

Abstract
Giant unilamellar vesicles (GUVs) have been widely used as a model membrane system to study membrane organization, dynamics, and protein-membrane interactions. Most recent studies have relied on imaging methods, which require good contrast for image resolution. Multiple sequential image processing only detects slow components of membrane dynamics. We have developed a new fluorescence correlation spectroscopy (FCS) technique, termed scanning FCS (i.e., SFCS), which performs multiple FCS measurements simultaneously by rapidly directing the excitation laser beam in a uniform (circular) scan across the bilayer of the GUVs in a repetitive fashion. The scan rate is fast compared to the diffusion of the membrane proteins and even small molecules in the GUVs. Scanning FCS outputs a “carpet” of timed fluorescence intensity fluctuations at specific points along the scan. In this study, GUVs were assembled from rat kidney brush border membranes, which included the integral membrane proteins. Scanning FCS measurements on GUVs allowed for a straightforward detection of spatial-temporal interactions between the protein and the membrane based on the diffusion rate of the protein. To test for protein incorporation into the bilayers of the GUVs, antibodies against one specific membrane protein (NaPi II cotransporter) were labeled with ALEXA-488. Fluorescence images of the GUVs in the presence of the labeled antibody showed marginal fluorescence enhancement on the GUV membrane bilayers (poor image contrast and resolution). With the application of scanning FCS, the binding of the antibody to the GUVs was detected directly from the analysis of diffusion rates of the fluorescent antibody. The diffusion coefficient of the antibody bound to NaPi II in the GUVs was ∼200-fold smaller than that in solution. Scanning FCS provided a simple, quantitative, yet highly sensitive method to study protein-membrane interactions.
INTRODUCTION
The plasma membrane serves as the interface between the interior of the cell and the extracellular fluid that bathes all cells. It protects the cell and serves as an information relay center. The membrane proteins anchored on the plasma membrane together with specific lipids are known to be involved in many important biological processes such as signal transduction, molecule transportation, intracellular communication, etc. (Arbuzova et al., 1998; Gonzalez-Gaitan, 2003; Murata et al., 1995; Russell, 2000). Consequently, molecule interactions on the membrane have always been important in understanding membrane structure, dynamics, and function. These interactions include protein-lipid, protein-membrane protein, or ligand-receptor, etc. A variety of artificial systems have been used to study these membrane interactions in vitro, such as small, large, and giant unilamellar vesicles (SUVs, LUVs, and GUVs) and multilamellar vesicles, as well as the planar bilayers (Mattjus et al., 1999; Sanchez et al., 2002; Slade et al., 2002; Wanaski et al., 2003). Among these systems, GUVs best represent the plasma membrane of mammalian cells, because their sizes are comparable to mammalian cells (average diameter of ∼30 μm) and the GUVs can be assembled from the same components as those of the plasma membrane (Bagatolli and Gratton, 1999). When GUVs are prepared with the electroformation method, their lipid and protein composition can be modulated to suit experimental needs. GUVs have been assembled from pure artificial lipid (Bagatolli and Gratton 1999), lipid mixtures (Bagatolli and Gratton 2000), raft mixtures (Dietrich et al., 2001), or even membrane extracts from cells. GUVs are convenient for studying lipid-lipid and lipid-protein interactions with optical microscopy techniques. Most current studies have relied on imaging methods, which require good contrast for image resolution. However, sequential multiple image processing can only detect slow events in protein-membrane interactions. We have developed a method to quantitatively study dynamics in membranes that exploit the advantages of the temporal FCS technique and spatial resolution of imaging simultaneously.
