

Abstract
We have developed a statistical-mechanical model of the effect of solution additives on protein association reactions. This model incorporates solvent radial distribution functions obtained from all-atom molecular dynamics simulations of particular proteins into simple models of protein interactions. In this way, the effects of additives can be computed along the entire association/dissociation reaction coordinate. We used the model to test our hypothesis that a class of large solution additives, which we term “neutral crowders,” can slow protein association and dissociation by being preferentially excluded from protein-protein encounter complexes, in a manner analogous to osmotic stress. The magnitude of this proposed “gap effect” was probed for two simple model systems: the association of two spheres and the association of two planes. Our results suggest that for a protein of 20 Å radius, an 8 Å additive can increase the free energy barrier for association and dissociation by as much as 3–6 kcal/mol. Because the proposed gap effect is present only for reactions involving multiple molecules, it can be exploited to develop novel additives that affect protein association reactions although having little or no effect on unimolecular reactions such as protein folding. This idea has many potential applications in areas such as the stabilization of proteins against aggregation during folding and in pharmaceutical formulations.
INTRODUCTION
Most protein solutions of interest are metastable and degrade by a variety of chemical and physical routes including aggregation, oxidation, deamidation, and hydrolysis (Wang, 1999). The most prevalent of these routes is aggregation, the assembly of individual protein molecules into amorphous, multimeric states. The driving force for aggregation is similar to that for protein folding—favorable free energy for the reduction of solvent-accessible area, particularly of hydrophobic groups (Dill, 1990). Because aggregation and proper protein folding are linked in this way, whenever the hydrophobic groups of a protein are exposed to the solvent, kinetic competition arises between folding and aggregation (Fig. 1). An unfolded or partially unfolded protein molecule (U) may either fold into the native form (N) or associate with another unfolded molecule to form a dimer (A2), the first step in the aggregation process (Zettlmeissl et al., 1979; Kiefhaber et al., 1991):
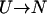 |
(1) |
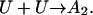 |
(2) |
FIGURE 1.

A free energy diagram along the folding/aggregation reaction coordinate for a globular protein is shown. The first small aggregate (A2) is formed from an unfolded or partially unfolded state, U, where a hydrophobic patch is exposed to the solvent. After formation of the first small aggregate (A2), further association reactions may take place to form higher-order aggregates (not shown).
Similarly, it is generally believed that a protein molecule that is initially in the native state (N) passes through a partially unfolded state in which some of its hydrophobic groups are exposed (U) before proceeding to form an aggregate (A2) (Lumry and Eyring, 1954):
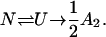 |
(3) |
The presence of such an intermediate is implied by the first-order kinetics observed in accelerated aggregation studies (Kendrick et al., 1998; Webb et al., 2001), and in some cases this intermediate has been characterized (Cleland, 1991).
The tendency of proteins to aggregate is an especially grave problem in biotechnology and the pharmaceutical industry where it is desired to synthesize, process, and store proteins at the highest possible concentrations and over long periods of time. Because proteins that are in the aggregated state generally do not have the same biological activity as proteins in the native state, can often be immunogenic, and may even have acute toxic effects in vivo, it is essential to develop strategies for preventing aggregation in these applications.
One such strategy is to add to protein solutions a compound that hinders aggregation. Molecules that have been shown to slow protein aggregation in different applications include urea, guanidinium chloride, amino acids (predominantly arginine and glycine), sugars, polyols, polymers (such as polyethylene glycol and cyclodextrins), surfactants, and antibodies. In most of these cases, the mechanisms by which the additives prevent aggregation are not known; therefore there is no potential to develop a rational methodology of additive selection, and additives are currently chosen by extensive, heuristic, experimental screening procedures.
In a few cases, however, qualitative mechanistic models have been developed to describe how a particular additive deters aggregation. Polyethylene glycol has been shown to inhibit the aggregation of carbonic anhydrase, interferon-γ, tissue plasminogen activator, and deoxyribonuclease during refolding (Cleland, 1991; Cleland et al., 1992) by binding to the unfolded protein and folding intermediates through hydrophobic interactions (Fig. 2). This binding decreases the free energy of the unfolded protein and refolding transition state, increases the activation energy for aggregation, slows the rate of aggregation, and increases the final yield of active protein.
FIGURE 2.

In refolding of carbonic anhydrase, interferon-γ, and DNase from the unfolded state (U), Cleland (1991) showed that polyethylene glycol (PEG) binds selectively to the unfolded protein and folding intermediates. This slows aggregation and increases the yield of native protein. The free energy in the absence of additive is shown as a solid line and, in the presence of PEG, as a dotted line.
Sucrose and other molecules that are preferentially excluded from the protein-solvent interface have been shown to stabilize native protein molecules against unfolding (Lee and Timasheff, 1981; Arakawa and Timasheff, 1982, 1985). Via an analogous mechanism, sucrose has been shown to deter the aggregation of interferon-γ from the native state in aqueous solution (Kendrick et al., 1998). Sucrose increases the free energy barrier of unfolding, which decreases the population of the intermediate (I), and therefore slows aggregation (Fig. 3).
FIGURE 3.

