

Abstract
Chimeras consisting of the homologous skeletal dihydropyridine receptor (DHPR) β1a subunit and the heterologous cardiac/brain β2a subunit were used to determine which regions of β1a were responsible for the skeletal-type excitation-contraction (EC) coupling phenotype. Chimeras were transiently transfected in β1 knockout myotubes and then voltage-clamped with simultaneous measurement of confocal fluo-4 fluorescence. All chimeras expressed a similar density of DHPR charge movements, indicating that the membrane density of DHPR voltage sensors was not a confounding factor in these studies. The data indicates that a β1a-specific domain present in the carboxyl terminus, namely the D5 region comprising the last 47 residues (β1a 478–524), is essential for expression of skeletal-type EC coupling. Furthermore, the location of β1aD5 immediately downstream from conserved domain D4 is also critical. In contrast, chimeras in which β1aD5 was swapped by the D5 region of β2a expressed Ca2+ transients triggered by the Ca2+ current, or none at all. A hydrophobic heptad repeat is present in domain D5 of β1a (L478, V485, V492). To determine the role of this motif, residues in the heptad repeat were mutated to alanines. The triple mutant β1a(L478A/V485A/V492A) recovered weak skeletal-type EC coupling (ΔF/Fmax = 0.4 ± 0.1 vs. 2.7 ± 0.5 for wild-type β1a). However, a triple mutant with alanine substitutions at positions out of phase with the heptad repeat, β1a(S481A/L488A/S495A), was normal (ΔF/Fmax = 2.1 ± 0.4). In summary, the presence of the β1a-specific D5 domain, in its correct position after conserved domain D4, is essential for skeletal-type EC coupling. Furthermore, a heptad repeat in β1aD5 controls the EC coupling activity. The carboxyl terminal heptad repeat of β1a might be involved in protein-protein interactions with ryanodine receptor type 1 required for DHPR to ryanodine receptor type 1 signal transmission.
INTRODUCTION
In skeletal muscle, the (dihydropyridine receptor) DHPR is responsible for activation of ryanodine receptor type 1 (RyR1) without the intervention of the L-type Ca2+ current (Dirksen and Beam, 1999; Ahern et al., 2001a; Sheridan et al., 2003b). Ca2+ transients persist in low external Ca2+, in the presence of the L-type Ca2+ channel antagonist nifedipine, and when a nonconducting pore mutant, α1S E1014K, replaces wild-type α1S (Dirksen and Beam, 1999; Ahern et al., 2001a; Sheridan et al., 2003a,b). From an experimental perspective, the main characteristic of skeletal-type excitation-contraction (EC) coupling is a sigmoidal relationship between the amplitude of the Ca2+ transient and the magnitude of the depolarization, with the largest Ca2+ transients occurring at large positive potentials (Garcia et al., 1994; Ahern et al., 2001b). By contrast, Ca2+ transients triggered by the DHPR Ca2+ current have a bell-shaped relationship between the amplitude of the Ca2+ transient and the magnitude of the depolarization, with the largest Ca2+ transients occurring at voltages that activate the maximum Ca2+ current (Garcia et al., 1994; Sheridan et al., 2003b). The voltage dependence of skeletal-type EC coupling is a consequence of the close physical proximity of DHPR and RyR1 channels across the gap separating the transverse tubules and the junctional sarcoplasmic reticulum. At this location, groups of four DHPRs arranged in tetrads are lined up with the foot structure of RyR1. This anatomical feature is only present in skeletal muscle (Franzini-Armstrong and Protasi, 1997). Tetrads are thought to function as surrogate “gating particles”, which, after acquiring the correct conformation, open the RyR1 channel (Rios et al., 1993). However, the molecular basis of the DHPR to RyR1 triggering mechanism is unknown.
Insights into the EC coupling triggering mechanism have been provided by studies in which skeletal DHPR subunits are replaced by heterologous counterparts. A change from a voltage-dependent skeletal-type EC coupling to a Ca2+-dependent EC coupling was initially described in skeletal dysgenic (α1S-null) myotubes expressing α1C, the cardiac pore isoform, instead of α1S, the endogenous isoform (Tanabe et al., 1990). This observation was used to identify domains of the α1S pore subunit essential for skeletal-type EC coupling. The expression of chimeras of α1S and α1C demonstrated that a domain within the cytosolic loop linking repeats II and III of α1S, the 720–765 region, was essential for skeletal-type EC coupling (Nakai et al., 1998a). However, this domain turned out to be insufficient. Using a deletion strategy, Ahern et al. (2001a) showed that ∼20% of the skeletal-type Ca2+ transient persisted after elimination of residues 671–690 and 720–765 from the α1S II-III loop. Therefore, other regions of the DHPR must participate directly in opening RyR1. Questions then arise as to where else in the DHPR to look for skeletal EC coupling domains, and what strategies to use to identify them.
We have used cultured primary myotubes from knockout (KO) mice lacking the DHPR β1 gene, which encodes the β1a isoform expressed in skeletal muscle, to identify possible EC coupling determinants present in this subunit (Beurg et al., 1997, 1999a,b; Sheridan et al., 2003a,b). DHPR β-subunits are tightly bound to α1 pore subunits, and are inextricably involved in all aspects of Ca2+ channel gating (Birnbaumer et al., 1998). Most important among them is the observation that β-subunits tighten the coupling between charge movement and Ca2+ channel opening (Neely et al., 1993; Olcese et al., 1996). Hence, β-subunits might either directly participate in voltage-sensing, or may respond to structural changes initiated by the voltage sensor. Both possibilities are highly relevant to the mechanism of EC coupling. β1 KO myotubes are phenotypically null for DHPR Ca2+ current and EC coupling; however, the wild-type (WT) phenotype can be quantitatively recovered by transient expression of the missing β1a subunit (Beurg et al., 1997). Studies in β1 KO myotubes by Sheridan et al. (2003a) showed that serial truncation of the carboxyl terminus of DHPR β1a modifies EC coupling, transforming it from a process controlled by voltage to a much weaker coupling process controlled by the Ca2+ current. This observation is significant since before this work, only exchanges of α1C for α1S were known to produce Ca2+-dependent EC coupling in skeletal myotubes. Hence, manipulations of α1S and β1a subunits each independently produced similar outcomes. As a function of carboxyl terminus truncation length, there was an overall decrease in the amplitude of Ca2+ transients with evident changes in the shape of the Ca2+ fluorescence versus voltage relationship and the kinetics of the Ca2+ transient. For the most severe truncations, we also observed a dependence of Ca2+ transients on external Ca2+. These observations are consistent with the emergence of Ca2+-dependent EC coupling, whereby Ca2+ entering the cell via the DHPR induces sarcoplasmic reticulum Ca2+ release, presumably by Ca2+-dependent activation of RyR1 (Sheridan et al., 2003b).
Ca2+-dependent EC coupling was also promoted in skeletal myotubes by pairing up the heterologous β2a variant with the skeletal α1S pore subunit (Sheridan et al., 2003b). The variance noise characteristics of Ca2+ currents expressed by the heterologous α1S/β2a pair are indistinguishable from those expressed by the homologous α1S/β1a pair (Beurg et al., 1999a). However, β2a overrides critical EC coupling determinants present in α1S, producing a loss in voltage-dependent EC coupling (Sheridan et al., 2003b). The latter was inferred by the drastic reduction in maximum Ca2+ fluorescence at large positive potentials (ΔF/Fmax) in double dysgenic/β1 KO myotubes overexpressing both the pore mutant α1S(E1014K) and β2a (Sheridan et al., 2003b). Hence, critical interactions of the DHPR with RyR1 are controlled by conformational states in both α1S and β1a subunits. Sequence comparison between β1a and β2a reveals two conserved central regions amounting to more than half of the total peptide sequence (domains D2 and D4), a nonconserved linker between the two conserved domains (D3), a nonconserved amino terminus (D1), and a nonconserved carboxyl terminus (D5) (Perez-Reyes and Schneider, 1994). Thus, there are three β1a-specific domains, namely D1, D3, and D5, that could be required for skeletal-type EC coupling. In this study, we narrowed the EC coupling domain of β1a using chimeras of β1a and β2a, and determined that a chimera possessing only β1aD5 on a β2a D1–D4 background is sufficient to recapitulate skeletal-type EC coupling quantitatively. Furthermore, we provide evidence for the involvement of a heptad repeat in the β1aD5 domain. Functional studies suggest this heptad repeat may be an intricate component of the interaction between the DHPR complex and RyR1. Part of this work has been previously published in abstract form (Sheridan et al., 2004).
MATERIALS AND METHODS
Identification of genotypes
We used polymerase chain reaction (PCR) assays to screen for the WT and mutant alleles of the DHPR β1 gene in mice with targeted disruption of the CANCB1 gene (Gregg et al., 1996). Tail samples were digested with Proteinase K (Sigma, St. Louis, MO), and the DNA was then isolated following the Puregene animal tissue protocol (Gentra Systems, Minneapolis, MN). The PCR reactions for each sample were composed of 11.7 μL distilled water, 1 μL of each 20 μM primer, 3.2 μL of 1.25 mM dNTPs (Stratagene, Cedar Creek, TX), 2 μL 10× PCR buffer (Qiagen, Valencia, CA), 1.2 μL Taq polymerase (Qiagen), and 1 μL of the DNA sample (∼100 μg/mL). PCR primers 5′ gag aga cat gac aga ctc agc tcg gag a 3′ and 5′ aca ccc cct gcc agt ggt aag agc 3′ were used to amplify a 250 bp fragment of the WT β1 allele. PCR primers 5′ aca ccc cct gcc agt ggt aag agc 3′ and 5′ aca ata gca ggc atg ctg ggg atg 3′ were used to amplify a 197 bp fragment of the KO β1 allele. The following conditions apply for the PCR of both DHPR β1 alleles: 1), 2′ at 94°C; 2), 30 s at 94°C; 3), 45 s at 60°C; 4), 1′ at 72°C; 5), cycle through steps 2–4 for 30 times; and 6) 10′ at 72°C.