Fluorescence correlation spectroscopy was introduced by Webb and co-workers in 1972 (Magde et al., 1972). It has been widely used to study particle diffusion (Fahey et al., 1977; Koppel et al., 1976), chemical kinetics (Haupts et al., 1998; Starr and Thompson, 2001), and molecular aggregation (Palmer and Thompson, 1987; Qian and Elson, 1990) in solution. Coupled with advances in conjugate green fluorescence proteins, FCS has also been used in cellular systems (Dittrich et al., 2001; Nomura et al., 2001; Ruan et al., 2002). FCS has evolved into a powerful method to study molecule dynamics at the single molecule level (Medina and Schwille, 2002). In addition, based on the principles of FCS, scanning FCS has been developed to expand the application of FCS. Scanning FCS was first described in the literature some 30 years ago. In the 1970s, Weissman and co-workers used scanning FCS to determine the molecular weights of macromolecules by measuring spontaneous concentration fluctuations (Weissman et al., 1976). In the 1980s, Petersen and co-workers used scanning FCS to examine particle aggregation in samples in which diffusion or flow was slow (Petersen, 1986; Petersen et al., 1986). Subsequently, Koppel further demonstrated the power of scanning FCS by measuring the diffusion rates of fluorescently labeled DNA molecules in solution and colloidal gold-tagged lipids in a planar bilayer with a confocal laser microscope (Koppel et al., 1994). However, in these previous applications, the sample was homogeneous and all points in the scanning orbit were considered equivalent. Therefore, only the temporal correlation was calculated. In 1993, Petersen and co-workers further expanded scanning FCS with a scanning confocal microscope and developed a method called image correlation spectroscopy (ICS) (Petersen et al., 1993). With this method, the translational motion of transferrin receptors in the membrane within the image could be determined by the temporal correlation function (Srivastava and Petersen, 1998). In 2000, Wiseman and co-workers introduced two-photon image correlation spectroscopy. Using a video-rate-capable multiphoton microscope, they demonstrated a cellular application of two-photon ICS for measurements of slow diffusion of green fluorescent protein/adhesion receptor constructs within the basal membrane of live CHO fibroblast cells (Wiseman et al., 2000). By exploring the spatial correlation of the ICS measurements, the number of dendritic spines in brain tissue slices were counted (Wiseman et al., 2002, 2000). ICS has become a rapidly growing area within the FCS field. However, since it takes a considerable amount of time to acquire one image for the spatial analysis, it is only suited for the study of samples with slow diffusion rates (∼1 μm2/s) or immobile samples. By contrast, we are now proposing to recover both the spatial and temporal correlation intrinsic to the scanning FCS measurement for samples that diffuse relatively fast. In our system, the excitation laser beam is rapidly directed in a uniform (e.g., circular) scan across the sample in a repetitive fashion. Scanning FCS then outputs a “carpet” of timed fluorescence intensity fluctuations at specific points along the scan. Our scanning FCS measurements are well suited for measuring the diffusion rate of large molecules in solution or particles interacting with a lipid membrane.
In this study, we are focused on the detection of antibody-antigen interactions on the membranes of GUVs with scanning FCS. Scanning FCS shares the same principle as FCS, and therefore has the same instrumentation requirement. With a slight modification of the previously designed GUV growing chamber, GUVs can be grown by the electroformation method within the focus of a high numerical aperture objective. Since the reconstitution of cell membranes into GUVs is a novel approach in studying biological membrane systems, it is important to establish that the GUVs represent the actual membrane composition, which includes both membrane lipids and proteins. In previous studies with GUVs, immunostaining was utilized to determine whether integral membrane proteins were incorporated into GUVs assembled from membrane fractions. The immunostaining experiments involved fluorescently labeled primary and secondary antibodies against specific membrane proteins that remained intact in the GUVs made from brush border membranes of the renal proximal tubular cells. The resulting immunofluorescence images indicated antibody binding to the membrane through an increase in the intensity of the vesicle border upon the addition of the labeled antibody. However, these detection methods were image-based and qualitative, thus providing very little information regarding the dynamics of protein-membrane interactions. On the other hand, our scanning FCS method can quantitatively reflect the dynamics of molecules on biological membranes.
MATERIALS AND METHODS
Design of the chamber for growing GUVs for FCS measurements
Chamber for growing GUVs was slightly modified from previous designs (Bagatolli and Gratton, 1999) to adapt for FCS measurements. Fig. 1 shows the sketch of the preparation chamber. Two platinum wires were fit into the bottom of the chamber so that the GUVs tethered on the wire are within the working distance of a high numerical aperture microscope objective (1.2 NA, WD = 0.2 mm). An exterior water-bath was connected to the inlet and outlet on the side of the chamber, which kept the chamber at the desired temperature.
FIGURE 1.

Design of a GUV chamber for FCS measurements.
GUVs generation
GUVs were prepared by using a modified version of the electroformation method developed by Angelova and Dimitrov (1986). Brush border (BBM) membrane from the renal cortical tissue of adult Sprague-Dawley rats was isolated by differential centrifugation, magnesium precipitation, and discontinuous sucrose gradient method (Levi et al., 1993; Molitoris and Simon, 1985). The resulting pellet was resuspended in a buffer of 300 mM mannitol, 16 mM HEPES, pH 7.50 with TRIS buffer to achieve a protein concentration of 5–10 mg protein/ml. Maltase (BBM-bound enzyme) activity was measured in cortical homogenate and BBM fraction to determine enrichment or purity (specific activity in membrane fraction/specific activity in homogenate) of each membrane preparation and the BBM fraction was found to be at least 12-fold enriched or purified compared to the cortical homogenate. Before vesicle formation, the brush border membranes were resuspended in 10 mM TRIS. GUV formation took place in a thermostatic chamber (Fig. 1) through the hydration of the membrane fragments in the presence of an electric field. GUV formation was performed at 43°C for the integral membrane fragments so as to avoid denaturation of the membrane proteins, but at a temperature greater than the transition temperature of the membrane lipids.
