intensities of the 19F spinning sidebands in the two systems. This result, therefore, justifies the average position for the K3 19F label on the methyl group C3 axis that was used in the REDOR dephasing calculations described in the Methods section.
Aggregation of K3 chains in the lipid bilayer
An 15N→13C transferred-echo double-resonance (TEDOR) experiment (Hing et al., 1992) on a mixture of labeled K3s (Fig. 1, left) results in a single peak at 177 ppm (Fig. 1, middle right). The TEDOR coherence transfer was optimized for a one-bond 13C-15N coupling so that this peak arises exclusively from the carbonyl carbon of Ala10. The appearance of a sizeable 15N → 13C{19F} TEDOR-REDOR difference signal for [1-13C]Ala10-(KIAGKIA)3-NH2 (Fig. 1, top right) unambiguously proves the proximity of peptide chains in the bilayer.
FIGURE 1.

(Left) Positions of the 13C, 15N, and 19F labels in the two K3 analogs ([1-13C]Ala10-[15N]Gly11-(KIAGKIA)3-NH2 and [2-2H, 3-19F]Ala10-(KIAGKIA)3-NH2) used in the REDOR experiments to detect aggregation of peptide chains. (Right) 50.3-MHz 15N → 13C{19F} TEDOR-REDOR NMR spectra of a 50-50 mixture KIA)3-NH2 and [2-2H, 3-19F]Ala10-(KIAGKIA)3-NH2 incorporated into synthetic MLVs of DPPG/DPPC (1:1) at a lipid/peptide molar ratio of 20, after 48 rotor cycles of dipolar evolution with magic-angle spinning at 5000 Hz. The 48-Tr full-echo spectrum (So) is shown at the bottom of the figure. The middle spectrum resulted from a 4-Tr 15N → 13C TEDOR coherence transfer followed by 48 additional rotor cycles with high power proton decoupling. The 48-Tr REDOR ΔS spectrum of the TEDOR-selected signal is shown at the top of the figure.
Once proximity has been established, a simpler method can be used for quantitation. Because K3 exists exclusively in an all α-helical conformation, the resonance frequency of the labeled carbonyl carbon of Ala10 is partially resolved from that of the lipid carbonyl carbon, and can be totally resolved by deconvolution. Therefore the determination of REDOR dephasing is possible without the preceding TEDOR selection. This simpler scheme has higher sensitivity because the inherent losses in the TEDOR coherence transfer are eliminated (Hing et al., 1992). Dephasing values obtained through deconvolution and natural-abundance correction were compared to those obtained through TEDOR-filtering for several different dipolar evolution times. The two methods were found to be in good agreement (0.165 vs. 0.154, 0.190 vs. 0.196, and 0.278 vs. 0.261 at dipolar evolution times of 9.6, 16, and 22.4 ms, respectively).
The aggregation experiments described above were carried out on lyophilized samples with 20% trehalose as a lyoprotectant included (Crowe and Crowe, 1984; Rudolph and Crowe, 1985). Fig. 2 shows a comparison of the results of 48 rotor cycle (9.6 ms) REDOR experiments on lyophilized and on frozen, fully hydrated samples at a lipid/peptide molar ratio of 20. The spectra are normalized with respect to the full-echo intensity of the peptide carbonyl peak at 177 ppm. Although the signal/noise ratio for the hydrated sample is much lower, the REDOR dephasing for the two samples is the same. Similar agreement was obtained after 32 and 80 rotor cycles of dipolar evolution (Fig. 3). These results indicate that freezing suppressed possible motional averaging of the 13C-19F dipolar coupling, and that K3 chain packing is similar in both sugar-protected lyophilized and frozen hydrated states.
FIGURE 2.

50.3-MHz 13C{19F} REDOR spectra of a 50-50 mixture of [1-13C]Ala10-[15N]Gly11-(KIAGKIA)3-NH2 and [2-2H, 3-19F]Ala10-(KIAGKIA)3-NH2 incorporated into synthetic MLVs of DPPG/DPPC (1:1) at a lipid/peptide molar ratio of 20 after 48 rotor cycles of dipolar evolution with magic-angle spinning at 5000 Hz. (Top) Frozen, fully hydrated sample. (Bottom) Lyophilized sample (in a protecting trehalose matrix). The full-echo spectra are normalized with respect to the carbonyl peak at 177.2 ppm. The experimental temperature was ∼−10°C for the lyophilized sample and −25°C for the hydrated sample. Both samples weighed ∼300 mg. The spectrum of the hydrated sample involved the accumulation of 64K scans in 3 days, and the spectrum of the lyophilized sample, 32K scans, in 1.5 days.
FIGURE 3.

50.3-MHz 13C{19F} REDOR dephasing (ΔS/So) for a 50-50 mixture of [1-13C]Ala10-[15N]Gly11-(KIAGKIA)3-NH2 and [2-2H, 3-19F]Ala10-(KIAGKIA)3-NH2, incorporated into synthetic MLVs of DPPG/DPPC (1:1) at a lipid/peptide molar ratio of 20 (○) and 40 (▴). Root mean-square deviation errors are indicated by the vertical lines for the first six evolution times (L/P = 20). Both samples were lyophilized in a protecting sugar matrix. The crosses show dephasing for the frozen fully hydrated (L/P = 20) sample of Fig. 2 (top). The solid lines show the calculated dephasing assuming two distributions of 13C-19F pair distances. At L/P = 20, the two distributions are about equally populated and the difference in average dephasing rates for the two populations is more than a factor of two. Based on signal/noise ratios, the root mean-square deviation uncertainty in the dephasing (ΔS/So) is estimated at 5% (see footnote to Table 1). One distribution has a mean internuclear 13C-19F separation of 4.5 Å with a width of 0.7 Å, and the other distribution has a mean of 9.6 Å and a width of 3.0 Å. At L/P = 40, the population with the smaller mean and narrower distribution width decreases.