Primary cultures
Cultures of myotubes were prepared from hind limbs of E18 fetuses, as described previously (Beurg et al., 1997). Muscles dissected from the fetuses were treated with 0.125% (w/v) trypsin and 0.05% (w/v) pancreatin. After centrifugation, mononucleated cells were resuspended in plating medium containing 78% Dulbeccos's modified Eagle's medium with low glucose, 10% horse serum, 10% fetal bovine serum, and 2% chicken serum extract. Cells were plated on plastic culture dishes coated with gelatin at a density of ∼1 × 104 cells per dish. Cultures were grown at 37°C in 8% CO2 gas. After myoblast fusion (∼6 days), the medium was replaced with fetal bovine serum free medium, and CO2 was decreased to 5%.
cDNA transfection
cDNA transfection was performed during the myoblast fusion stage with the polyamine LT1 (Panvera, Madison, WI). Cells were exposed for 2–3 h to a transfection solution containing LT1 and cDNA at a 5:1 μg ratio. In addition to the cDNA of interest, cells were cotransfected with a plasmid encoding the T-cell protein CD8, which is used as a transfection marker. Transfected myotubes expressing CD8 were recognized by surface binding of polystyrene beads coated with a monoclonal antibody specific for an external CD8 epitope (Dynal ASA, Oslo, Norway). The efficiency of cotransfection of the marker and the cDNA of interest was ∼90%. Whole-cell analysis of Ca2+ currents and Ca2+ transients was performed 3–5 days after transfection.
cDNA constructs
cDNAs for mouse β1a (GenBank accession No. NM_031173), rat β2a (Genbank accession No. M80545), rat β3 (GenBank accession No. M88751), and rat β4 (GenBank accession No. L02315) were subcloned into the pCR-Blunt vector (Invitrogen, Carlsbad, CA), excised by digestion with AgeI and NotI, and cloned into the pSG5 vector in frame with the first 11 residues of the phage T7 gene 10 protein for antibody tagging. All constructs carry a T7 epitope tag at the amino terminus for determining relative levels of protein expression in transfected cells.
Chimeric cDNAs
Peptide sequence alignments have identified five critical regions in β-subunits: two central conserved regions (D2 and D4) flanked by three divergent regions that are heavily spliced (D1, D3, and D5) (Perez-Reyes and Schneider, 1994). For the construction of chimeras, the boundaries of domains D1–D5 of mouse β1a and rat β2a were defined on the basis of an alignment of the peptide sequences performed with DNASTAR software (DNASTAR, Madison, WI) using the Jotun-Hein method. Sequence similarities were 41.2%, 78.2%, 36.7%, 90.6%, and 21.7% for domains D1–D5, respectively. Residue coordinates were as follows: β1aD1, residues1–57; β1aD2, residues 58–198; β1aD3, residues 199–253; β1aD4, residues 254–477; β1aD5, residues 478–524; β2aD1, residues 1–16; β2aD2, residues 17–157; β2aD3, residues 158–205; β2aD4, residues 206–419; and β2aD5, residues 420–604. All cDNAs were made by two-step PCR techniques. Primers were designed to amplify the 5′ end of the chimera and to introduce an AgeI site at the 5′end of the PCR product. Separate primers were designed to amplify the 3′ end of the chimera and to introduce a NotI site at the 3′ end of the PCR product. The two sets of primers produced two PCR products with a 17–20 bp overlap of identical sequence. The two PCR products were electrophoresed on agarose gels, excised from the gel, and eluted using GenElute columns (SupelCo, Bellefonte, PA). The two PCR products were mixed in an equimolar ratio, denatured, allowed to reanneal, and used in a PCR reaction to amplify the full-length chimeric cDNA bracketed by AgeI and NotI sites. This cDNA was subcloned in a pCR 2.1 vector, cut with AgeI and NotI enzymes, and fused in frame to the first 11 amino acids of the phage T7 gene 10 protein in the pSG5 vector. Residue coordinates of chimeras are as follows: β2a(D1–D3)/β1a(D4, D5) has residues 1–207 of β2a fused to residues 254–524 of β1a. β1a(D1–D3)/β2a(D4, D5) has residues 1–253 of β1a fused to residues 208–604 of β2a. β2a(D1–D4)/β1aD5 has residues 1–419 of β2a fused to residues 478–524 of β1a. β1a(D1–D4)/β2aD5 has residues 1–477 of β1a fused to residues 430–604 of β2a. β1a(D1–D4)/β1aD5/β2aD5 has residues 1–524 of β1a fused to residues 430–604 of β2a. β2a(D1–D4)/β2aD5/β1aD5 has residues 1–604 of β2a fused to residues 478–524 of β1a. β1a(D1–D4)/β2aD5/β1aD5 has residues 1–477 of β1a fused to residues 430–604 of β2a fused to residues 478–524 of β1a. β2a(D1–D4)/β1aD5/β2aD5 has residues 1–419 of β2a fused to residues 478–524 of β1a fused to residues 430–604 of β2a.
Heptad repeat mutations
D5ALA has residues 1–524 of mouse β1a with substitutions L478A, V485A, and V492A. D5ALAc has substitutions S481A, L488A, and S495A. Constructs were made by two-step PCR techniques, and base changes for the alanine substitutions were introduced in a single primer. The PCR product was purified on QIAquick PCR purification columns (Qiagen) and diluted to 1:10. The amplified PCR product was subcloned into a pCR 2.1 vector, cut with MluI and SacII, and subcloned into a MluI/SacII digested pSG5-T7-β1a vector.
Whole-cell voltage clamp
Whole-cell recordings were performed with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). Effective series resistance was compensated up to the point of amplifier oscillation with the Axopatch circuit. All experiments were performed at room temperature. For Ca2+ currents and Ca2+ transients, the external solution was (in mM) 130 TEA methanesulfonate, 10 CaCl2, 1 MgCl2, 10−3 TTX, 10 HEPES titrated with TEA(OH) to pH 7.4. The pipette solution consisted of (in mM) 140 Cs aspartate, 5 MgCl2, 0.1 EGTA (when Ca2+ transients were recorded) or 5 EGTA (when only Ca2+ currents were recorded), and 10 MOPS titrated with CsOH to pH 7.2. Patch pipettes had a resistance of 1–3 MΩ when filled with the pipette solution. The limit of Ca2+ current detection was ∼20 pA/cell or ∼0.05 pA/pF for the smallest cells having the lowest capacitative noise. To obtain Ca2+ conductance curves, cells were maintained at a holding potential of −40 mV and depolarized in ascending order every 3 s. The pulse duration was 500 ms and was changed in 5 mV increments up to +85 mV. To obtain Ca2+ transient curves, cells were maintained at −40 mV and depolarized in descending order every 30 s. The pulse duration was 50 ms or 200 ms and was changed in 20 mV decrements from +90 mV to −30 mV. Between each depolarization, the cell was maintained at the resting potential for 30 s to permit recovery of the resting fluorescence. To obtain charge movement curves, we used a protocol with a long prepulse to inactivate Na+ channel ionic and gating currents (Ahern et al., 2001a,b; 2003). The pulse protocol was as follows: The command voltage was stepped from a holding potential of −80 mV to −30 mV for 698 ms, then to −50 mV for 5 ms, then to the test potential for 50 ms, then to −50 mV for 50 ms, and then to the −80 mV holding potential. Test potentials were applied in decreasing order every 10 mV from +100 or +110 mV to −80 mV. The intertest pulse period was 10 s. Online subtraction of the linear charge was done by a P/4 procedure. The P/4 pulses were delivered immediately before the pulse protocol from −80 mV in the negative direction. For charge movements, the internal solution was (in mM) 120 NMG (N-methyl glucamine)-glutamate, 10 HEPES-NMG, 10 EGTA-NMG pH 7.3 (Ahern et al., 2003). The external solution was supplemented with 0.5 mM CdCl2 and/or 0.5 mM LaCl3 to block the Ca2+ current, and 0.05 mM TTX to block residual Na+ current.
Confocal fluorescence microscopy
Confocal line scan measurements were performed at room temperature. Cells were loaded with 5 μM fluo-4 acetoxymethyl ester (Molecular Probes, Eugene, OR) for 60 min at room temperature. Cells were viewed with an inverted Olympus microscope with a 20× objective (N.A. = 0.4) and a Fluoview confocal attachment (Olympus, Melville, NY). A 488 nm spectrum line for fluo-4 excitation was provided by a 5 mW Argon laser attenuated to 6% with neutral density filters. Line scans consisted of 1,000 lines, each of 512 pixels were acquired at a speed of 2.05 milliseconds per line. The spatial dimension of the line scan was 30–60 microns, and covered the entire width of the myotube. Locations selected for line scans were devoid of nuclei and had a low resting fluorescence. Line scans were synchronized to start 100 ms before the onset of the depolarization. The time course of the space-averaged fluorescence intensity change and ΔF/F units were estimated as described elsewhere (Sheridan et al., 2003a,b). The peak-to-peak noise in the baseline fluorescence averaged ∼0.1 ΔF/F units. Since ΔF/F was spatially averaged, peak-to-peak noise varied with spatial inhomogeneities in fluo-4 fluorescence and cell size. To construct Ca2+ fluorescence versus voltage curves, we used the highest ΔF/F value attained between the onset and termination of the voltage pulse. Excessive photobleaching was avoided by limiting the number of line scans to 14 per cell (7 per each 200-ms and 50-ms curves). Image analyses were performed with NIH Image software (National Institutes of Health, Bethesda, MD).