Antibody labeling
The anti-Na-Pi type II cotransporter (NaPi-II) antibody was purified from the serum of an immunized rabbit through ammonium sulfate precipitation (Zajicek et al., 2001). The antibody was subsequently conjugated with the amine-reactive fluorescent probe Alexa using an Alexa Fluor 488 Protein Labeling Kit (Molecular Probes, Eugene, OR) and purified with an Affi-Gel Protein A column (Bio-Rad, Richmond, CA). Conjugates were labeled with an average of three dye molecules per antibody molecule.
Instrument and measurement setup
The two-photon excitation scanning fluorescence microscope used in these experiments was assembled in the Laboratory for Fluorescence Dynamics (LFD, University of Illinois, Urbana-Champaign, IL) and has been described in Ruan et al. (2002). A mode-locked titanium-sapphire laser with 80-MHz, 100-fs pulse width (Tsunami; Spectra-Physics, Mountain View, CA) was used as the excitation light source. The laser was guided into the microscope by x,y galvano-scanner mirrors (Model 6350; Cambridge Technology, Watertown, MA) to achieve beam scanning in both x- and y-directions. The scanner mirrors were moved by voltage generated in a computer card and the movement of the x-scanner mirror is independent from the y-scanner mirror. For the laser beam to move in a circular path, the x- and y-scanner mirrors were driven by two identical sinewaves with 90° phase shift. The radius and frequency of the circular scan was controlled by the amplitude and frequency of the sinewave. For a raster scan, the x- and y-scanner mirrors were driven by two sawtooth signals at different frequencies. A photomultiplier tube (Hamamatsu HC120-08, Hamamatsu City, Japan) was used for light detection in the photon-counting mode. A BG39 optical filter was placed before the photomultiplier for efficient suppression of infrared excitation light. A Zeiss 40× (1.2 NA) water immersion objective lens (Carl Zeiss, Thornwood, NY) was used for the measurement because of its exceptional long working distance (200 μm). The excitation wavelength used in the study was 785 nm, where the fluorophore (ALEXA488) was efficiently excited.
For two-photon excitation scanning fluorescence imaging, data were collected at the rate of 50 μs/pixel. Each frame had 256 × 256 pixels and each image was integrated from 10 frames. The scanning areas ranged from 100 μm × 100 μm to 5 μm × 5 μm.
For FCS measurements, regions of interest can be directly selected from the fluorescence image. The pixel sampling frequency used was 40 KHz, and each measurement lasted <5 min. The laser power at the sample was 10 mW. The average fluorescence intensity of the sample remained constant, indicating the fluorophore was not photobleached during the measurement. Due to the variation in the laser alignment from day to day, the waist (ω0) of the excitation beam was calibrated before each day's measurement. The calibration was achieved by measuring the autocorrelation curve of 10 nM fluorescein in 0.01 M NaOH, and fitted with a diffusion rate of 300 μm2/s. The typical values of ω0 were at the range of 0.3–0.35 μm.
For the scanning FCS measurement, the center of the circular scanning path was directly selected from the fluorescence image. The data acquisition frequency was set at 40 KHz, and the scanning frequency at 1 KHz. Therefore, 40 data points were collected in each scanning cycle.
Data analysis
For FCS measurement, the fluorescence intensity of the sample at the laser focus was collected as a function of time and saved as a long data string (F1, F2, F3, F4, F5…F11999998, F11999999, F12000000). The autocorrelation curves of the FCS measurements were calculated by applying the normalized autocorrelation function 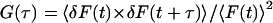 to the collected data set. The equation δF(t) = F(t)−〈F〉 expresses the fluctuation in fluorescence intensity at time t. The autocorrelation curves were then fit to the theoretical model using a Gaussian-Lorentzian beam profile (Berland et al., 1995) to recover the diffusion coefficient and the number of molecules in the excitation volume. The data were collected and calculated with SimFCS (software developed at the LFD) and analyzed with Globals (software developed in LFD). For scanning FCS measurements, all the data points were saved as a long data string in the same way as the FCS measurements, although they represent the fluorescence intensity of multiple sample regions as a function of time (F1,1, F1,2, F1,3, F1,4, F1,5,…F1,39, F1,40, F2,1, F2,2, F2,3, F2,4, F2,5,…F2,39, F2,40,…F5000000,1, F5000000,2,…F5000000,40). The first subscript number represents the scanning period, the second subscript number represents the sample position. For example, F2,39 represents the fluorescence intensity of sample position 39 measured during the second scanning period. For data analysis, it first undergoes a hyperspace transformation where the long string of data was segmented by each scanning circle. Data segments were then transposed to form a fluorescence intensity matrix as a function of time. Thus each vertical column contains information on the fluorescence intensity fluctuation as a function of time at a particular sample position. Autocorrelation curves can be calculated from each vertical column, each reflecting a specific position. The diffusion coefficient and G0 were recovered by fitting the autocorrelation curves with the theoretical model using a Gaussian-Lorentzian beam profile (Berland et al., 1995).