Because the critical concentration to induce lysis for Xenopus laevis peptides appears to be a lipid/peptide molar ratio of ∼30 (Westerhoff et al., 1989; Matsuzaki et al., 1991; Cruciani et al., 1992; Grant et al., 1992), we analyzed REDOR dephasing at two different lipid/peptide molar ratios, L/P = 20 and L/P = 40. At L/P = 20, the total dephasing is ∼40% after 32 ms of dipolar evolution with no clear signs of having reached a maximum (Fig. 3). In any mixture of 13C- and 19F-labeled peptides, isolated 13C-13C and 19F-19F pairs are not detected by 13C{19F} REDOR. Thus, the observed dephasing shows that 80% or more of the peptide chains have aggregated. About half of the dephasing occurs in the first 10 ms, which suggests a strong coupling and a 13C-19F distance of <5 Å. This coupling appears to be associated with dimers because of its simple dependence on change in location of the 13C and 19F labels, as described in detail in the next subsection. In addition, the coupling gives rise to differences in the dephasing rates of the [1-13C]Ala10 spinning sidebands (also described below), consistent with a preferred orientation for the 13C-19F vector relative to the carbonyl carbon shift tensor. This sort of specificity would be lost in an aggregate larger than a dimer. Although the REDOR results do not support the presence of tightly packed large aggregates, the results are consistent with loosely packed aggregates of dimers and monomers. That is, the fast dephasing of Fig. 3 is attributed to the strong coupling within dimers, and the slower dephasing to the weaker coupling between monomers, or between dimers and monomers.
The total dephasing reached a slightly higher value when the MLVs (L/P = 20) were formed in the presence of 100 mM NH4OAc (data not shown), meaning that at high salt concentration, a somewhat larger fraction of 13C labeled peptides are near 19F. On the other hand, there was no fast-dephasing “bulge” associated with a narrow distribution of short 13C-19F distances. Instead, the ΔS/So values gave the best fit to a single distribution of isolated 13C-19F pairs with a mean internuclear separation of 7.6 Å and a broad distribution width of 4.2 Å. The chemical shift of the carbonyl carbon at Ala10 was 177 ppm, indicating an α-helical conformation at high as well as low salt concentration.
Parallel arrangement of K3 chains in the dimer
A 50-50 mixture of [3-13C]Ala3-[15N]Gly4-[1-13C]Ala10-[2-13C]Gly11-[6-15N]Lys12-[1-13C]Ala17-[15N]Gly18-(KIAGKIA)3-NH2 and [2-2H, 3-19F]Ala14-(KIAGKIA)3-NH2 (Fig. 4) was incorporated into MLVs of DPPC/DPPG (1:1) at a lipid/peptide molar ratio of 20. The carbonyl 13C label of Ala17 was unambiguously selected by a four-rotor cycle 15N→13C TEDOR transfer. This transfer was optimized for a one bond 13C-15N coupling so that the peak at 177 ppm (Fig. 5, bottom) arises only from Ala17. There is no contribution from the carbonyl label of Ala10 or from natural-abundance 13C in peptide or lipid carbonyl carbons. As illustrated in Fig. 4, for an antiparallel arrangement of the peptide chains, the [1-13C]Ala17-[3-19F]Ala14 internuclear separation would be too large (at least 16–17 Å) to be detected by 13C{19F} REDOR. The appearance of a sizeable difference signal (Fig. 5, top) is an immediate indication of a parallel (or approximately parallel) arrangement of the peptide chains. It is reasonable to assume that only peptide chains that form tight dimers (at L/P = 20 about half of the chains) give rise to detectable Ala14-Ala17 interchain 13C-19F contact. Based on this assumption, in a 50-50 mixture of 13C- and 19F-labeled peptides, the 10% dephasing of the TEDOR-So signal after 128 rotor cycles (Fig. 5, top) corresponds to an interchain [1-13C]Ala17-[3-19F]Ala14 separation of 10.4 Å. This distance is consistent with the packing of two parallel K3 helices with a 4.5 Å separation between the labels of Ala10 (cf. below). Tightly packed aggregates larger than dimers would not show the simple dependence of dephasing on label position that is inferred from the results of Figs. 3 and 5. If tightly packed larger aggregates are not present in the lyophilized MLVs, they certainly are not present in fully hydrated MLVs.
FIGURE 4.

Relative positions of the 13C and 15N labels (A17) and 19F label (A14) in the two (KIAGKIA)3-NH2 versions of K3 used to determine whether K3 chains form parallel or antiparallel dimers.
FIGURE 5.

50.3-MHz 15N → 13C{19F} TEDOR-REDOR NMR spectra of a 50-50 mixture of ([3-13C]Ala3-[15N]Gly4-[1-13C]Ala10-[2-13C]Gly11-[6-15N]Lys12-[1-13C]Ala17-[15N]Gly18-(KIAGKIA)3-NH2 and [2-2H, 3-19F]Ala14-(KIAGKIA)3-NH2), incorporated into synthetic MLVs of DPPG/DPPC (1:1) at a lipid/peptide molar ratio of 20 after 128 rotor cycles of dipolar evolution with magic-angle spinning at 5000 Hz. The spectrum at the bottom resulted from a 4-Tr, 15N→13C TEDOR coherence transfer followed by 128 additional rotor cycles with high power proton decoupling. The 128-Tr REDOR ΔS spectrum of the TEDOR-selected signal is shown at the top of the figure.
Preferred orientation between K3 chains
Fig. 6 presents the 13C{19F} REDOR full-echo (So) and difference (ΔS) spectra of a 50-50 mixture of [1-13C]Ala10-[15N]Gly11-(KIAGKIA)3-NH2 and [2-2H, 3-19F]Ala10-(KIAGKIA)3-NH2 (see Fig. 1) incorporated into MLVs of DPPG/DPPC (1:1) at a lipid/peptide molar ratio of 20, after a dipolar evolution time of 8 ms at a magic-angle spinning speed of 2000 Hz. At this concentration, approximately half the peptide chains form tight dimers with an interchain 13C-19F distance of 4.5 Å (see above). For evolution times of 8 ms or less, the major contribution (∼95%) of the dephasing arises from these dimers, where the existence of a preferential orientation between the peptide chains is most likely. Such a preference would be manifested in differential dephasing rates for spinning sidebands (O'Connor and Schaefer, 2002).
FIGURE 6.