Curve fitting
The voltage dependence of the Ca2+ conductance and fluorescence versus voltage curves with a sigmoidal shape were fitted with a Boltzmann distribution
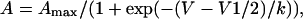 |
(1) |
where Amax is Gmax or ΔF/Fmax, V1/2 is the potential at which A = Amax/2, and k is the slope factor. For myotubes with a bell-shaped fluorescence versus voltage curve, the peak fluorescence was fit with a modified Boltzmann distribution
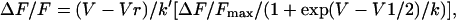 |
(2) |
where (V − Vr) is a factor that accounts for the decrease in Ca2+ current trigger at positive potentials and k′ is a scaling factor that varies with the magnitude of ΔF/Fmax (Sheridan et al., 2003a,b). Other parameters are the same as in Eq. 1. Parameters of a fit of averages of many cells (population average) are shown in the figures. Parameters of the fit of individual cells are shown in the tables. Analysis of variance (ANOVA) was performed with Analyze-it software (Analyze-it, Leeds, UK).
RESULTS
To identify which heterologous β-subunit would provide the most suitable background when chimerized with β1a, we performed a functional screen of variants from the four mammalian β-genes. Fig. 1 shows Ca2+ transients and Ca2+ currents at +30 mV, and Ca2+ conductance versus voltage curves expressed by four representative variants in β1 KO myotubes. In these experiments, the homologous mouse skeletal muscle β1a (Powers et al., 1992) served as a reference, and rat heart β2a (Perez-Reyes and Castellano, 1992), rat brain β3 (Castellano et al., 1993a), and rat brain β4 (Castellano et al., 1993b) were tested as possible chimeras. All variants, with the exception of β3, recovered a Ca2+ conductance density similar to that recovered by β1a. The β3 variant was unable to recover a normal charge movement density (1.2 ± 0.6 vs. 4.2 ± 0.7 fC/pF for β3 and β1a, respectively; see Table 1 for β1a and β2a), hence β3 was discarded. Only β2a, in addition to the homologous β1a, was capable of coupling depolarization to a cytosolic Ca2+ release. This was inferred from the partial recovery of Ca2+ transients observed after β2a overexpression in β1 KO myotubes. This result indicated that only β2a, among the heterologous variants, was capable of integrating into an EC coupling-competent skeletal DHPR. We have previously estimated that when β1a is replaced by β2a, voltage is ∼10-fold less effective as a trigger signal, and furthermore, the bulk of the Ca2+ transient induced by β2a is due to cytosolic Ca2+ increase triggered by the Ca2+ current (Sheridan et al., 2003b). Because there may be numerous reasons for the inability of β4 to recover EC coupling, we opted for the more conservative approach, which was to utilize β2a as the backbone for our chimeras.
FIGURE 1.

Recovery of Ca2+ current and Ca2+ transients by splice variants of the four β-genes expressed in β1 KO myotubes. Columns show representative β1 KO myotubes transfected with skeletal muscle β1a, cardiac/brain β2a, brain β3, and brain β4. GenBank identification numbers are indicated in Materials and Methods. Top trace corresponds to the spatial integral of the confocal Ca2+ transient in ΔF/F units in response to a 200 ms depolarization to +30 mV from a holding potential of −40 mV. The Ca2+ current during the 200-ms depolarization is shown expanded. Note change in amplitude scale for the time course of fluorescence. Bottom shows Ca2+ conductance versus voltage curves for population averages in response to a 500-ms depolarization in 5 mV increments from −35 to +60 mV. The shaded line in all figures shows the average Ca2+ conductance of nontransfected β1 KO myotubes. Curves were fit with Eq. 1 with the following parameters (Gmax in pS/pF, V1/2 in mV, and k in mV, respectively). For β1a: 194, 14.9, and 7.0; for β2a: 183, 11.6, and 5.2; for β3: 40, 40.2, and 28.1; and for β4: 171, 16.9, and 7.0.
TABLE 1.
Charge movements expressed by domain chimeras and heptad repeat mutants in β1 KO myotubes
|
Q-V
|
|||
|---|---|---|---|
| Qmax (fC/pF) | V1/2 (mV) | k (mV) | |
| WT β1a | 4.2 ± 0.7* (4) | 24.7 ± 5.2 | 20.1 ± 1.9 |
| WT β2a | 3.5 ± 0.6* (8) | 15.4 ± 7.8 | 19.0 ± 1.7 |
| β2a(D1–D3)/β1a(D4, D5) | 4.0 ± 0.7* (4) | 26 ± 6 | 16.6 ± 2.5 |
| β1a(D1–D3)/β2a(D4, D5) | 4.5 ± 0.4* (5) | 30 ± 2 | 19.7 ± 0.4 |
| β2a(D1–D4)/β1aD5 | 3.5 ± 0.1* (5) | 10 ± 11 | 16.8 ± 3.5 |
| β1a(D1–D4)/β2aD5 | 4.0 ± 0.1* (4) | 11 ± 6 | 17.9 ± 1.9 |
| D5ALA (L478A, V485A, V492A) | 3.8 ± 0.7* (4) | 15 ± 3 | 18.1 ± 2.5 |
| D5ALAc (S481A, L488A, S495A) | 3.9 ± 0.6* (4) | 13 ± 8 | 17.3 ± 2.6 |
| Nontransfected β1 KO | 1.8 ± 0.16† (5) | 16 ± 5 | 11.6 ± 3.1 |
Mean ± SEM of Boltzmann parameters fitted to each cell with number of cells in parenthesis. Qmax, V1/2, and k are parameters of the Boltzmann fit to each cell with Eq. 1.
Parameters in each column compared to nontransfected cells with one-way ANOVA significance p < 0.05.
Parameters in each column compared to WT β1a with one-way ANOVA significance p < 0.05. Data for WT β1a and WT β2a are from Ahern et al. (2003).
Chimeras are identified by their domain content (β1a or β2a domains D1–D5), listing first the domain content of the amino terminus followed by the domain content nearest to the carboxyl terminus. The boundaries of domains D1–D5 of both variants, and the amino acid coordinates of each chimera, appear in Materials and Methods. We tested two complementary chimeras with a C-terminal half belonging to β1a or β2a, namely β2a(D1–D3)/β1a(D4, D5) and β1a(D1–D3)/β2a(D4, D5), and two complementary chimeras with domain D5 belonging to β1a or β2a, namely β2a(D1–D4)/β1aD5 and β1a(D1–D4)/β2aD5. The interaction between the pore-forming subunit of the Ca2+ channel and the β-subunit promotes trafficking of the DHPR complex to the plasma membrane (Chien et al., 1995; Bichet et al., 2000). To estimate the density of DHPR voltage sensors expressed by each chimera, we used a charge movement protocol. The top panels of Fig. 2 show charge movements expressed by the four tested chimeras using a protocol that isolated the charge movements produced by the expressed DHPR from those generated by other voltage-gated channels present in myotubes (Ahern et al., 2001a,b). The graphs in Fig. 2 show charge versus voltage relationships for the four tested constructs obtained by integration of the OFF component, which usually was less contaminated by ionic current than the ON component. The voltage dependence of the mean charge was fit in each case to a Boltzmann equation (Eq. 1) indicated by the solid line. The experimental maximum charge detected at large positive potentials was within 10% of the fitted Qmax, indicating that the chosen range of test pulses was adequate to detect the bulk of the nonlinear charge movements. In all cases, the Qmax was significantly larger than the background Qmax of nontransfected cells, as confirmed by ANOVA (Table 1). High significance was attached to the fact that the mean Qmax of cells expressing the different chimeras were not significantly different from the mean Qmax expressed by WT β1a (Table 1). This indicated that the β1a or β2a content of the chimeras did not selectively affect the voltage-sensing properties of the DHPR, or the trafficking of the DHPR complex to the cell surface.
FIGURE 2.

Charge movements and Ca2+ conductance expressed by β1a-β2a chimeras in β1 KO myotubes. Chimeras were identified according to the source, β1a or β2a, of the five domains (D1, D2, D3, D4, and D5). Boundaries for each domain were obtained from a sequence lineup described in Materials and Methods. In the block diagram representation of the chimeras, shaded is β2a and black is β1a. The shaded line in all graphs shows the average charge movement or Ca2+ conductance of nontransfected β1 KO myotubes. Charge movement curves were fit with Eq. 1 with the following parameters (Qmax in fC/pF, V1/2 in mV, and k in mV, respectively). For β2a(D1–D3)/β1a(D4, D5): 4.0, 32.5, and 19.7; for β1a(D1–D3)/β2a(D4, D5): 4.7, 33.3, and 20.9; for β2a(D1–D4)/β1aD5: 3.5, −6.8, and 11.5; for β1a(D1–D4)/β2aD5: 3.9, 9.3, and 18.2; and for nontransfected β1 KO myotubes: 1.8, 5.2, and 17. Ca2+ conductance curves were fit with Eq. 1 with the following parameters (Gmax in pS/pF, V1/2 in mV, and k in mV, respectively). For β2a(D1–D3)/β1a(D4, D5): 204, 9.9, and 6.0; for β1a(D1–D3)/β2a(D4, D5): 63, 15.2, and 5.3; for β2a(D1–D4)/β1aD5: 181, 5.5, and 5.0; for β1a(D1–D4)/β2aD5: 42, 14.6, and 6.2; and for nontransfected β1 KO myotubes: 20, 20, and 14.