to the collected data set. The equation δF(t) = F(t)−〈F〉 expresses the fluctuation in fluorescence intensity at time t. The autocorrelation curves were then fit to the theoretical model using a Gaussian-Lorentzian beam profile (Berland et al., 1995) to recover the diffusion coefficient and the number of molecules in the excitation volume. The data were collected and calculated with SimFCS (software developed at the LFD) and analyzed with Globals (software developed in LFD). For scanning FCS measurements, all the data points were saved as a long data string in the same way as the FCS measurements, although they represent the fluorescence intensity of multiple sample regions as a function of time (F1,1, F1,2, F1,3, F1,4, F1,5,…F1,39, F1,40, F2,1, F2,2, F2,3, F2,4, F2,5,…F2,39, F2,40,…F5000000,1, F5000000,2,…F5000000,40). The first subscript number represents the scanning period, the second subscript number represents the sample position. For example, F2,39 represents the fluorescence intensity of sample position 39 measured during the second scanning period. For data analysis, it first undergoes a hyperspace transformation where the long string of data was segmented by each scanning circle. Data segments were then transposed to form a fluorescence intensity matrix as a function of time. Thus each vertical column contains information on the fluorescence intensity fluctuation as a function of time at a particular sample position. Autocorrelation curves can be calculated from each vertical column, each reflecting a specific position. The diffusion coefficient and G0 were recovered by fitting the autocorrelation curves with the theoretical model using a Gaussian-Lorentzian beam profile (Berland et al., 1995).
FCS simulation
To better understand the range limitation of scanning FCS, we used the Monte Carlo method to simulate the random diffusion of fluorescent particles in a closed box. For each simulation, the particles diffuse at a given rate (10, 15, 20, 30, 40 μm2/s). The fluorescence intensities at a fixed position inside the box were recorded and used to calculate the autocorrelation curves. These simulation conditions were close to the real FCS experimental conditions. For the scanning FCS simulation, the fluorescent intensities at multiple fixed positions inside the box were repeatedly recorded. The sampling and repetition rates were the same as the real scanning FCS experimental conditions. The data sets obtained were then analyzed in the same manner as described in the previous section.
RESULTS
FCS and scanning FCS measurements in solution
The GUVs prepared in the newly designed chambers (Fig. 1) were generated in the same manner as previously described; however, the position of the GUVs were close to the coverslip on the bottom of the chamber. This new geometry allowed for the use of a 40× water objective (1.2 NA) and facilitates FCS measurements on the GUVs.
As a control experiment, scanning FCS measurements were first carried out in solution. The results were compared with conventional FCS measurements carried out on the same sample. Fig. 2 shows the autocorrelation curve of the ALEXA-488 labeled antibody (against NaPi II cotransporter) in aqueous solution obtained from FCS measurements. The curve could be best fitted with a two-component model—one component corresponded to the labeled antibody, and the other to the free fluorophore. The graph in the lower panel shows the fitting residues. The presence of free fluorophore in the sample solution was a common technical difficulty due to the incomplete removal of the fluorophore after labeling. The diffusion coefficient of labeled antibody was 21.4 ± 1.3 μm2/s. On the other hand, the autocorrelation curve obtained from scanning FCS after hyperspace transformation and calculation (Fig. 3) could be fit with a one-component diffusion model. For a homogeneous sample, all the sample positions on the scanning path were equivalent and therefore yielded the same autocorrelation curve. The diffusion coefficient from scanning FCS was 20.6 ± 1.1 μm2/s. The reason that the fast component (free fluorophore) was not reflected in the autocorrelation curve from scanning FCS was because its fluorescence intensity fluctuation was in a faster time window than that measured by the particular condition of this scanning FCS experiment. From this control experiment it was clear that scanning FCS could be used to measure the diffusion rate of molecules with the same precision as conventional FCS measurements. In short, the data analysis method (hyperspace transformation) was equivalent to the conventional FCS data analysis method in providing all the temporal information.
FIGURE 2.

Autocorrelation curves of ALEXA-488 labeled antibody from conventional FCS measurement. The open circles were obtained directly from the measurement. The light line represents the fit with a one-component diffusion model. The darker line represents the fit with a two-component diffusion model. The diffusion rate of the antibody obtained from the two-component diffusion model is 21.4 μm2/s.
FIGURE 3.

Autocorrelation curves of ALEXA-488-labeled antibody from scanning FCS measurement. The open circles were obtained directly from the measurement. The curve represents the fit with a one-component diffusion model. The diffusion rate of the antibody was 20.6 μm2/s.