50.3-MHz 13C{19F} REDOR spectra of a 50-50 mixture of [1-13C]Ala10-[15N]Gly11-(KIAGKIA)3-NH2 and [2-2H, 3-19F]Ala14-(KIAGKIA)3-NH2, incorporated into synthetic MLVs of DPPG/DPPC (1:1) at a lipid/peptide molar ratio of 20 after 8 ms of dipolar evolution at a magic-angle spinning speed of 2000 Hz.
Values of the experimental centerband and sideband dephasings (ΔS/So) are listed in Table 1 for two short evolution times. The dephasings relative to the total dephasing (in parentheses) are also given. Because the −2 and +2 sidebands were noisy in the difference spectra, only the centerband and the −1 and +1 sidebands were used in the analysis of the orientation of tightly packed K3 dimers. The observed differences in dephasing rates of 15–20% are outside experimental error. These differences are consistent with the presence of a preferred dimer orientation with α = 50° and β = 85° (Fig. 7), where α and β are the azimuthal and polar angles, respectively, of the 13C-19F dipolar vector in the CSA principal axis system of the carbonyl 13C label in Ala10 (Fig. 8). Due to the D2h symmetry of the relative CSA-dipolar orientations, orientations characterized by {α, β} = {50°, 85°}, {−α, β} = {−50°, 85°}, {180° − α, β} = {130°, 85°}, {180° + α, β} = {230°, 85°}, {α, 180° − β} = {50°, 95°}, {−α, 180° − β} = {−50°, 95°}, {180°−α, 180°−β} = {130°, 95°}, and {180° + α, 180° − β} = {230°, 95°} have the same error function and are equally favorable.
TABLE 1.
13C{19F} REDOR sideband dephasing* (ΔS/So) after 4 and 8 ms of dipolar evolution
| Sideband No.
|
||||||
|---|---|---|---|---|---|---|
| Evolution time[ms] | +2 | +1 | 0 | −1 | −2 | σ |
| 4 | 0.132 | 0.063 | 0.072 | 0.065 | 0.072 | |
| (0.93) | (1.05) | (0.96) | ||||
| Calculated | (0.92) | (1.06) | (1.02) | 0.03 | ||
| 8 | 0.079 | 0.082 | 0.102 | 0.089 | 0.123 | |
| (0.86) | (1.06) | (0.92) | ||||
| Calculated | (0.94) | (1.08) | (0.98) | 0.06 | ||
Values of ΔS/So scaled relative to the total dephasing are shown in parentheses for the centerband and first spinning sidebands. Calculated values together with root mean-square deviations (σ) for each dephasing time are shown in italics. Magic-angle spinning was at 2 kHz.
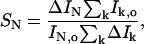 |
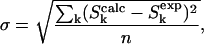 |
FIGURE 7.

Contour plots of the minimum root mean-square deviation error values for the 2000-Hz REDOR data of Table 1 as a function of the angles α and β, defined in Fig. 8. Each contour represents a 50% increase in error, with the darkest region representing the best fit.
FIGURE 8.

Definition of the 13C carbonyl CSA tensor used in the sideband analysis. The σ33 axis is perpendicular to the peptide plane, and the σ22 and σ11 axes are in the peptide plane. The σ22 axis is close to the C=O bond vector; the σ11 axis makes an angle of ∼40° with the C-N bond vector and is approximately perpendicular to the C=O bond; α and β are the azimuthal and polar angles, respectively, that define the orientation of the C-X dipolar vector in the principal axis system. In our application X = 19F, β is the angle between the most shielded element (σ33) and the dipolar vector, C-F, and α is the angle between the projection of the C-F bond on the σ11-σ22 plane and the σ11 axis. Adopted from Wei et al. (2001).
Structural model for the dimer
The orientational preferences revealed by the sideband analysis, and the two interchain 13C-19F distances from total REDOR dephasing, were used together as restraints on molecular modeling. The fluorinated helix was rotated about its 19F-labeled methyl-Cα bond, restrained by the orientation of the carbonyl CSA tensor of Ala10 and the 13C-19F interhelical dipolar vector suggested by the REDOR sideband data, until a [1-13C]Ala17-[3-19F]Ala14 separation of 10.4 ± 0.5 Å was achieved. The position of the 13C-labeled helix was held fixed during this process. Some of the helix-helix arrangements showed severe steric clashes and were discarded. Energy minimization of the remaining structures resulted in a dimer in which the helical axes were tilted at a 15–20° angle with respect to each other (Fig. 9). The interfacial area between the two helices appears to be made up largely of alanines and isoleucines, whereas the lysine side chains project in a number of directions on the two opposite sides of the dimer (Fig. 10). The positions of these side chains seem to offer a possibility of extensive contact between peptide chains in the bilayer and lipid headgroups, while at the same time minimizing the repulsive forces between each other and their contact with hydrophobic residues in the neighboring chain. Examination of the dimer reveals favorable interhelical van der Waals contacts between Ile6-Ala3, Ile6-Ala7, and Ile13-Ala14.
FIGURE 9.

Model of the dimerized K3 chains obtained from the combination of distance and angular information. After energy minimization, the [1-13C]Ala10-[3-19F]Ala10 and the [1-13C]Ala17-[3-19F]Ala14 interchain distances are 4.5 Å and 10.5 Å, respectively. The REDOR determined distances are 4.5 Å and 10.4 Å. The helices intersect at an approximate cross-angle of 20° (inset).
FIGURE 10.

(Top) Side groups of the K3 dimer. Only the labeled residues (Ala10 and Ala17 of one chain, and Ala10 and Ala14 of the other) as well as the lysine side chains are shown. The 13C and 19F labels are shown as black and green balls, respectively, and the amino nitrogens as blue balls. The approximate tilt angle of the two helices is 20°. (Bottom) Surface representation of the K3 dimer. The amino-terminus of the peptide and the terminal amino groups of the protruding lysine side chains are blue.