Ca2+ conductance versus voltage curves expressed by the chimeras are shown in the bottom panels of Fig. 2, and a statistical analysis of this data is shown in Table 2. We found that the chimeras with a carboxyl terminus belonging to β1a expressed a high Ca2+ conductance density similar to that expressed by WT β1a or WT β2a (see Table 2). In contrast, the reverse chimeras with a carboxyl terminus belonging to β2a, namely β1a(D1–D3)/β2a(D4, D5), and β1a(D1–D4)/β2aD5, expressed a Ca2+ conductance three- to fourfold lower than the chimeras that included β1aD5. Even though the behavior of the low-conductance chimeras was different from that of both parent variants, the results were consistent with a previous study of the structural determinants present in β-subunits for Ca2+ current expression in skeletal myotubes (Ahern et al., 2003). We have shown that WT β2a derives its ability to express high-density Ca2+ currents from a unique double cysteine motif present in domain D1. In contrast, WT β1a derives its ability to express high-density Ca2+ currents from domain D5, and deletion of this domain leads to a significant loss in Ca2+ current expression (Ahern et al., 2003; Sheridan et al., 2003a). Chimeras β1a(D1–D3)/β2a(D4, D5), and β1a(D1–D4)/β2aD5 have neither domains β2aD1 nor β1aD5, and thus, Ca2+ current expression by these chimeras is predicted to be minimal due to the absence of the required structural elements. Chimeras β2a(D1–D3)/β1a(D4, D5) and β2a(D1–D4)/β1aD5 have both domains required for Ca2+ current expression, hence Ca2+ current expression by these chimeras was predicted to be entirely normal.
TABLE 2.
Ca2+ conductance expressed by domain chimeras and heptad repeat mutants in β1 KO myotubes
|
G-V
|
|||
|---|---|---|---|
| Gmax (pS/pF) | V1/2 (mV) | k (mV) | |
| WT β1a | 195 ± 23 (10) | 15 ± 2 | 4.5 ± 0.6 |
| WT β2a | 184 ± 36 (6) | 11 ± 1 | 5.1 ± 0.3 |
| β2a(D1–D3)/β1a(D4, D5) | 223 ± 17 (19) | 12 ± 1 | 4.2 ± 0.4 |
| β1a(D1–D3)/β2a(D4, D5) | 63 ± 6* (20) | 16 ± 1 | 4.9 ± 0.3 |
| β2a(D1–D4)/β1aD5 | 158 ± 31 (9) | 7 ± 2 | 4.4 ± 0.4 |
| β1a(D1–D4)/β2aD5 | 48 ± 12* (7) | 21 ± 3 | 6.1 ± 0.8 |
| β2a(D1–D4)/β2aD5/β1aD5 | 193 ± 38 (8) | 17 ± 2 | 4.9 ± 0.6 |
| β2a(D1–D4)/β1aD5/β2aD5 | 174 ± 28 (7) | 11 ± 2 | 3.1 ± 0.4 |
| β1a(D1–D4)/β1aD5/β2aD5 | 180 ± 20 (7) | 18 ± 2 | 5.4 ± 0.6 |
| β1a(D1–D4)/β2aD5/β1aD5 | 71 ± 14 (6) | 20 ± 2 | 5.0 ± 0.3 |
| D5ALA (L478A, V485A, V492A) | 121 ± 18 (8) | 23 ± 4 | 4.4 ± 0.3 |
| D5ALAc (S481A, L488A, S495A) | 134 ± 16 (8) | 23 ± 2 | 5.0 ± 0.2 |
| Nontransfected β1 KO | 20 ± 5* (10) | 20.3 ± 7 | 14 ± 2.2* |
Mean ± SEM of Boltzmann parameters fitted to each cell with number of cells in parenthesis. Gmax, V1/2, and k are parameters of the Boltzmann fit to each cell with Eq. 1.
Parameters in each column compared to WT β1a with one-way ANOVA significance p < 0.05. Data for WT β1a and WT β2a are from Sheridan et al. (2003b).
Fig. 3 shows Ca2+ fluorescence versus voltage curves of the four chimeras investigated determined by two separate pulse protocols. Prior studies showed that Ca2+ transients can be readily evoked in cultured myotubes by a 50-ms depolarization, which is adequate for completion of charge movements in the DHPR (Ahern et al., 2001a,b). However, the L-type Ca2+ current expressed by the DHPR evolves much more slowly and is not fully activated at the end of a 50 ms pulse. To determine a possible contribution of the Ca2+ current to the EC coupling recovered by the chimeras, we compared Ca2+ transients activated by depolarizations lasting 50 ms (solid symbols) and 200 ms (open symbols). As shown in the top row of Fig. 3, the two chimeras possessing domain D5 of β1a produced Ca2+ transients that increased in a sigmoidal manner with voltage, with the amplitude peaking at potentials more positive than +30 mV in both pulse protocols. Both features, namely the absence of pulse duration dependence and persistence at large positive potentials, are hallmarks of skeletal-type EC coupling, and have been verified for the case of β1 KO myotubes expressing WT β1a (Table 3). In contrast, the chimera that included domains D4 and D5 of β2a shown in the bottom row of Fig. 3, namely β1a(D1–D3)/β2a(D4, D5), expressed Ca2+ transients with a drastically reduced ΔF/Fmax. Ca2+ transients of a small magnitude were detected at ∼+30 mV with the 200 ms depolarization, but not with the 50 ms stimulus. Furthermore, the largest Ca2+ transients coincided with the maximum Ca2+ current. The bell-shaped fluorescence curve observed with the 200 ms depolarization is indicative of Ca2+-dependent EC coupling previously described for WT β2a (Sheridan et al., 2003b). However, the reduced magnitude of the Ca2+ transients at all potentials prevented us from further testing this possibility. The chimera that included domain D5 of β2a on a background of domains D1–D4 of β1a, labeled β1a(D1–D4)/β2aD5, was entirely inactive. This result is consistent with the fact that β1a(D1–D4)/β2aD5 expressed minimal Ca2+ current, and thus, Ca2+-dependent EC coupling expressed by this chimera might be too small to be detected. In summary, the behavior of the chimeras demonstrated the critical involvement of the D5 region of β1a in skeletal-type EC coupling. Absence of β1aD5 leads to severe changes in the magnitude and voltage dependence of Ca2+ transients, or in the case of β1a(D1–D4)/β2aD5, to a complete loss of Ca2+ transients activated by depolarization. Moreover, other unique domains of β1a, namely D1 and D3, do not appear to be directly required for this signaling mechanism since they could be swapped by β2a(D1–D3) without impinging on the voltage-dependent characteristics of the fluorescence versus voltage curve. Fig. 4 shows that the EC coupling expressed by the two chimeras with domain D5 belonging to β1a, namely β2a(D1–D3)/β1a(D4, D5) and β2a(D1–D4)/β1aD5, persisted after the Ca2+ current was blocked by nifedipine. In these experiments, the pulse duration was 200 ms, and the same myotube was subjected to stimulation in control external solution, and in external solution supplemented with nifedipine. In all cases, perfusion of cells with external solution containing 2.5 μM nifedipine abolished the Ca2+ current to a level below detection (shaded traces), which was ∼20 pA/cell. In β1 KO myotubes overexpressing WT β1a, ∼40% of the Ca2+ transient was resistant to nifedipine, consistent with previous studies in normal cultured skeletal myotubes (Nakai et al., 1998a; Wilkens et al., 2001). The fluorescence versus voltage plots show that the Ca2+ transients in the β1aD5-containing chimeras were resistant to nifedipine by an equal or better margin. Nifedipine has been shown to negatively effect charge movement in the DHPR (Rios and Brum, 1987), and this could explain the partial decrease in Ca2+ transient amplitude seen in WT β1a and the two chimeras. The observations confirmed the presence of voltage-dependent skeletal-type EC coupling in the β2a(D1–D3)/β1a(D4, D5) and β2a(D1–D4)/β1aD5 chimeras.
FIGURE 3.

Ca2+ transients expressed by β1a-β2a chimeras in β1 KO myotubes. Open symbols correspond to peak ΔF/F obtained with a 200-ms depolarization and solid symbols were obtained with a 50-ms depolarization. Except for β1a(D1–D4)/β2aD5, the lines correspond to a Boltzmann fit of the mean (peak ΔF/F) for the population of cells. For β1a(D1–D4)/β2aD5, the lines are an interpolation of the mean at each potential. All fluorescence versus voltage curves were fit with Eq. 1, except those obtained with the 200 ms depolarization in the bottom row, which were fit with Eq. 2. Curves were fit with the following parameters (ΔF/Fmax in ΔF/F units, V1/2 in mV, and k in mV, respectively). For β2a(D1–D3)/β1a(D4, D5): 2.5, −5.5, 8.4 (50 ms), and 2.9, −7.1, 8.0 (200 ms); for β2a(D1–D4)/β1aD5: 2.3, 2.8, 5.8 (50 ms) and 2.5, −1.5, 5.5 (200 ms); and for β1a(D1–D3)/β2a(D4, D5): 0.2, −16.3, 13.4 (50 ms) and 0.9, 11.1, 1.9 (200 ms).
TABLE 3.