Scanning FCS on GUVs
GUVs were made from rat kidney brush border membranes. To test for membrane protein incorporation into the bilayers of the GUVs, antibodies against one specific membrane protein (NaPi II cotransporter) were labeled with ALEXA-488. Fig. 4 a shows the image of background fluorescence of GUVs, and Fig. 4 b shows the fluorescence image of a GUV after the addition of the ALEXA-488 labeled antibody. The images showed no discernable fluorescence enhancement on the GUV membrane bilayers border. Two possible explanations emerged: Either 1), the membrane protein did not incorporate into the GUVs, and consequently the fluorescently labeled antibody could not detect it on the GUV perimeter or 2), the poor image contrast and resolution limited detection. By performing scanning FCS across the GUV membrane (circle in Fig. 4 b), the binding of the antibody to the NaPi II cotransporter was reflected by its different diffusion rate on the GUVs as opposed to the bulk solution. This result confirmed the incorporation of membrane proteins into the bilayers of the GUVs when prepared by electroformation methods. Fig. 5 shows the hyperspace transformation of the raw scanning FCS data. The fluorescence intensity was displayed in pseudo color. The red section (region I) represents ALEXA-488 labeled antibodies in solution; the blue section (region II) corresponds to the interior of the vesicles; the membranes of the GUV appeared at the interfaces of the two sections. In each circular scan, the GUV membrane is bisected twice at two different locations. Fig. 6 shows the autocorrelation curves calculated line by line from Fig. 5. Lines 0–10 corresponded to the diffusion of the antibody in solution; lines 12–26 corresponded to the diffusion inside the GUV; lines 28–38 corresponded to the diffusion of the antibody in solution; and lines 11 and 27 corresponded to the diffusion of the antibody on the membrane of the GUV. The autocorrelation curves decayed slower on the membrane than in the solution. Fig. 7 shows examples of the fitting of autocorrelation curves at selected positions from Fig. 6. The diffusion coefficient of ALEXA-488 labeled antibody was 20 μm2/s in solution (line 2), in agreement with the control experiment in Fig. 2, whereas on the membrane of the GUV (line 11), the diffusion coefficient was reduced to 0.11 μm2/s. There are three components in the system (the free fluorophore, the fluorophore-labeled antibody in solution, and the labeled antibody bound to the membrane of the GUVs), which would yield diffusion coefficients at different timescales. However, because of the relatively slow circular scanning, the diffusion of the free fluorophore would not be reflected in the autocorrelation curve, although this component could be detected by measuring the scanning FCS and acquiring data at a high sampling rate. Therefore, the autocorrelation curve was fit with a two-component model, with the fast component (70%) corresponding to the labeled antibody in solution and the slow component to the labeled antibody bound to the membrane of the GUVs. Inside the GUV (line 20), no autocorrelation was observed because there were no fluorescent molecules inside the GUV. The diffusion coefficient of the antibody on the membrane of the GUVs was 200-fold smaller than that in solution, which indicated that the antibody was bound to NaPi II, thereby confirming that NaPi II cotransporter was incorporated into the GUVs. To eliminate the possibility of nonspecific binding of the antibody to the GUVs, GUVs made from pure lipids (POPC) without NaPi II cotransporter were used to study its interaction with the antibody as a control experiment. The diffusion coefficient of the antibody on the membrane of GUVs was similar to that in solution. No slow diffusion component was observed, indicating the absence of nonspecific antibody interactions with the GUVs (data not shown).
FIGURE 4.

Fluorescence images of GUVs before (a) and after (b) the addition of ALEXA-488-labeled antibody. The red circle represents the scanning orbit for scanning FCS.
FIGURE 5.

Hyperspace transformation of the raw scanning FCS data performed on GUVs shown in Fig. 4 b. Each row corresponds to one scanning circle and each column of the data matrix contains the fluorescence intensity for the same spatial location at different times. The red sections represent ALEXA-488 labeled antibodies in solution, the blue sections represent the interior of the vesicle, and the interface of the two sections show the membranes of the GUV.
FIGURE 6.

Autocorrelation curve calculated from Fig. 5. Lines 0–10 correspond to the diffusion of the antibody in solution; lines 12–26 correspond to the diffusion inside the GUV; lines 28–38 correspond to the diffusion of the antibody in solution; and lines 11 and 27 correspond to the diffusion of the antibody on the membrane of the GUV. It is clear the autocorrelation curves decay much more slowly on the membrane than in the solution.
FIGURE 7.

Examples of the fitting of autocorrelation curves at some positions from Fig. 6. The diffusion coefficient of the antibody in solution (a) was 20 μm2/s. On the GUV (b), the diffusion coefficient for the slow component was 0.11 μm2/s. There was no correlation inside the GUV (c).