Static 19F NMR of K3 in oriented DMPC bilayers
The 19F NMR spectra of [3-19F, 2-2H]Ala10-(KIAGKIA)3-NH2 (the deuterium label was not used in this experiment) in fully hydrated liquid crystalline dimyristoylphosphatidylcholine (DMPC) bilayers aligned on glass plates are shown in Fig. 11 as a function of peptide concentration and orientation of the membrane normal relative to the static magnetic field. At low concentration (L/P = 200), the peptide is well oriented and undergoes fast rotation about the membrane normal, as a narrow peak is observed at both 0° and 90° sample orientations. The high mobility suggests that the peptide probably exists as a monomer (Salgado et al., 2001; Grage et al., 2002; Afonin et al., 2004). At high peptide concentration (L/P = 20), a relatively narrow signal is observed at 0° tilt, but with a distinctly different resonance frequency from that at L/P = 200. This result shows that the peptides are still well oriented but have changed their alignment with respect to the bilayer plane. Moreover, the signal at 90° tilt covers nearly the full width of the static CSA tensor of the crystalline amino acid (Fig. 11, bottom). Hence, the CSA of the 19F-label is no longer averaged by rotation of the peptide about the membrane normal, presumably because it is laterally associated in an oligomeric state in the membrane. The spectra at high peptide concentration reveal a small but distinct signal corresponding to a population of mobile peptide monomers, which can be estimated to be ∼5–10% of the total. The monomer-oligomer populations of K3 therefore do not exchange on the timescale of the experiment.
FIGURE 11.

470-MHz 19F NMR spectra of [3-19F, 2-2H]Ala10-(KIAGKIA)3-NH2 in hydrated, liquid crystalline DMPC bilayers aligned on glass plates, as a function of the lipid/peptide molar ratio and orientation of the membrane normal relative to the static magnetic field. The spectrum at the bottom is of a dry powder of crystalline L-[3-19F, 2-2H]alanine.
In an attempt to allow better comparison with the REDOR data of the sugar-protected lyophilized samples, spectra were acquired below the phase transition of DMPC (not shown). Whereas the signals remained narrow at 0° tilt and shifted slightly by only a few ppm, they disappeared at 90° tilt, presumably due to extensive broadening (increased 1H couplings), or unfavorable T2 relaxation in the gel state of the lipid bilayer. These spectral changes were completely reversible at the phase transition temperature of the lipids, when the sample was slowly heated back to 35°C. In another sample with an intermediate peptide concentration of L/P = 100, a temperature-dependent transition between the two signals was observed close to the chain-melting transition of DMPC (Tm = 23°C). Below 22°C and above 34°C, we only observed a signal at 217–221 ppm, as in the L/P = 200 sample. At temperatures between 24°C and 32°C, however, we also found a signal at 206–210 ppm. At 28°C, this signal, which corresponds to the peptide orientation in the L/P = 20 sample, had 70–80% of the total intensity. The 31P spectrum of the sample showed a well-ordered lamellar structure with approximately equal signals of fluid and gel-phase lipids.
Fluorescence leakage experiments on fluorinated and nonfluorinated lipid bilayers
To examine K3-induced membrane permeabilization, calcein (6-carboxyfluorescein) leakage from small unilamellar vesicles of DPPG and DPPC (1:1, molar) was monitored fluorometrically as the decrease in self-quenching at different lipid/peptide molar ratios. Leakage is given as the difference in the fluorescence intensity (ΔF) before (Fo) and after the treatment (F) with the peptide; the leakage percentages are normalized values with respect to leakage observed after treatment with 200 μL 10% (v/v) Triton X-100. The initial leakage rate (leakage after 1 min of peptide-lipid incubation) as a function of peptide concentration, at several different total lipid concentrations, is shown in Fig. 12. The solid lines are second-order polynomials that give the best fit in the observed leakage range of 0–80%. Measurements were made in the range of the same lipid/peptide molar ratios that were used in the NMR experiments. Similarly to other magainin-like peptides, leakage approximately starts at a lipid/peptide molar ratio of ∼100 (for example, 83 μM lipid and 0.5 μM peptide, Fig. 12, solid diamonds). This is in good agreement with the concentration range in which pore formation of K3 has previously been reported in acidic bilayers (Blazyk et al., 2001).
FIGURE 12.

Fractional release of calcein from SUVs of DPPG/DPPC (1:1) 1 min after the addition of (KIAGKIA)3-NH2 in the presence (open symbols) and in the absence (solid symbols) of 2.5 mol % F-lipid as a function of total (bound + free) peptide concentration. The lipid concentrations were 83 μM (♦), 166 μM (▴), 174 μM (▵), 332 μM (•), and 348 μM (○). The fluorinated lipid vesicles (open symbols) were indistinguishable from the nonfluorinated ones (solid symbols). Leakage is normalized with respect to that observed after treatment with 200 μL Triton X-100.
DISCUSSION
REDOR detection of aggregates
In our investigation of (KIAGKIA)3-NH2, REDOR provides a direct structural proof of the aggregation of peptide chains within the bilayer between lipid/peptide molar ratios of 40 and 20. This concentration range matches that where similar, magainin-like peptides were found to reorient themselves about the membrane normal (Ludtke et al., 1994), and where pore formation has been observed (Ludtke et al., 1996; Huang, 2000; Blazyk et al., 2001). The most tightly packed K3 chains appear to be dimers, based on the distance dependence of dipolar couplings between labels at Ala10, Ala14, and Ala17 (Fig. 9). At L/P = 20, REDOR detects a close interchain Ala10 (13C=O)-Ala10 (19FCH2) separation of 4.5 Å with a narrow distribution width (0.7 Å), indicating a highly specific interaction that appears to be present for about half of the peptide chains (Fig. 3). At L/P = 40, this short-distance population makes a much smaller contribution to dephasing. The concentration-dependent dimerization of K3 chains is consistent with the sigmoidal nature of binding isotherms of similar, magainin-like antimicrobial peptides to negatively charged lipid vesicles (Matsuzaki et al., 1994, 1998a,b,c). It is also in agreement with a recent solution-state NMR investigation of a magainin 2 analog in which long-range transferred nuclear Overhauser enhancement peaks suggested the dimerization of peptide chains in the presence of phosphatidylcholine vesicles (Wakamatsu et al., 2002).
In addition to the short interchain Ala10-Ala10 distance, a second population of peptide chains is found at both concentrations with a mean interchain Ala10 (13C=O)-Ala10 (19FCH2) separation of ∼10 Å. The longer distances are associated with substantially larger distribution widths (3.0 Å) than those associated with the 4.5 Å distance, suggesting nonspecific interactions for the longer distances.