Ca2+ transients expressed by domain chimeras and heptad repeat mutants in β1 KO myotubes
| 50 ms F-V
|
200 ms F-V
|
|||||
|---|---|---|---|---|---|---|
| ΔF/Fmax | V1/2 (mV) | k (mV) | ΔF/Fmax | V1/2 (mV) | k (mV) | |
| WT β1a | 2.7 ± 0.5 (10) | −3 ± 2 | 7.7 ± 0.5 | 3.4 ± 0.6 (10) | −8 ± 2 | 5.8 ± 1.1 |
| WT β2a | 0.9 ± 0.2* (10) | −2 ± 3 | 7.4 ± 1.5 | 1.7 ± 0.4* (10) | 6 ± 3 | 7.6 ± 1.9 |
| β2a(D1–D3)/β1a(D4, D5) | 2.8 ± 0.6 (8) | −2 ± 3 | 7.8 ± 1.1 | 2.9 ± 0.4 (9) | −7 ± 3 | 5.9 ± 1.2 |
| β1a(D1–D3)/β2a(D4, D5) | 0.2 ± 0.1* (7) | 24 ± 11* | 23.0 ± 4.1* | 0.7 ± 0.1* (6) | 14 ± 2* | 6.5 ± 2.1 |
| β2a(D1–D4)/β1aD5 | 2.4 ± 0.5 (8) | 4 ± 3 | 8.2 ± 1.8 | 2.6 ± 0.6 (9) | −4 ± 2 | 4.8 ± 1.3 |
| β1a(D1–D4)/β2aD5 | − (5) | − | − | − (5) | − | − |
| β2a(D1–D4)/β2aD5/β1aD5 | 0.3 ± 0.1* (11) | 6 ± 4 | 15.2 ± 3.2 | 1.0 ± 0.3* (11) | 12 ± 3* | 7.6 ± 1.2 |
| β2a(D1–D4)/β1aD5/β2aD5 | 2.2 ± 0.4 (5) | 18 ± 5 | 9.6 ± 2.1 | 2.5 ± 0.8 (5) | 8 ± 6 | 7.2 ± 2.7 |
| β1a(D1–D4)/β1aD5/β2aD5 | 1.9 ± 0.5 (9) | 12 ± 3 | 12.0 ± 1.7 | 2.2 ± 0.2 (7) | 11 ± 4 | 10.2 ± 1.6 |
| β1a(D1–D4)/β2aD5/β1aD5 | 0.4 ± 0.1 (3/7) | 17 ± 11 | 19.2 ± 1.7 | 0.5 ± 0.1* (5) | 11 ± 4 | 9.4 ± 2.1 |
| D5ALA (L478A, V485A, V492A) | 0.4 ± 0.1* (8) | 12 ± 3 | 20.0 ± 4.3 | 0.7 ± 0.2* (8) | 15 ± 5 | 11.4 ± 2.2 |
| D5ALAc (S481A, L488A, S495A) | 2.1 ± 0.4 (7) | 14 ± 2 | 7.2 ± 1.7 | 2.6 ± 0.5 (7) | 16 ± 5* | 9.1 ± 3.4 |
Mean ± SEM of Boltzmann parameters fitted to each cell with number of cells in parentheses. Parameters of fluorescence versus voltage curves are shown for 50 ms and 200 ms depolarizations. ΔF/Fmax, V1/2, and k are parameters of the Boltzmann fit to each cell. All data was fit with Eq. 1 except fluorescence data in response to 200 ms in cells expressing β1a(D1–D3)/β2a(D4, D5), β2a(D1–D4)/β2aD5/β1aD5, and β1a(D1–D4)/β2aD5/β1aD5, which were fit with Eq. 2.
Parameters in each column compared to WT β1a with one-way ANOVA significance p < 0.05. Data for WT β1a and WT β2a are from Sheridan et al. (2003b).
FIGURE 4.

Nifedipine-insensitive Ca2+ transients expressed by β1a-β2a chimeras in β1 KO myotubes. Columns show representative β1 KO myotubes expressing full-length WT β1a, β2a(D1–D3)/β1a(D4, D5), and β2a(D1–D4)/β1aD5. Myotubes were depolarized for 200 ms from a holding potential of −40 mV to +30 mV in standard external solution containing 10 mM Ca2+. Ca2+ transients and Ca2+ currents were measured in the same myotube before and after (shaded traces) inhibition of the Ca2+ current by 2.5 μM nifedipine added to the external solution. Ca2+ currents during the 200 ms depolarization are shown expanded. Graphs show peak ΔF/F versus voltage relationships before and after (shaded symbols) nifedipine inhibition.
Since chimeras with domain D5 of β2a had weak Ca2+ current expression and lacked skeletal-type EC coupling function, we investigated whether β2aD5 had a dominant negative effect on the function of the subunit. This was achieved by fusing a tandem of β1aD5 and β2a D5 to the carboxyl terminus of domains D1–D4. Fig. 5 shows the Ca2+ conductance and EC coupling behavior of four chimeras designed on the basis of the order of β1aD5 and β2aD5 in the tandem and the source of domains D1–D4. A chimera with β2aD5 was fused to the carboxyl terminus of full-length β1a, labeled β1a(D1–D4)/β1aD5/β2aD5, expressed a normal Ca2+ conductance, and furthermore, the fluorescence versus voltage curve had a sigmoidal shape with a ΔF/Fmax only slightly lower than that expressed by WT β1a control and β2a(D1–D4)/β1aD5 (Table 3). In these experiments, we used the protocol described in Fig. 3 consisting of depolarizations for 50 ms (solid symbols) and 200 ms (open symbols). We found that for the β1a(D1–D4)/β1aD5/β2aD5 chimera, pulse duration had no impact in the shape of the fluorescence versus voltage relationship. Hence, fusion of the β2aD5 region to an otherwise full-length β1a subunit did not alter the skeletal-type EC coupling behavior of the β1a subunit. Conversely, domain D5 of β1a could have a dominant positive effect on β2a, which would lead to the expression of skeletal-type EC coupling. A chimera with β1aD5 was fused to the carboxyl terminus of full-length β2a, labeled β2a(D1–D4)/β2aD5/β1aD5, expressed a high-density Ca2+ conductance; however, skeletal-type EC coupling was not recovered. The bell-shaped fluorescence versus voltage curve expressed by β2a(D1–D4)/β2aD5/β1aD5 was consistent with Ca2+-dependent EC coupling, and was similar to that expressed by full-length β2a (Sheridan et al., 2003b). Hence, neither β1aD5 nor β2aD5 could significantly alter the function of an intact β-subunit when the heterologous D5 domain was fused downstream from the homologous D5 domain. These results ruled out a simple dominant negative effect of β2aD5, or a dominant positive effect of β1aD5. However, the position of β1aD5 next to D4 turned out to be an important factor for the EC coupling function of the subunit. This was inferred from the behavior of chimeras in which the order of β1aD5 and β2aD5 in the tandem was changed. The chimera labeled β2a(D1–D4)/β1aD5/β2aD5 consisted of a β2a(D1–D4) backbone with β1aD5 fused next to β2aD4 followed by the β2aD5 domain. Fig. 5 shows that this chimera expressed a normal Ca2+ current density, and furthermore, the fluorescence versus voltage characteristics were sigmoidal in shape irrespective of pulse duration. Thus, by moving β1aD5 from the carboxyl terminus of β2a to a location next to β2aD4, we observed a gain of skeletal EC coupling function. The chimera labeled β1a(D1–D4)/β2aD5/β1aD5 consisted of a β1a(D1–D4) backbone and the heterologous β2aD5 domain fused to the carboxyl terminus followed by the homologous β1aD5 domain. This chimera expressed minimal Ca2+ current and EC coupling. Thus, by simply moving β1aD5 away from its homologous location next to β1aD4, we observed a loss of skeletal EC coupling function. From these domain swapping approaches, we concluded that β1aD5 derives its ability to dictate skeletal-type EC coupling function from its position in the linear sequence immediately downstream from domains D1–D4. However, the composition of D1–D4, either from β1a or β2a, is not critical.
FIGURE 5.

Ca2+ conductance and Ca2+ transients expressed by β1a-β2a chimeras with domain D5 in different positions. Columns show the functional behavior chimeras with a C-terminus corresponding to a tandem of β1aD5 and β2aD5 domains expressed in β1 KO myotube. Ca2+ conductance curves were fit with Eq. 1 with the following parameters (Gmax in pS/pF, V1/2 in mV, and k in mV, respectively). For β2a(D1–D4)/β2aD5/β1aD5: 192, 16.7, and 5.5; for β2a(D1–D4)/β1aD5/β2aD5: 175, 11.9, and 4.4; for β1a(D1–D4)/β2aD5/β1aD5: 71, 19.4, and 5.3; and for β1a(D1–D4)/β1D5/β2aD5: 177, 18.5, and 6.8. For fluorescence curves, open symbols correspond to peak ΔF/F obtained with a 200-ms depolarization, and solid symbols were obtained with a 50-ms depolarization. The lines correspond to fit of the mean peak ΔF/F. All fluorescence versus voltage curves were fit with Eq. 1 except those obtained with the 200 ms depolarization in myotubes expressing β1a(D1–D4)/β2aD5/β1aD5 and β2a(D1–D4)/β2aD5/β1aD5, which were fit with Eq. 2. Curves were fit with the following parameters (ΔF/Fmax in ΔF/F units, V1/2 in mV, and k in mV, respectively). For β1a(D1–D4)/β1aD5/β2aD5:1.9, 7.0, and 9.4 (50 ms), and 2.0, 8.9, and 10.5 (200 ms). For β1a(D1–D4)/β2aD5/β1aD5: 0.4, 20.3, and 21.4 (50 ms), and 0.5, 11.1, and 7.8 (200 ms). For β2a(D1–D4)/β2aD5/β1aD5: 0.3, 4.9, and 12.5 (50ms), and 0.9, 16.9, and 9.5 (200 ms). For β2a(D1–D4)/β1aD5/β2aD5: 2.2, 16.3, and 9.9 (50 ms), and 2.3, 5.8, and 6.3 (200 ms).
We further determined specific amino acids in β1aD5 that contributed to skeletal-type EC coupling. A prior study based on serial truncation of the D5 region suggested that β1a residues 464–503 were critical (Sheridan et al., 2003a). This region contains a hydrophobic heptad repeat L478-V485-L492, which is a protein motif typically encountered in protein-protein interactions affecting gating (McCormack et al., 1991; Garcia et al., 1997) and RyR1 channel kinase/phosphatase interactions (Marx et al., 2001). Furthermore, this heptad repeat is not present in D5 domains of other β-subunit genes (Perez-Reyes and Schneider, 1994). The top sequences in Fig. 6 show the mutation scheme utilized to test the functional significance of the heptad repeat of β1aD5. We mutated the three positions in the heptad repeat to alanines (L478A/V487A/L492A), and as a control, introduced alanines at three positions out of step with the heptad repeat (S481A/L488A/S495A). The middle row shows Ca2+ conductance versus voltage curves for the triple heptad repeat mutant (D5ALA) and the control triple mutant (D5ALAc). The D5ALA and control mutations produced a slight reduction in Ca2+ conductance compared to WT β1a (shaded line). However, the difference was not statistically significant (see Table 2). Furthermore, charge movements expressed by D5ALA and the control triple mutant were not significantly different from the charge movements expressed by WT β1a (see Table 1). The bottom row of Fig. 6 shows that D5ALAc recovered nearly normal Ca2+ transient compared to WT β1a. The Boltzmann fit shown by the shaded line corresponds to the fit of WT β1a from the left panel of Fig. 6. Open symbols correspond to Ca2+ transients in response to a 200 ms depolarization, and solid symbols are in response to 50 ms. D5ALAc displayed a lack of pulse length dependence, typical of skeletal-type EC coupling. In contrast, D5ALA expressed Ca2+ transients with a drastically reduced voltage-sensitivity to both pulse protocols. In response to large positive potentials, the 200 ms pulse produces Ca2+ transients that were ∼6-fold smaller than those of WT β1a and ∼3-fold less than those of D5ALAc. From these results, we concluded that positions L478-V485-L492 in domain D5 play a critical role in skeletal-type EC coupling.