The results of the simulated data were listed in Table 1. The first row represents the assigned diffusion coefficient of the particles in simulation. The second row represents the recovered diffusion coefficient from the autocorrelation curve in FCS simulation. The third row represents the recovered diffusion coefficient in the scanning FCS simulation, which could reflect the limitation for scanning FCS. With the FCS simulation, the recovered diffusion constant is within 95% accuracy of the simulated value. With the scanning FCS simulation, the recovered diffusion constant has the same accuracy as that of FCS simulation when the diffusion coefficient is no higher than 20 μm2/s. The accuracy of the recovered diffusion constant decreased to <70% when the simulated diffusion coefficient is 30 μm2/s. The accuracy degrades as the diffusion constant of the particle increase to a higher value.
TABLE 1.
FCS simulation
| Simulation value (μm2/s) | 10.0 | 15.0 | 20.0 | 30.0 | 40.0 |
| Recovered value (μm2/s) from FCS | 10.0 | 15.0 | 20.9 | 31.5 | 39.0 |
| Recovered value (μm2/s) from scanning FCS | 11.7 | 16.7 | 22.0 | 39.5 | 50.2 |
DISCUSSION
With the combination of currently available techniques (GUV electroformation, FCS, and scanning FCS) and some hardware and software modifications, we have developed a new method to quantitatively study protein-membrane interactions. Fluorescence recovery after photobleaching (FRAP) has been the conventional method to study molecular dynamics on the membrane surface; however, it was not suitable for the protein-membrane interactions when the Kd of binding was relatively high. The fluorescence signal from labeled protein in solution would overshadow any small fluorescence intensity recovery occurred on the membrane. Therefore, the only way to study the protein-membrane interactions would be based on the fluorescence image. In a previous study, fluorescence images of the GUVs in the presence of fluorophore-labeled primary antibody against NaPi II cotransporter were used to test the incorporation of membrane proteins in the membrane of the GUVs. However, due to the scarcity of the membrane protein on the membrane of GUVs, the fluorescence signal of the labeled primary antibody on the membrane of the GUV was weak. The image contrast was poor; there was no discernable fluorescence enhancement on the GUV membrane bilayer. To visualize the membrane protein, a fluorescently labeled secondary antibody was introduced into the system. The fluorescence signal on the membrane of the GUVs was enhanced because multiple secondary antibodies bound to one primary antibody. This image confirmed the presence of NaPi II cotransporter in the membrane of the GUVs. With the application of scanning FCS, the interaction of the primary antibody with the membrane protein was sufficient to detect the complex based on the diffusion coefficient of the antibody on the membrane of the GUVs without the addition of secondary antibody. The diffusion rate of the primary antibody (0.11 μm2/s) bound to the membrane was ∼200-fold slower than that in solution (20 μm2/s). In principle the relative concentration of the bound and free species could be determined from the G0 of the bound and free species, respectively. However, the labeling of the antibodies is not uniform. The labeling ratio is ∼3, but there is a distribution of labeled species. If we can assume that the distribution of labeled species is not affected by the binding equilibrium, then the ratio of the G0 of the two species should give us the ratio of the two concentrations. However, there is another important assumption in using this estimation, namely that the receptor protein is not clustered. To prove the existence of clustering will require intensity analysis photon counting histogram, which was not performed on our data.
Scanning FCS can be used not only to detect the interaction of the protein with the membrane, but also to investigate the nature of the interaction. When the protein interacts with a membrane protein on the membrane, two types of processes are expected. One is the protein bound to the membrane protein tightly undergoing a lateral diffusion on the membrane; the diffusion rate of this type of motion should be mainly affected by the dynamics of the membrane. It should be comparable to the diffusion of the lipids on the membrane, if not slower. The other type of process could be the on/off rate of the protein. If the interaction between the protein and the membrane was not strong enough to keep the protein bound to the membrane continuously, the protein might dissociate/associate from the membrane and the apparent diffusion rate obtained from the autocorrelation curve would actually reflect its on/off rate. Based on the actual diffusion coefficient obtained in the experiment, we believe the antibody has a strong binding with the NaPi II cotransporter, and together the complex underwent lateral diffusion. FRAP experiments on different systems showed that the diffusion coefficients for proteins in a cell membrane are 5–100 times lower than the values for proteins in an artificial bilayer (Saxton and Jacobson, 1997). The diffusion coefficient of the antibody on the membrane of the GUVs is comparable with the diffusion coefficient of adenylate kinase, another membrane protein measured on the cell membrane (Ruan et al., 2002).
Scanning FCS does not lose any dynamic information about the molecule of interest as long as the molecule's diffusion constant is slower than the orbit rate. Our simulation data showed that the recovered diffusion constant from scanning FCS is comparable to conventional FCS as long as the diffusion coefficient is below 20 μm2/s. For particles diffusing faster than 20 μm2/s, the apparent diffusion rate is biased (Saffarian and Elson, 2003) because the sampling rate is no longer compatible with the diffusion rate. To obtain the real diffusion rate, corrections need to be included in the calculation. This topic will be discussed in future manuscripts. The diffusion coefficient of EGFP in living cells is ∼15–20 μm2/s, thus scanning FCS is suitable for most of the cellular measurements. Another advantage of scanning FCS was that it would cause less photodamage to the sample, because the laser beam was not situated or “parked” at one position for an extended length of time.