Comparison of 13C{19F} REDOR dephasing at low and high ionic strengths indicates that high salt concentration loosened the contact between K3 chains. We suspect that the negative influence of salt on the dimerization of K3 chains is due to a close association of salt ions with the peptide, which prevents a deeper penetration into the hydrophobic region of the membrane, a possible requirement for the formation of tight K3 dimers. The negative effect of high salt concentration is consistent with the absence of large tightly packed K3 aggregates.
Orientation of K3 dimers
The analysis of REDOR sideband dephasing rates indicates the existence of a preferential orientation between the carbonyl CSA tensor of Ala10 and the interchain Ala10 (13C=O)-Ala10 (19FCH2) dipolar vector of the tightly packed dimers. Molecular modeling restrained by the combination of available distance and orientation information results in a dimer structure in which K3 chains are interacting at an interhelical angle of 15–20° (Fig. 9, inset). This is a frequently occurring packing mode between α-helices, which occurs, for example, in four-helix bundle structures. When ridges formed by residues separated by three (four) in the amino acid sequence fit into grooves formed by residues separated by four (three), a tilt angle of ∼20° results (Branden and Tooze, 1999). In the repetitive sequence of K3, each of the lysine, alanine, and isoleucine residues is systematically separated from the previous or next lysine, alanine, or isoleucine residue by either three or four amino acids. Glycines are separated from each other by seven residues (almost two complete helical turns). The favorable van der Waals interactions between Ile6-Ala3, Ile6-Ala7, and Ile13-Ala14 (Fig. 10) are in good agreement with recent findings that small hydrophobic as well as β-branched amino acids play major roles in the packing of helical membrane proteins (Javadpour et al., 1999; Eilers et al., 2000; Popot and Engelman, 2000).
A recent solution-state NMR study of a magainin 2-analog (F5Y, F16W magainin 2) has indicated intertwined α-helices, forming a short coiled-coil region (Wakamatsu et al., 2002), a structure not observed for K3 either in solution or in bilayers. The interface of the magainin dimer appeared to be composed of CβH2 of Lys4, Tyr5, Leu6, Ala9, Phe12, Gly13, and Trp16, suggesting favorable aromatic-aromatic interactions between the adjacent helices. Comparison of the sequence of F5Y, F16W magainin 2 (GIGKYLHSAKKFGKAWVGEIMNS) to that of K3 suggests that the positioning of lysine residues in K3 (Fig. 10) may play a role in preventing the peptide from forming a coiled-coil structure.
In our investigation of K3, a parallel (N–N, C–C) arrangement of the peptide chains was found within the tightly packed K3 dimers. Although the dipole moment of α-helices generally makes antiparallel helix dimers more stable than parallel dimers, there are several examples of parallel helix aggregates for antimicrobial peptides. For example, magainin 2 was found to form parallel heterodimers with PGLa, another antimicrobial peptide from the skin secretions of Xenopus (Soravia et al., 1988; Hara et al., 2001b). The heterodimer exhibited an order of magnitude higher membrane permeabilization activity than magainin 2 or PGLa monomers alone, suggesting dimer formation as an explanation for the widely observed synergism between magainin 2 and PGLa (Williams et al., 1990; Westerhoff, 1995; Vaz Gomez et al., 1993; Matsuzaki et al., 1998c).
K3 alignment by oriented-sample 19F NMR
Solid-state NMR experiments on macroscopically oriented membrane samples are conveniently used to determine the alignment of individual labeled molecular segments with respect to the bilayer normal. Changes in the peptide alignment or dynamics may thus be detected straightaway. In cases where the orientation of the CSA tensor is known within the molecular framework, it is even possible to deduce the structure of a larger peptide from a number of orientational constraints (Salgado et al., 2001; Afonin et al., 2004). In this case, however, the latter possibility cannot yet be pursued due to incomplete information on the 19F CSA tensor in 19FCH2-Ala.
Fig. 11 shows that the 19F chemical shift at high peptide concentration (−208 ppm at L/P = 20) differs significantly from that at low concentration (−217 ppm at L/P = 200). This result indicates that the time-averaged orientation of the 19F-labeled Ala10 segment changed with the lipid/peptide molar ratio. The most likely explanation for this concentration-dependent effect is a realignment of the entire K3 helix in the membrane, possibly as a result of pore formation. Alternatively, the difference in chemical shifts could be caused by a major change in the local Ala10 side-chain dynamics. However, our 19F magic angle spinning analysis confirmed that the 19FCH2-group undergoes fast C3 rotation at L/P = 20. In addition, a preliminary calculation shows that a putative isotropic order parameter of Smol = 1.0 for the oligomers would have to decrease below Smol = 0.2 for the monomers to account for the respective chemical shifts relative to the isotropic frequency at 219 ppm. This seems unlikely. Hence, we attribute the observed change to a realignment of virtually the whole peptide population in liquid crystalline membranes over the concentration range between L/P = 200 and 20. Small units of monomeric (or possibly dimeric) species are observed to rotate about the membrane normal at low peptide concentration, whereas at L/P = 20, virtually the whole population is converted into an oligomeric state. These observations support our interpretation that the highly specific K3 dimers reported by REDOR at L/P = 20 correspond to oligomeric species that are presumably arranged in a membrane pore.
At intermediate L/P = 100, the state of the peptide depends on temperature. The preferential formation of the oligomeric species around the phase transition of the lipid suggests that the presence of defects may stimulate a reorientation and immersion of the peptide. As magainin peptides have been found in proximity to lipid headgroups (Bechinger et al., 1993; Hirsh et al., 1996; Toke et al., 2004) at concentrations where pore-formation takes place (Ludtke et al., 1996; Huang, 2000), the extensive polar surface area on the two opposite sides of the K3 dimer (Fig. 10) probably plays a significant role in pore formation, and may be one of the reasons for the superior activity of the peptide relative to naturally occurring magainins.