FIGURE 6.

Ca2+ conductance and Ca2+ transients expressed by heptad repeat mutants in β1 KO myotubes. β1a positions L478, V485, and V492 were mutated to alanine (β1a L478A/V485A/V492A). This triple mutation is labeled D5ALA. β1a positions S481, L488, and S495 were mutated to alanine (β1a S481A/L488A/S495A). This triple control mutation is labeled D5ALAc. Ca2+ conductance curves expressed by WT β1a, D5ALA, and D5ALAc were fit with Eq. 1 with the following parameters (Gmax in pS/pF, V1/2 in mV, and k in mV, respectively). For WT β1a (see Fig. 1); for D5ALA: 123, 24.9, and 5.3; for D5ALAc: 106, 23.0, and 10.7. The shaded line in the top right curve corresponds to WT β1a. In fluorescence curves, open symbols correspond to peak ΔF/F obtained with a 200-ms depolarization and solid symbols were obtained with a 50-ms depolarization. All fluorescence versus voltage curves were fit with Eq. 1 with the following parameters (ΔF/Fmax in ΔF/F units, V1/2 in mV, and k in mV, respectively). For WT β1a: 2.8, −6.1, and 7.8 (50 ms), and 3.2, −10.2, and 5.7 (200 ms). For D5ALA: 0.4, 14.6, and 14.4 (50 ms), and 0.7, 18.1, and 12.1 (200 ms). For D5ALAc: 2.1, 13.7, and 7.9 (50 ms), and 2.4, 9.1, and 4.3 (200 ms). The shaded lines in bottom right curve correspond to WT β1a used as a reference. Conductance versus voltage curve for WT β1a shown in this figure is the same as in the left panel of Fig. 1. Ca2+ transient versus voltage curves from WT β1a are from Sheridan et al. (2003b).
DISCUSSION
The main conclusions of this work are that i), carboxyl terminal amino acids 478–524 of β1a comprising the last tenth of the sequence, herein identified as domain β1aD5, are essential for skeletal-type EC coupling; ii), β1aD5 is functionally active when next to conserved domain D4 (amino acids 254–477 of β1a); and iii), the heptad repeat motif L478-V485-L492 present in β1aD5 is a critical determinant of the EC coupling function. Domain D5 of β1a is present exclusively in β1a and β1c, and only these two variants, among variants tested from the four mammalian β-genes (Fig. 1), are capable of restoring skeletal-type EC coupling (see Beurg et al., 1999b, for β1c, and Sheridan et al., 2003b, for β1a). A third splice variant of the β1-gene, namely β1b, is entirely devoid of EC coupling function (Cheng et al., 2004), and D5 domain of β1b bears no sequence homology with β1aD5 (Powers et al., 1992). The data also excluded participation of β1aD1 and β1aD3, which are two additional divergent regions present in β1a. In other β-variants, domains D1 and D3 modulate the kinetics of activation and inactivation of the Ca2+ current. However, until now no function had been attributed to divergent domain D5 in any variant (Qin et al., 1996; Restituto et al., 2000; Ahern et al., 2003). The identification of β1aD5 underscores the fact that β-subunits, far from ancillary, have critical tissue-specific functions that can only be identified using homologous cell expression systems.
Our results are consistent with proposed mechanisms of EC coupling and models of DHPR-RyR1 domain organization. Mechanisms of EC coupling have been influenced by a mechanical coupling model introduced earlier by Chandler et al. (1976) and later refined as allosteric coupling (Rios et al., 1993). In this model, the outward movement of the voltage sensors during depolarization is coupled to Ca2+ release from the sarcoplasmic reticulum by mechanical torque exerted by the voltage sensors on the foot structure of RyR1. The actions attributed to the α1S II-III loop borrow extensively from this idea (Garcia et al., 1994; Wilkens et al., 2001). However, observations accumulated in recent years suggest that models based on a single DHPR domain such as the α1S II-III loop interacting with RyR1, are no longer tenable. First, the proposed signaling role of the α1S II-III loop has been seriously questioned (Ahern et al., 2001a). Second, the biochemical evidence indicates that multiple domains of α1S interact with RyR1 (El Hayek et al., 1995; Leong and MacLennan, 1998; Sencer et al., 2001; Proenza et al., 2002). Finally, a physical contact between β1a and RyR1 is almost certain, based on three-dimensional reconstructions of skeletal DHPR particles (Sherysheva et al., 2002; Wolf et al., 2003), and the observation that recombinant β1a specifically binds to the 3490–3523 region of RyR1 (Cheng et al., 2004). A β-binding site at this location is consistent with the position of the β-subunit on the foot structure of RyR1 according to the Wolf-Grigorieff model (Wolf et al., 2003). The putative β1a binding region is just upstream of the CaM binding domain (Yamaguchi et al., 2001; Zhang et al., 2003), and half the way between the clamp region and transmembrane pore domains (Samso and Wagenknecht, 2002). Binding of β1a at this location in RyR1 could assist DHPR tetrad formation, could strengthen the docking of the DHPR to RyR1, or could serve to funnel conformational changes initiated in the voltage sensors into the pore-forming region of RyR1. None of these possibilities are mutually exclusive. However, a specific proposal on the hierarchy of the signals generated by different domains of α1S or β1a cannot be specified at this time. Signals generated in separate DHPR domains or subunits could converge onto a single RyR1 domain, or separate DHPR domains could activate separate RyR1 domains. In summary, the biochemical and structural data, taken together with the data in this report, suggest that multiple DHPR domains might be physically docked to RyR1, and might be responsible for transmission of the trigger signal from the DHPR voltage sensor to RyR1.
Our data indicate that the location of β1aD5 next to D4 a critical determinant of the skeletal EC coupling phenotype. We have attempted to explain this result based on a proposed model of the domain organization of β-subunits. β-subunits belong to the MAGUK superfamily composed of a tandem of PDZ, SH3, and GK domains (Anderson, 1996; Craven and Bredt, 1998). In all β-variants, a prominent SH3 domain is present in conserved domain D2, and a nonfunctional GK domain is prominent in domain D4. PDZ domains are present in domain D1 of some variants but are weakly represented in members of the β1 gene (Hanlon et al., 1999). The atomic structure of the SH3-GK core of the MAGUK protein PSD-95 has been elucidated at a 2.3 Å resolution (McGee et al., 2001; Tavares et al., 2001). In PSD-95, the SH3 and GK domains interact extensively via hydrophobic residues lined up across a cleft separating the two domains. Our data show that ∼90% of the β1a sequence is “generic” in the sense that it can be replaced by sequences present in the heterologous β2a variant. This generic backbone includes the D2 and D4 domains, that house SH3 and GK, respectively. We believe that it is highly unlikely that the domain organization of the D2–D4 core would be altered in any of the tested chimeras since this core, according to the PSD-95 model, is held together by hydrophobic interactions involving multiple conserved residues in D2 and D4. However, the location of D5 relative to the D2–D4 core could change depending on the composition of D5, and this could produce a change in the EC coupling phenotype. The β1aD5 domain differs from the β2aD5 domain in several respects. The β2aD5 tail is bulkier than the β1aD5 tail (185 vs. 47 residues; see Materials and Methods for sequence alignment), and is predicted to be more hydrophilic (Kyte-Doolitle index +3.5), and richer in turns (high Chou-Fasman, Garnier-Robson indices) than β1aD5. For these reasons, an “EC coupling-permissive” conformation may not be achievable when β2aD5 replaces β1aD5. Fig. 7 shows models of the domain organization of β1aD5 (in red) and β2aD5 (in blue) that could account for the change observed in the EC coupling phenotype. A β1aD5 domain tightly bound to the D2–D4 core (Fig. 7 A) might facilitate interaction of the subunit, and the DHPR complex as a whole, with RyR1. Furthermore, the location of β1aD5 next to D4 might direct sequences fused to the carboxyl terminus of β1aD5, such as β2aD5, away from the DHPR-RyR1 binding loci (Fig. 7 B). In contrast, the more flexible and bulkier β2aD5 domain could hinder interaction of the subunit with RyR1 (Fig. 7 C), and fusion of β1aD5 to the carboxyl terminus of β2aD5 might not correct this situation (Fig. 7 D). The model is consistent with the expected constancy of the D2–D4 core structure, and could also explain why heterologous sequences fused to the carboxyl terminus of β1a, such as green fluorescent proteins, do not alter skeletal excitation-contraction coupling (Neuhuber et al., 1998; Bhattacharya, et al., 2004; Leuranguer et al., 2004). Finally, it is important to acknowledge that the proposed model of the domain organization of β1a pertains to an EC coupling permissive state achieved when β1aD5 is next to D4. It is entirely possible that the EC coupling permissive state reflects a preferential folding pattern of the β1a subunit that facilitates binding of the DHPR to RyR1 regardless of whether β1a is tightly bound to RyR1 or not. The latter possibility remains to be confirmed by in vitro binding approaches (Cheng et al, 2004).
FIGURE 7.