Conventional FCS measurements on the membrane were difficult because of the small contrast between the solution and membrane when the Kd was relatively high. In addition, the slightest movement of the GUV could shift the point of measurement, resulting in the laser focus shifting to the inside or the outside of the GUVs, instead of on the membrane of the GUVs. With scanning FCS, based on the images generated by hyperspace transformation, any GUV movement can be compensated for with a shift of the hyperspace image.
From the view of emerging GUV applications, it is also a significant finding that the membrane proteins are incorporated into the GUVs when prepared with electroformation methods. The GUVs prepared with electroformation methods can better represent a cell membrane in terms of its composition and structure. It can be used to characterize membrane proteins in a well-controlled model membrane system.
In conclusion, scanning FCS provided a simple, quantitative, yet highly sensitive method to study particle-membrane interactions.
Acknowledgments
All the experiments reported in this study were performed at the Laboratory for Fluorescence Dynamics in the Department of Physics of the University of Illinois at Urbana-Champaign.
The Laboratory for Fluorescence Dynamics is funded by the National Institutes of Health (NIH RR03155) and University of Illinois at Urbana-Champaign.
References
- Angelova, M. I., and D. S. Dimitrov. 1986. Liposome electroformation. Faraday Discuss. Chem. Soc. 81:303–311. [Google Scholar]
- Arbuzova, A., D. Murray, and S. McLaughlin. 1998. MARCKS, membranes, and calmodulin: kinetics of their interaction. Biochim. Biophys. Acta. 1376:369–379. [DOI] [PubMed] [Google Scholar]
- Bagatolli, L. A., and E. Gratton. 1999. Two-photon fluorescence microscopy observation of shape changes at the phase transition in phospholipid giant unilamellar vesicles. Biophys. J. 77:2090–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagatolli, L. A., and E. Gratton. 2000. Two photon fluorescence microscopy of coexisting lipid domains in giant unilamellar vesicles of binary phospholipid mixtures. Biophys. J. 78:290–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berland, K. M., P. T. So, and E. Gratton. 1995. Two-photon fluorescence correlation spectroscopy: method and application to the intracellular environment. Biophys. J. 68:694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich, C., L. A. Bagatolli, Z. N. Volovyk, N. L. Thompson, M. Levi, K. Jacobson, and E. Gratton. 2001. Lipid rafts reconstituted in model membranes. Biophys. J. 80:1417–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittrich, P., F. Malvezzi-Campeggi, M. Jahnz, and P. Schwille. 2001. Accessing molecular dynamics in cells by fluorescence correlation spectroscopy. Biol. Chem. 382:491–494. [DOI] [PubMed] [Google Scholar]
- Fahey, P. F., D. E. Koppel, L. S. Barak, D. E. Wolf, E. L. Elson, and W. W. Webb. 1977. Lateral diffusion in planar lipid bilayers. Science. 195:305–306. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Gaitan, M. 2003. Signal dispersal and transduction through the endocytic pathway. Nat. Rev. Mol. Cell Biol. 4:213–224. [DOI] [PubMed] [Google Scholar]
- Haupts, U., S. Maiti, P. Schwille, and W. W. Webb. 1998. Dynamics of fluorescence fluctuations in green fluorescent protein observed by fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA. 95:13573–13578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppel, D. E., D. Axelrod, J. Schlessinger, E. L. Elson, and W. W. Webb. 1976. Dynamics of fluorescence marker concentration as a probe of mobility. Biophys. J. 16:1315–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppel, D. E., F. Morgan, A. E. Cowan, and J. H. Carson. 1994. Scanning concentration correlation spectroscopy using the confocal laser microscope. Biophys. J. 66:502–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi, M., P. V. Wilson, O. J. Cooper, and E. Gratton. 1993. Lipid phases in renal brush border membranes revealed by Laurdan fluorescence. Photochem. Photobiol. 57:420–425. [DOI] [PubMed] [Google Scholar]
- Magde, D., E. Elson, and W. W. Webb. 1972. Thermodynamics fluctuations in a reacting system: measurement by fluorescence correlation spectroscopy. Phys. Rev. Lett. 29:705–708. [Google Scholar]
- Mattjus, P., J. G. Molotkovsky, J. M. Smaby, and R. E. Brown. 1999. A fluorescence resonance energy transfer approach for monitoring protein-mediated glycolipid transfer between vesicle membranes. Anal. Biochem. 268:297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina, M. A., and P. Schwille. 2002. Fluorescence correlation spectroscopy for the detection and study of single molecules in biology. Bioessays. 24:758–764. [DOI] [PubMed] [Google Scholar]