Summary of results relevant to K3 mode of action
Fig. 1 of I shows the most important features of the three general models that have been suggested for the mode of action of antimicrobial peptides. Based on REDOR and other solid-state NMR results for K3 described in I and II, we conclude that i), the α-helical structure of K3 is consistent with all three models; ii), aggregation of K3 is consistent with all three models, but iii), a mix of monomers and dimers in the aggregates is not consistent with the barrel-stave model; iv), phospholipid headgroup contact with the middle of the K3 chain is not consistent with the barrel-stave model; v), a change in orientation of the K3 helix relative to the bilayer normal with increasing peptide concentration is not consistent with the carpet model; and vi), all of the results just cited are consistent with the toroidal-pore model.
Pore model
Based on all the information obtained from REDOR, as well as from nonspinning 19F and 31P NMR experiments, we envision a pore structure that is shown in Fig. 13. A similar toroidal pore model was first proposed by Huang and co-workers a few years ago for magainin 2 (Huang, 2000) and subsequently for melittin (Yang et al., 2001) and synthetic analogs of magainin 2 (Hallock et al., 2003).
FIGURE 13.

Cartoons of the pore model: cross section (top) and top view (bottom). (KIAGKIA)3-NH2 monomers and dimers are shown in yellow and red, respectively. The lysine side chains are indicated by blue. Peptide chains and lipid headgroups together line the wall of the torus-like pore. A few of the kinked lipid chains have 19F-labeled tails (in green) near the phospholipid headgroups.
The most important feature of the model is that layers of phospholipids bend continuously from one membrane leaflet to the other, forming a surface like the inside of a doughnut. Peptide chains that were lying on the membrane surface already submerged within the bilayer are pulled together with the lipid molecules, resulting in a pore in which peptide chains and lipid headgroups together line the wall. The K3 dimers in Fig. 13 are shown approximately parallel to the bilayer normal (away from the pore) but could be at a less severe angle. The lipid headgroups have an important role in screening the electrostatic repulsion between the highly positively charged peptide chains. Their incorporation into the pore allows substantially larger pore sizes (Ludtke et al., 1996) than a conventional barrel-stave pore model (see Fig. 1 in I). In the original toroid pore proposed by Huang, magainin chains were envisioned as monomers (Huang, 2000). In a more recent report, a pentameric toroid-type magainin pore was proposed that consisted of one dimer and three monomers. This model was based on the leakage activity of magainin analogs (Hara et al., 2001a).
The longer distances (10 Å) inferred from the slower dephasing component of Fig. 3 can be associated with spacings between peptide chains that are separated by a line of phospholipid molecules in the bilayer. (From now on we will refer to these spacings as “indirect” contacts.) We suspect that most of the indirect contacts occur between monomers since they are more flexible than dimers, although monomer-dimer neighbors may make a contribution to long-range 13C{19F} dephasing. However, in the case of two neighboring dimers, in which the Ala10 residues are all pointing toward the interior of the dimer they belong to, no detectable indirect 13C-19F dipolar coupling is expected.
The stoichiometry of the pore can be estimated from the ratio of the populations of peptide chains that are associated with the short versus long interchain separation (Fig. 3 legend). Based on the above discussion, indirect contacts are exclusively attributed to monomer-monomer and monomer-dimer neighbors separated by a line of lipid molecules. We consider a large number of dimers or monomers in the vicinity of each other but scattered outside the pores highly unlikely. Based on these assumptions, the L/P = 20 REDOR data (with about half the chains in tightly packed dimers) are consistent with a pore that consists of 2 dimers and 3–4 monomers. This stoichiometry would satisfy the ∼30–35 Å inner diameter of magainin-like pores detected by neutron diffraction in the same L/P concentration range (Ludtke et al., 1996; Huang, 2000; Yang et al., 2001), as well as with the prediction that there are 4–7 peptide chains present in the pore (Ludtke et al., 1996). We should note that in conductivity experiments, the structure of magainin-like pores appeared to be continuously variable (Duclohier et al., 1989), which may be an indication that there is more than one type of pore in the membrane. The coexistence of K3 dimers and monomers in membrane pores reported by REDOR is consistent with fluorescent studies (Hara et al., 2001a), in which magainin monomers and disulfide-dimerized magainin 2 analogs exhibited a synergistic effect in membrane permeabilization.
At lower peptide concentration (L/P = 40 rather than 20), the ratio of the peptide populations associated with the short versus long interchain distances is different, with a larger concentration of the population associated with the longer distances. This change suggests smaller pores for L/P = 40 resulting from more monomers than dimers within the pore. A reduction in pore size is consistent with the observed reduced leakage rate at L/P = 40 (Fig. 12) and is in agreement with fluorescent experiments on phospholipid vesicles containing antimicrobial peptides like melittin that indicated decreasing pore size with decreasing peptide concentration (Matsuzaki et al., 1997; Ladokhin et al., 1997).
Acknowledgments
This work was supported by National Institutes of Health grant EB02058 to J.S., and by a Deutsche Forschungsgemeinschaft grant (TPB6/SFB 604) to A.S.U.
References
- Afonin, S., U. H. N. Dürr, R. W. Glaser, and A. S. Ulrich. 2004. “Boomerang”-like insertion of a fusogenic peptide in a lipid membrane revealed by solid state 19F-NMR. Magn. Reson. Chem. 42:195–203. [DOI] [PubMed] [Google Scholar]
- Andreu, D., and L. Rivas. 1998. Animal antimicrobial peptides: an overview. Biopolymers. 47:415–433. [DOI] [PubMed] [Google Scholar]
- Bechinger, B., M. Zasloff, and S. J. Opella. 1993. Structure and orientation of the antibiotic peptide magainin in membranes by solid-state NMR spectroscopy. Protein Sci. 2:2077–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazyk, J., R. Wiegand, J. Klein, J. Hammer, R. M. Epand, R. F. Epand, W. L. Maloy, and U. P. Kari. 2001. A novel linear amphipathic β-sheet cationic antimicrobial peptide with enhanced selectivity for bacterial lipids. J. Biol. Chem. 276:27899–27906. [DOI] [PubMed] [Google Scholar]
- Branden, C.-I., and J. Tooze. 1999. Introduction into Protein Science. Garland Publishing, New York.