Proposed model of the domain organization of β1a. An EC coupling “permissive” domain organization of β1a (diagrams A and B) requires domain β1aD5 (red) to be present immediately downstream from conserved D4 (yellow). Nonpermissive domain organizations (diagrams C and D) come about when the bulkier β2aD5 domain (blue) hinders interaction of the subunit and critical binding partners such as RyR1 (gray). A core of tightly bound D2 (green) and D4 (yellow) is conserved among all tested chimeras, consistent with the homology of β-subunits to MAGUK proteins and the structure of the MAGUK protein PSD-95 (McGee et al., 2001; Tavares et al., 2001). For the sake of clarity, β1aD5 has been drawn next to D4. However, according to the PSD-95 structure, β1aD5 may be located closer to the cleft between D2 and D4 domains. EC coupling permissive states with β1aD5 next to D4 (A and B) could bind preferentially to RyR1 (gray), or could facilitate binding of other domains of the DHPR to RyR1.
Evidence that β1aD5 could be engaged in protein-protein interactions relates to the finding of a hydrophobic heptad repeat motif in that region, which when modified, drastically altered the EC coupling phenotype. Secondary structure predictions using SOMP, PROFPRED, HNN, and other web-based programs indicated that some of the heptad repeat positions fall in α-helical regions, although none of the programs made predictions for the entire 14-residue stretch covering the three positions in the repeat. Hence, we are not entirely sure at this point if the identified positions have the secondary structure of canonical leucine zippers (Landschulz et al., 1988). We believe this may be the case since mutation of the three positions in the heptad repeat produced a drastic reduction in Ca2+ transient amplitude; however, mutation of 3 positions out of step with the heptad repeat were phenotypically neutral. The control mutations did not affect the magnitude of the Ca2+ transients relative to WT β1a, and furthermore, charge movement densities were the same in D5ALA, the control triple mutant D5ALAc, and WT β1a (Table 1). Thus, explanations for the change in phenotype based on a mistargeting of DHPRs to sites away from the cell surface are unlikely. Leucine-valine heptad repeats are well-known motifs engaged in protein-protein interactions and oligomerization (Simmerman et al., 1996; Surks et al., 1999). Studies have shown the presence of functionally significant leucine/isoleucine heptad repeats downstream from S4 in each of the four internal repeats of α1S, and at several locations in RyR1 (Garcia et al., 1997; Marx et al., 2001). Inspection of the RyR1 sequence indicates two five-position hydrophobic heptad repeats in region R9 and three four-position repeats in region R10 (see Hakamata et al., 1992, for sequence comparison). RyR1 regions R9 and R10 have been implicated in skeletal-type excitation-contraction coupling (Nakai et al., 1998b). In addition, a highly conserved heptad repeat motif is present in domain D2 of all β-variants (see Perez-Reyes and Schneider, 1994, for a sequence comparison). Thus, in principle, there could be three potential targets for the β1aD5 “hemi-zipper”: the voltage sensor, domain D2 in the same subunit, or RyR1. Interactions with the voltage sensor itself are unlikely since the D5-ALA mutant does not affect charge movements, and furthermore, the heptad repeat is not a molecular determinant of Ca2+ current expression (see Table 2; Ahern et al., 2003). However, interactions with D2 or RyR1 may be equally significant in light of the proposed domain organization model (Fig. 7). Hence, the heptad repeat may be required for intramolecular SH3-GK interactions, or may form part of a “molecular hook” engaged in binding other partners. Whether the heptad repeat is the sole motif in β1aD5 required for EC coupling recovery was not resolved in this study. Further studies are required to clarify if the heptad repeat acts alone or in concert with other motifs present in β1aD5.
Acknowledgments
Supported by National Institutes of Health grants AR46448, HL47053, and T32 HL07936; a training grant predoctoral fellowship to L.C., and a predoctoral fellowship from the Wisconsin Heart Association to D.C.S.
References
- Ahern, C. A., D. Bhattacharya, L. Mortenson, and R. Coronado. 2001a. A component of excitation-contraction coupling triggered in the absence of the T671–L690 and L720–Q765 regions of the II–III loop of the dihydropyridine receptor α1S pore subunit. Biophys. J. 81:3294–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahern, C. A., J. Arikkath, P. Vallejo, C. A. Gurnett, P. A. Powers, K.P. Campbell, and R. Coronado. 2001b. Intramembrane charge movements and excitation-contraction coupling expressed by two-domain fragments of the Ca2+ channel. Proc. Natl. Acad. Sci. USA. 98:6935–6940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahern, C. A., D. C. Sheridan, W. Cheng, L. Mortenson, P. D. Allen, and R. Coronado. 2003. Ca2+current and charge movements in skeletal myotubes promoted by the β-subunit of the dihydropyridine receptor in the absence of ryanodine receptor type 1. Biophys. J. 84:942–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, J. M. 1996. Cell signaling: MAGUK magic. Curr. Biol. 6:382–384. [DOI] [PubMed] [Google Scholar]
- Bhattacharya, D., G. Marriott, and R. Coronado. 2004. Intramolecular FRET signal from the DHPR beta subunit in cultured myotubes. Biophys. J. 86:64a. (Abstr.) [Google Scholar]
- Beurg, M., M. Sukhareva, C. Strube, P. A. Powers, R. G. Gregg, and R. Coronado. 1997. Recovery of Ca2+ current, charge movements, and Ca2+ transients in myotubes deficient in dihydropyridine receptor β1 subunit transfected with β1 cDNA. Biophys. J. 73:807–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurg, M., M. Sukhareva, C. A. Ahern, M. W. Conklin, E. Perez-Reyes, P. A. Powers, R. G. Gregg, and R. Coronado. 1999a. Differential regulation of skeletal muscle L-type Ca2+ current and excitation-contraction coupling by the dihydropyridine receptor β-subunit. Biophys. J. 76:1744–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurg, M., C. A. Ahern, P. Vallejo, M. Conklin, P. A. Powers, R. G. Gregg, and R. Coronado. 1999b. Involvement of the carboxy-terminus region of the dihydropyridine receptor β1a subunit in excitation-contraction coupling of skeletal muscle. Biophys. J. 77:2953–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bichet, D., V. Cornet, S. Geib, E. Carlier, S. Volsen, T. Hoshi, Y. Mori, and M. De Waard. 2000. The I–II loop of the Ca2+ channel α1 subunit contains an endoplasmic reticulum retention signal antagonized by the β subunit. Neuron. 25:177–190. [DOI] [PubMed] [Google Scholar]
- Birnbaumer, L., N. Qin, and R. Olcese. 1998. Tareilus, E., Platano, D., Constantin, J., and Stefani, E. Structures and functions of calcium channel beta subunits. J. Bioenerg. Biomembr. 30:357–375. [DOI] [PubMed] [Google Scholar]
- Castellano, A., X. Wei, L. Birnbaumer, and E. Perez-Reyes. 1993a. Cloning and expression of a third calcium channel beta subunit. J. Biol. Chem. 268:3450–3455. [PubMed] [Google Scholar]
- Castellano, A., X. Wei, L. Birnbaumer, and E. Perez-Reyes. 1993b. Cloning and expression of a neuronal calcium channel beta subunit. J. Biol. Chem. 268:12359–12366. [PubMed] [Google Scholar]
- Chandler, W. K., R. F. Rakowski, and M. F. Schneider. 1976. Effects of glycerol treatment and maintained depolarization on charge movement in skeletal muscle. J. Physiol. 254:285–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, W. L., Carbonneau, L. Keys, X. Altafaj, M. Ronjat, and R. Coronado. 2004. Positive charges in the 3495–3502 region of RyR1 are required for physical interaction with the skeletal the skeletal DHPR beta subunit. Biophys. J. 86:220a. (Abstr.) [Google Scholar]
- Chien, A. J., X. L. Zhao, R. E. Shirokov, T. S. Puri, C. F. Chang, K. Sun, E. Rios, and M. M. Hosey. 1995. Roles of a membrane-localized β subunit in the formation and targeting of functional L-type Ca2+ channels. J. Biol. Chem. 270:30036–30044. [DOI] [PubMed] [Google Scholar]
- Craven, S. E., and D. S. Bredt. 1998. PDZ domains organize synaptic signaling pathways. Cell. 93:495–498. [DOI] [PubMed] [Google Scholar]
- Dirksen, R. T., and K. G. Beam. 1999. Role of calcium permeation in dihydropyiridine receptor function. Insights into channel gating and excitation-contraction coupling. J. Gen. Physiol. 114:393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hayek, R., B. Antoniu, J. Wang, S. L. Hamilton, and N. Ikemoto. 1995. Identification of a calcium release-triggering and blocking regions of the II–III loop of the skeletal muscle DHPR. J. Biol. Chem. 270:22116–22118. [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong, C., and F. Protasi. 1997. Ryanodine receptors of striated muscles: a complex channel capable of multiple interactions. Physiol. Rev. 77:699–729. [DOI] [PubMed] [Google Scholar]