- Molitoris, B. A., and F. R. Simon. 1985. Renal cortical brush-border and basolateral membranes: cholesterol and phospholipid composition and relative turnover. J. Membr. Biol. 83:207–215. [DOI] [PubMed] [Google Scholar]
- Murata, M., J. Peranen, R. Schreiner, F. Wieland, T. V. Kurzchalia, and K. Simons. 1995. VIP21/caveolin is a cholesterol-binding protein. Proc. Natl. Acad. Sci. USA. 92:10339–10343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura, Y., H. Tanaka, L. Poellinger, F. Higashino, and M. Kinjo. 2001. Monitoring of in vitro and in vivo translation of green fluorescent protein and its fusion proteins by fluorescence correlation spectroscopy. Cytometry. 44:1–6. [PubMed] [Google Scholar]
- Palmer, A. G., and N. L. Thompson. 1987. Molecular aggregation characterized by high order autocorrelation in fluorescence correlation spectroscopy. Biophys. J. 52:257–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, N. O. 1986. Scanning fluorescence correlation spectroscopy. I. Theory and simulation of aggregation measurements. Biophys. J. 49:809–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, N. O., P. L. Hoddelius, P. W. Wiseman, O. Seger, and K. E. Magnusson. 1993. Quantitation of membrane receptor distributions by image correlation spectroscopy: concept and application. Biophys. J. 65:1135–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, N. O., D. C. Johnson, and M. J. Schlesinger. 1986. Scanning fluorescence correlation spectroscopy. II. Application to virus glycoprotein aggregation. Biophys. J. 49:817–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian, H., and E. L. Elson. 1990. Distribution of molecular aggregation by analysis of fluctuation moments. Proc. Natl. Acad. Sci. USA. 87:5479–5483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan, Q., Y. Chen, E. Gratton, M. Glaser, and W. W. Mantulin. 2002. Cellular characterization of adenylate kinase and its isoform: two-photon excitation fluorescence imaging and fluorescence correlation spectroscopy. Biophys. J. 83:3177–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, J. M. 2000. Sodium-potassium-chloride cotransport. Physiol. Rev. 80:211–276. [DOI] [PubMed] [Google Scholar]
- Saffarian, S., and E. L. Elson. 2003. Statistical analysis of fluorescence correlation spectroscopy: the standard deviation and bias. Biophys. J. 84:2030–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez, S. A., L. A. Bagatolli, E. Gratton, and T. L. Hazlett. 2002. A two-photon view of an enzyme at work: Crotalus atrox venom PLA2 interaction with single-lipid and mixed-lipid giant unilamellar vesicles. Biophys. J. 82:2232–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton, M. J., and K. Jacobson. 1997. Single-particle tracking: applications to membrane dynamics. Annu. Rev. Biophys. Biomol. Struct. 26:373–399. [DOI] [PubMed] [Google Scholar]
- Slade, A., J. Luh, S. Ho, and C. M. Yip. 2002. Single molecule imaging of supported planar lipid bilayer–reconstituted human insulin receptors by in situ scanning probe microscopy. J. Struct. Biol. 137:283–291. [DOI] [PubMed] [Google Scholar]
- Srivastava, M., and N. O. Petersen. 1998. Diffusion of transferrin receptor clusters. Biophys. Chem. 75:201–211. [DOI] [PubMed] [Google Scholar]
- Starr, T. E., and N. L. Thompson. 2001. Total internal reflection with fluorescence correlation spectroscopy: combined surface reaction and solution diffusion. Biophys. J. 80:1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanaski, S. P., B. K. Ng, and M. Glaser. 2003. Caveolin scaffolding region and the membrane binding region of SRC form lateral membrane domains. Biochemistry. 42:42–56. [DOI] [PubMed] [Google Scholar]
- Weissman, M., H. Schindler, and G. Feher. 1976. Determination of molecular weights by fluctuation spectroscopy: application to DNA. Proc. Natl. Acad. Sci. USA. 73:2776–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman, P. W., F. Capani, J. A. Squier, and M. E. Martone. 2002. Counting dendritic spines in brain tissue slices by image correlation spectroscopy analysis. J. Microsc. 205:177–186. [DOI] [PubMed] [Google Scholar]
- Wiseman, P. W., J. A. Squier, M. H. Ellisman, and K. R. Wilson. 2000. Two-photon image correlation spectroscopy and image cross-correlation spectroscopy. J. Microsc. 200:14–25. [DOI] [PubMed] [Google Scholar]
- Zajicek, H. K., H. Wang, K. Puttaparthi, N. Halaihel, D. Markovich, J. Shayman, R. Beliveau, P. Wilson, T. Rogers, and M. Levi. 2001. Glycosphingolipids modulate renal phosphate transport in potassium deficiency. Kidney Int. 60:694–704. [DOI] [PubMed] [Google Scholar]