- Crowe, J. H., and L. M. Crowe. 1984. Preservation of membranes in anhyrobiotic organisms: the role of trehalose. Science. 223:701–704. [DOI] [PubMed] [Google Scholar]
- Cruciani, R. A., J. L. Barker, S. R. Durell, G. Raghunathan, H. R. Guy, M. Zasloff, and E. F. Stanley. 1992. Magainin 2, a natural antibiotic from frog skin, forms ion channels in lipid bilayer membranes. Eur. J. Pharmacol. 226:287–296. [DOI] [PubMed] [Google Scholar]
- Duclohier, H., G. Molle, and G. Spach. 1989. Antimicrobial peptide magainin 1 from Xenopus skin forms anion-permeable channels in planar lipid bilayers. Biophys. J. 56:1017–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers, M., S. C. Shekar, T. Shieh, S. O. Smith, and P. J. Fleming. 2000. Internal packing of helical membrane proteins. Proc. Natl. Acad. Sci. USA. 97:5796–5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epand, R. M., and H. J. Vogel. 1999. Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta. 1462:11–28. [DOI] [PubMed] [Google Scholar]
- Goetz, J. M., J. H. Wu, A. F. Yee, and J. Schaefer. 1998. Two-dimensional transferred-echo double resonance study of molecular motion in a fluorinated polycarbonate. Solid State Nucl. Magn. Reson. 12:87–95. [DOI] [PubMed] [Google Scholar]
- Grage, S. L., J. Wang, T. A. Cross, and A. S. Ulrich. 2002. Structure analysis of fluorine-labeled tryptophan side-chains in gramicidin A by solid state 19F-NMR. Biophys. J. 83:3336–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant, E., T. J. Beeler, K. M. P. Taylor, K. Gable, and M. A. Roseman. 1992. Mechanism of magainin 2a induced permeabilization of phospholipid vesicles. Biochemistry. 31:9912–9918. [DOI] [PubMed] [Google Scholar]
- Gullion, T., and J. Schaefer. 1989a. Rotational echo double-resonance NMR. J. Magn. Reson. 81:196–200. [DOI] [PubMed] [Google Scholar]
- Gullion, T., and J. Schaefer. 1989b. Detection of weak heteronuclear dipolar coupling by rotational-echo double resonance. Adv. Magn. Reson. 13:57–83. [Google Scholar]
- Hallock, K. J., D.-K. Lee, and A. Ramamoorthy. 2003. MSI-78, an analogue of the magainin antimicrobial peptides, disrupts lipid bilayer structure via positive curvature strain. Biophys. J. 84:3052–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock, R. E. W., and G. Diamond. 2000. The role of cationic antimicrobial peptides in innate host defenses. Trends Microbiol. 8:402–410. [DOI] [PubMed] [Google Scholar]
- Hara, T., H. Kodama, M. Kondo, K. Wakamatsu, A. Takeda, T. Tachi, and K. Matsuzaki. 2001a. Effects of peptide dimerization on pore formation: antiparallel disulfide-dimerized magainin 2 analogue. Biopolymers. 58:437–446. [DOI] [PubMed] [Google Scholar]
- Hara, T., Y. Mitani, K. Tanaka, N. Uematsu, A. Takakura, T. Tachi, H. Kodama, M. Kondo, H. Mori, A. Otaka, F. Nobutaka, and K. Matsuzaki. 2001b. Heterodimer formation between the antimicrobial peptides magainin 2 and PGLa in lipid bilayers: a cross-linking study. Biochemistry. 40:12395–12399. [DOI] [PubMed] [Google Scholar]
- Hing, A. W., S. Vega, and J. Schaefer. 1992. Transferred-echo double-resonance NMR. J. Magn. Reson. 96:205–209. [Google Scholar]
- Hirsh, D. J., J. Hammer, W. L. Maloy, J. Blazyk, and J. Schaefer. 1996. Secondary structure and location of a magainin analog in synthetic phospholipid bilayers. Biochemistry. 35:12733–12741. [DOI] [PubMed] [Google Scholar]
- Holl, S. M., G. R. Marshall, D. D. Beusen, K. Kociolek, A. S. Redlinski, M. T. Leplawy, R. A. McKay, S. Vega, and J. Schaefer. 1992. Determination of an 8-Å interatomic distance in a helical peptide by solid-state NMR spectroscopy. J. Am. Chem. Soc. 114:4830–4833. [Google Scholar]
- Huang, H. W. 2000. Action of antimicrobial peptides: two-state model. Biochemistry. 39:8347–8352. [DOI] [PubMed] [Google Scholar]
- Javadpour, M. M., M. Eilers, M. Groesbeek, and S. O. Smith. 1999. Helix packing in polytopic membrane proteins: role of glycine in transmembrane helix association. Biophys. J. 77:1609–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kates, M. 1986. Techniques of Lipidology: Isolation, analysis and identification of lipids, 2nd revised ed. In Laboratory Techniques in Biochemistry. R. H. Burdon and P. H. van Knippenberg, editors. Elsevier, Amsterdam and New York. 114–115.
- Ladokhin, A. S., M. E. Selsted, and S. H. White. 1997. Sizing membrane pores in lipid vesicles by leakage of co-encapsulated markers: pore formation by melittin. Biophys. J. 72:1762–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtke, S. J., K. He, W. T. Heller, T. A. Harroun, L. Yang, and H. W. Huang. 1996. Membrane pores induced by magainin. Biochemistry. 33:13723–13728. [DOI] [PubMed] [Google Scholar]
- Ludtke, S. J., K. He, Y. Wu, and H. W. Huang. 1994. Cooperative membrane insertion of magainin correlated with its cytolytic activity. Biochim. Biophys. Acta. 1190:181–184. [DOI] [PubMed] [Google Scholar]
- Maloy, L. M., and U. P. Kari. 1995. Structure-activity studies on magainins and other host defense peptides. Biopolymers. 37:105–122. [DOI] [PubMed] [Google Scholar]
- Matsuzaki, K., D. Chapman, and P. Haris. 1998a. Biomembrane Structures. IOS Press, Amsterdam. 205–227.