- Garcia, J., T. Tanabe, and K. G. Beam. 1994. Relationship of calcium transients to calcium currents and charge movements in myotubes expressing skeletal and cardiac dihydropyridine receptors. J. Gen. Physiol. 103:125–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia, J., J. Nakai, K. Imoto, and K. G. Beam. 1997. Role of S4 segments and the leucine heptad motif in the activation of an L-type calcium channel. Biophys. J. 72:2515–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg, R. G., A. Messing, C. Strube, M. Beurg, R. Moss, M. Behan, M. Sukhareva, S. Haynes, J. A. Powell, R. Coronado, and P. A. Powers. 1996. Absence of the β subunit (CCHB1) of the skeletal muscle dihydropyridine receptor alters expression of the α1 subunit and eliminates excitation-contraction coupling. Proc. Natl. Acad. Sci. USA. 93:13961–13966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakamata, Y., J. Nakai, H. Takeshima, and K. Imoto. 1992. Primary structure and distribution of a novel ryanodine receptor/calcium release channel from rabbit brain. FEBS Lett. 312:229–235. [DOI] [PubMed] [Google Scholar]
- Hanlon, M. R., N. S. Berrow, and A. C. Dolphin. 1999. and B.A. Wallace. Modeling of a voltage-dependent Ca2+ channel β subunit as a basis for understanding its functional properties. FEBS Lett. 445:366–370. [DOI] [PubMed] [Google Scholar]
- Landschulz, W. H., P. F. Johnson, and S. L. McKnight. 1988. The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science. 240:1759–1764. [DOI] [PubMed] [Google Scholar]
- Leong, P., and D. H. MacLennan. 1998. The cytoplasmic loops between domains II and III and domains III and IV in the skeletal muscle dihrydropyridine receptor bind to a contiguous site in the skeletal muscle ryanodine receptors. J. Biol. Chem. 273:29958–29964. [DOI] [PubMed] [Google Scholar]
- Leuranguer, V., S. Papadopoulos, and K. G. Beam. 2004. Insights into DHPR-RyR1 interactions using CFP-YFP tandem as a FRET probe. Biophys. J. 86:220a. (Abstr.) [Google Scholar]
- Marx, S. O., S. Reiken, Y. Hisamatsu, M. Gaburjakova, and J. Gaburjakova, Y-M Yang, N. Rosemblit, and A. R. Marks. 2001. Phosphorylation-dependent regulation of ryanodine receptors: a novel role for leucine/isoleucine zippers. J. Cell Biol. 153:699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack, K., M. A. Tanouye, L. E. Iverson, J. W. Lin, M. Ramaswami, T. McCormack, J. T. Campanelli, M. K. Mathew, and B. Rudy. 1991. A role for hydrophobic residues in the voltage-dependent gating of Shaker K+ channels. Proc. Natl. Acad. Sci. USA. 88:2931–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee, A. W., S. R. Dakoji, O. Olsen, D. S. Bredt, W. A. Lim, and K. E. Prehoda. 2001. Structure of the SH3-Guanylate kinase module from PSD-95 suggests a mechanism for regulated assembly of MAGUK scaffolding proteins. Mol. Cell. 8:1291–1301. [DOI] [PubMed] [Google Scholar]
- Nakai, J., R. T. Dirksen, H. T. Nguyen, I. N. Pessah, K. G. Beam, and P. D. Allen. 1996. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature. 380:72–75. [DOI] [PubMed] [Google Scholar]
- Nakai, J., T. Tanabe, T. Konno, B. Adams, and K. G. Beam. 1998a. Localization in the II–III loop of the dihydropyridine receptor of a sequence critical for excitation-contraction coupling. J. Biol. Chem. 273:24983–24986. [DOI] [PubMed] [Google Scholar]
- Nakai, J., N. Sekiguchi, T. A. Rando, P. D. Allen, and K. G. Beam. 1998b. Two regions of the ryanodine receptor involved in coupling with L-type Ca2+ channels. J. Biol. Chem. 273:13403–13406. [DOI] [PubMed] [Google Scholar]
- Neely, A., X. Wei, R. Olcese, L. Birnbaumer, and E. Stefani. 1993. Potentiation of the β subunit of the ratio of the ionic current to the charge movement in the cardiac calcium channel. Science. 262:575–578. [DOI] [PubMed] [Google Scholar]
- Neuhuber, B., U. Gester, J. Mitterdorfer, H. Glossmann, and B. E. Flucher. 1998. Differential effects of Ca2+ channel β1a and β2a subunits on complex formation with α1S and on current expression in tsA201 cells. J. Biol. Chem. 273:9110–9118. [DOI] [PubMed] [Google Scholar]
- Olcese, R., A. Neely, N. Qin, X. Wei, L. Birnbaumer, and E. Stefani. 1996. Coupling between charge movement and pore opening in vertebrate neuronal α1E calcium channels. J. Physiol. 497:675–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Reyes, E., and A. Castellano. 1992. Kim. H.S., Bertrand, P. Baggstrom, E., Lacerda, A.E., Wei, X.Y., and Birnbaumer, L. Cloning and expression of a cardiac/brain beta subunit of the L-type Ca2+ channel. J. Biol. Chem. 267:1792–1797. [PubMed] [Google Scholar]
- Perez-Reyes, E., and T. Schneider. 1994. Calcium channels: structure, function, and classification. Drug Development Res. 33:295–318. [Google Scholar]
- Powers, P. A., S. Liu, K. Hogan, and R. G. Gregg. 1992. Skeletal muscle and brain isoforms of a beta subunit of human voltage-dependent calcium channels are encoded by a single gene. J. Biol. Chem. 267:22967–22972. [PubMed] [Google Scholar]
- Proenza, C., J. O'Brien, J. Nakai, S. Mukherjee, P. D. Allen, and K. G. Beam. 2002. Indentification of a region of RyR1 that participates in allosteric coupling with the α1S (CaV1.1) II–III loop. J. Biol. Chem. 277:6530–6535. [DOI] [PubMed] [Google Scholar]
- Qin, N., R. Olcese, J. Zhou, O. A. Cabello, L. Birnbaumer, and E. Stefani. 1996. Identification of a second region of the β subunit involved in regulation of calcium channel inactivation. Am. J. Physiol. 271:C1539–C1545. [DOI] [PubMed] [Google Scholar]
- Restituto, S., T. Cens, C. Barrere, S. Geib, S. Galas, M. De Waard, and P. Charnet. 2000. The β2a subunit is a molecular groom for the Ca2+ channel inactivation gate. J. Neurosci. 20:9046–9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios, E., and G. Brum. 1987. Involvement of dihydropyridine receptors in excitation-contraction coupling in skeletal muscle. Nature. 325:717–720. [DOI] [PubMed] [Google Scholar]
- Rios, E., M. Karhanek, J. Ma, and A. Gonzalez. 1993. An allosteric model of the molecular organization of excitation-contraction coupling in skeletal muscle. J. Gen. Physiol. 102:449–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samso, M., and T. Wagenknecht. 2002. Apocalmodulin and Ca2+-calmodulin bind neighboring locations on the ryanodine receptor. J. Biol. Chem. 277:1349–1353. [DOI] [PubMed] [Google Scholar]
- Sencer, S., R. V. L. Papineni, and D. B. Halling, P. Pate, J. Krol, J.-Z. Zhang, and S.L. Hamilton. 2001. Coupling of RyR1 and L-type calcium channels via calmodulin binding domains. J. Biol. Chem. 276:38237–38241. [DOI] [PubMed] [Google Scholar]
- Sheridan, D. C., W. Cheng, C. A. Ahern, L. Mortenson, D. Alsammarae, P. Vallejo, and R. Coronado. 2003a. Truncation of the carboxyl terminus of the dihydropyridine receptor β-subunit promotes Ca2+ dependent excitation contraction coupling in skeletal myotubes. Biophys. J. 84:220–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan, D. C., L. Carbonneau, C. A. Ahern, P. Nataraj, and R. Coronado. 2003b. Ca2+ dependent excitation-contraction coupling triggered by the heterologous cardiac/brain DHPR β2a subunit in skeletal myotubes. Biophys. J. 85:3739–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan, D. C., W. Cheng, and R. Coronado. 2004. A heptad repeat in the C-terminal domain of the DHPR β1a subunit provides an active signal for fast skeletal-type EC coupling. Biophys. J. 86:63a. (Abstr.) [Google Scholar]
- Sherysheva, I. I., S. J. Ludtke, M. R. Backer, W. Chui, and S. L. Hamilton. 2002. 3D structure of the voltage-gated L-type Ca2+ channel by electron cryomicroscopy. Proc. Natl. Acad. Sci. USA. 99:10370–10375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmerman, H. K., Y. M. Kobayashi, J. M. Autry, and L. R. Jones. 1996. A leucine zipper stabilizes the pentameric membrane domain of phospholamban and forms a coiled-coil pore structure. J. Biol. Chem. 271:5941–5946. [DOI] [PubMed] [Google Scholar]
- Surks, H. K., N. Mochizuki, Y. Kasai, S. P. Georgescu, M. Tang, M. Ito. T. M. Lincoln, and M. E. Mendelsohn. 1999. Regulation of myosin phosphatase by a specific interaction with cGMP-dependent protein kinase I alpha. Science. 286:1583–1587. [DOI] [PubMed] [Google Scholar]
- Tanabe, T., K. G. Beam, B. A. Adams, T. Niidome, and S. Numa. 1990. Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature. 346:567–569. [DOI] [PubMed] [Google Scholar]
- Tavares, G. A., E. H. Panepucci, and A. T. Brunger. 2001. Structural characterization of the intramolecular interaction between the SH3 and guanylate kinase domains of PSD-95. Mol. Cell. 8:1313–1325. [DOI] [PubMed] [Google Scholar]
- Wilkens, C. M., N. Kasielke, B. E. Flucher, K. G. Beam, and M. Grabner. 2001. Excitation-contraction coupling is unaffected by drastic alteration of the sequence surrounding residues L-720–L764 of the α1S II–III loop. Proc. Natl. Acad. Sci. USA. 98:5892–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf, M., A. Eberhart, H. Glossmann, J. Striessnig, and N. Grigorieff. 2003. Visualization of the domain structure of an L-type Ca2+ channel using electron cryo-microscopy. J. Mol. Biol. 332:171–182. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, N., C. Xin, and G. Meissner. 2001. Identification of apocalmodulin and Ca2+-calmodulin regulatory domain in skeletal muscle Ca2+ release channel, ryanodine receptor. J. Biol. Chem. 276:22579–22585. [DOI] [PubMed] [Google Scholar]
- Zhang, H., J.-Z. Zhang, C. I. Danila, and S. L. Hamilton. 2003. A noncontiguous, intersubunit binding site for calmodulin on the skeletal muscle Ca2+ release channel. J. Biol. Chem. 278:8348–8355. [DOI] [PubMed] [Google Scholar]