- Matsuzaki, K. 1998b. Magainins as paradigm for the mode of action of pore forming polypeptides. Biochim. Biophys. Acta. 1376:391–400. [DOI] [PubMed] [Google Scholar]
- Matsuzaki, K., M. Harada, S. Funakoshi, N. Fujii, and K. Miyajima. 1991. Physicochemical determinants for the interactions of magainins 1 and 2 with acidic lipid bilayers. Biochim. Biophys. Acta. 1063:162–170. [DOI] [PubMed] [Google Scholar]
- Matsuzaki, K., Y. Mitani, K. Akada, O. Murase, S. Yoneyama, M. Zasloff, and K. Miyajima. 1998c. Mechanism of synergism between antimicrobial peptides magainin 2 and PGLa. Biochemistry. 37:15144–15153. [DOI] [PubMed] [Google Scholar]
- Matsuzaki, K., O. Murase, and K. Miyajima. 1995. Kinetics of pore formation by an antimicrobial peptide, magainin 2, in phospholipid bilayers. Biochemistry. 34:12553–12559. [DOI] [PubMed] [Google Scholar]
- Matsuzaki, K., O. Murase, H. Tokuda, S. Funakoshi, N. Fujii, and K. Miyajima. 1994. Orientational and aggregational states of magainin 2 in phospholipid bilayers. Biochemistry. 33:3342–3349. [DOI] [PubMed] [Google Scholar]
- Matsuzaki, K., S. Yoneyama, and K. Miyijima. 1997. Pore formation and translocation of melittin. Biophys. J. 73:831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller, K. T. 1995. Analytic solutions for the time evolution of dipolar-dephasing NMR signal. J. Magn. Reson. Series A. 113:81–93. [Google Scholar]
- O'Connor, R. D., and J. Schaefer. 2002. Relative CSA-dipolar orientation from REDOR sidebands. J. Magn. Reson. 154:46–52. [DOI] [PubMed] [Google Scholar]
- O'Connor, R. D., B. Poliks, D. H. Bolton, J. M. Goetz, J. A. Byers, K. L. Wooley, and J. Schaefer. 2002. Chain-packing in linear phenol-polycarbonate by 13C{2H} REDOR. Macromolecules. 35:2608–2617. [Google Scholar]
- Popot, J.-L., and D. M. Engelman. 2000. Helical membrane protein folding, stability, and evolution. Annu. Rev. Biochem. 69:881–922. [DOI] [PubMed] [Google Scholar]
- Rudolph, A. S., and J. J. Crowe. 1985. Membrane stabilization during freezing: the role of two natural cryoprotectants, trehalose and proline. Cryobiology. 22:367–377. [DOI] [PubMed] [Google Scholar]
- Salgado, J., S. L. Grage, L. H. Kondojewski, R. S. Hodges, R. N. McElhany, and A. S. Ulrich. 2001. Membrane-bound structure and alignment of the antimicrobial β-sheet peptide gramicidin S derived from angular and distance constraints by solid state 19F NMR. J. Biomol. NMR. 21:191–208. [DOI] [PubMed] [Google Scholar]
- Soravia, E., G. Martini, and M. Zasloff. 1988. Antimicrobial properties of peptides from Xenopus granular gland secretions. FEBS Lett. 228:337–340. [DOI] [PubMed] [Google Scholar]
- Toke, O., W. L. Maloy, S. J. Kim, J. Blazyk, and J. Schaefer. 2004. Secondary structure and lipid contact of a peptide antibiotic in phospholipid bilayers by REDOR. Biophys. J. 87:662–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich, A. S., M. P. Heyn, and A. Watts. 1992. Structure determination of the cyclohexane ring of retinal in bacteriorhodopsin by solid state deuterium NMR. Biochemistry. 31:10390–10399. [DOI] [PubMed] [Google Scholar]
- Schumann, M., M. Dathe, T. Wieprecht, M. Beyermann, and M. Bienert. 1997. The tendency of magainin to associate upon binding to phospholipid bilayers. Biochemistry. 36:4345–4351. [DOI] [PubMed] [Google Scholar]
- Shai, Y. 1999. Mechanisms of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta. 1462:55–70. [DOI] [PubMed] [Google Scholar]
- van't Hof, W., E. C. Veerman, E. J. Helmerhorst, and A. V. N. Amerongenm. 2001. Antimicrobial peptides: properties and applicability. Biol. Chem. 382:597–619. [DOI] [PubMed] [Google Scholar]
- Vaz Gomes, A., A. de Waal, J. A. Berden, and H. V. Westerhoff. 1993. Electric potentiation, cooperativity, and synergism of magainin peptides in protein-free liposomes. Biochemistry. 32:5365–5372. [DOI] [PubMed] [Google Scholar]
- Wakamatsu, K., A. Takeda, T. Tachi, and K. Matsuzaki. 2002. Dimer structure of magainin 2 bound to phospholipid vesicles. Biopolymers. 64:314–327. [DOI] [PubMed] [Google Scholar]
- Wei, Y., D.-K. Lee, and A. Ramamoorthy. 2001. Solid-state 13C NMR chemical shift anisotropy tensors of polypeptides. J. Am. Chem. Soc. 123:6118–6126. [DOI] [PubMed] [Google Scholar]
- aWesterhoff, H. V., D. Juretic, R. W. Hendler, and M. Zasloff. 1989. Magainins and the disruption of membrane-linked free-energy transduction. Proc. Natl. Acad. Sci. USA. 86:6597–6601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerhoff, H. V., M. Zasloff, J. L. Rosner, R. W. Hendler, A. de Waal, A. Vaz Gomes, P. M. Jongsma, A. Riethorst, and D. Juretic. 1995. Functional synergism of the magainins PGLa and magainin-2 in Escherichia coli, tumor cells and liposomes. Eur. J. Biochem. 228:254–267. [PubMed] [Google Scholar]
- Williams, R. W., V. Starman, K. M. P. Taylor, K. Gable, T. Beeler, M. Zasloff, and D. Covell. 1990. Raman-spectroscopy of synthetic antimicrobial frog-peptides magainin-2a and PGLa. Biochemistry. 29:4490–4496. [DOI] [PubMed] [Google Scholar]
- Yang, L., T. A. Harroun, T. M. Weiss, L. Ding, and H. W. Huang. 2001. Barrel-stave model or toroidal model? A case study on melittin pores. Biophys. J. 81:1475–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasloff, M. 1987. Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. USA. 84:5449–5453. [DOI] [PMC free article] [PubMed] [Google Scholar]