In studies of interferon-γ aggregation from the native state, Kendrick et al. (1998) showed that sucrose decreases the concentration of the aggregation-competent intermediate (I) and therefore slows aggregation. The free energy in the absence of additive is shown as a solid line and, in the presence of sucrose, as a dotted line.
Another prevalent anti-aggregation additive which appears to operate via a distinct mechanism is the amino acid arginine. Arginine has very little effect on the folding equilibrium (Arakawa and Tsumoto, 2003; Taneja and Ahmad, 1994; Shiraki et al., 2002) yet it facilitates refolding of several proteins from the unfolded state (Rudolph et al., 1985; Arora and Khanna, 1996; Armstrong et al., 1999; Rinas et al., 1990; Buchner and Rudolph, 1991). Although a mechanism which can explain fully how arginine functions has not been proposed (Lilie et al., 1998), these results suggest that arginine selectively increases the barrier for protein-protein association. Such observations led us to search for a mechanism that was consistent with the above experimental observations and energetic arguments with the objective of using this understanding to design new anti-aggregation additives.
To affect the kinetics of protein association reactions selectively without affecting protein folding and solution phase structure, there must be a unique feature of the association transition state that can be exploited by a binding interaction with an additive. The emerging picture of protein association/dissociation transition states indicates that each protein in the encounter complex is still mostly solvated but near the orientation in the final complex (Selzer and Schreiber, 2001; Schreiber, 2002). Because the complex is still mostly solvated but the two protein molecules are in close proximity to one another, there is the potential for a “gap effect” to arise in a mixed solvent if the additive is significantly larger than the primary solvent (Fig. 4). This gap effect is analogous to osmotic stress (Rand, 1992). In such a situation, the large additive will be excluded from solvating the gap between the protein molecules for steric reasons. This, in turn, results in an increase in the free energy of the protein-protein encounter complex.
FIGURE 4.

If a protein molecule (P) contains narrow channels too small for a large additive (solid circles) to enter as in a, the cosolvent exerts an osmotic stress effect that favors the collapse of these channels and the release of the water (shaded circles) they contain. An analogous effect can occur in any protein state which contains gaps, such as the protein-protein encounter complex shown in b. In this case, the “gap effect” caused by the large additive selectively increases the free energy of encounter complexes that contain such gaps. The gap effect therefore slows isomerization between the associated and dissociated protein states.
For an additive to reduce the rate of aggregation without affecting the folding rate and equilibrium, in addition to being preferentially excluded from encounter complexes, it is necessary for the additive not to interact with isolated protein molecules differently than water does. We call additives that exhibit both of these properties “neutral crowders.” We hypothesize that a neutral crowder would affect a refolding/aggregation reaction coordinate as shown in Fig. 5.
FIGURE 5.

The hypothesized effect of a neutral crowder on the free energy of protein states along the refolding/aggregation reaction coordinate is shown. The free energy in the absence of additive is shown as a solid line and, in the presence of a neutral crowder, as a dotted line. The neutral crowder is preferentially excluded from the gap between the protein molecules in the association transition state (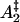 ), increasing the free energy of this state.
), increasing the free energy of this state.
With this gap effect hypothesis in mind, the specific objectives of this study are to
Develop and justify the use of a simple model for protein-protein association reactions in the presence of additives. This model should take into account protein-additive binding interactions over the entire association reaction coordinate, and extend prior theoretical work on additive effects on protein thermodynamics; for example, that of Mahadevan and Hall (1990).
Use this model to test the gap effect hypothesis and evaluate the potential magnitude of any observed gap effect.
Evaluate the potential of neutral crowders as anti-aggregation additives.
We expect that our study should have impact in protein processing and protein formulation (Wang, 1999; Cleland et al., 1993).
THEORETICAL APPROACH
To test our gap effect hypothesis, we compute the effects of various solution additives on the association and dissociation rate constants for a suitable protein-protein model system. If the only effect of an additive is to alter the free energy barrier to the transition state, the change in a rate constant can be expressed as
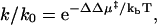 |
(4) |
where k is the rate constant in the presence of the additive, k0 is the rate constant in the absence of the additive, ΔΔμ‡ is the change in the activation free energy induced by the additive, kb is Boltzmann's constant, and T is the absolute temperature. Thus, to compute the relative rate constants, we must compute the shift in activation free energy for association and dissociation induced by the additives. Our approach to this is to
Define two simple, limiting models of a protein-protein association reaction, i), the association of two parallel planes, and ii), the association of two spheres. We also propose with our models suitable reaction coordinates and compute the free energy as a function of reaction coordinate in each case.
Compute the perturbations to each free-energy diagram induced by an additive. These perturbations are obtained by using data determined from explicit all-atom molecular dynamics simulations and incorporating them into our simple models.
The details of each of these steps are described in the following sections.
Two-model association reactions
As two limiting models of proteins undergoing an association and dissociation reaction, we choose:
The reaction of two spheres, each 20 Å in radius. This is roughly the size of a 25-kDa protein.
The reaction of two parallel, planar plates, each with 400 πÅ2 of area on a face. This area was selected to make the change in protein solvent-accessible area of reaction the same for the cases of the two spheres and two planes (the reaction coordinates are explained below). The thermodynamics properties of these plates are obtained by calculating the property per unit of surface area of a pair of infinite plates and then multiplying by the area above. Thus, edge effects are ignored.
These two geometries can be considered as extreme cases by which associating proteins approach one another. Because of the symmetry of these simple model proteins, the reaction coordinates, x, can be simply defined as the shortest distance between the planes and the distance between the centers of the spheres. We are then free to choose any representative free energy of the complex as a function of this reaction coordinate, μP, 0(x). For convenience, we set the reference states as the monomers (x → +∞), define the dimer states to be 8 kcal/mol more stable than monomer states, and place a modest 2 kcal/mol free energy barrier for association between the two states. Arbitrarily, we select x = 20 Å as the dimer state for the spheres, and x = 1.5 Å as the dimer state for the planes. The resulting reaction coordinates can be modeled as
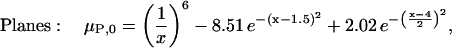 |
(5) |
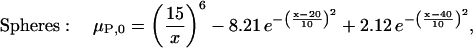 |
(6) |
which employ Gaussians for the energy minimum at the dimer state and for the energy maximum at the transition state, and an inverse sixth-power function for repulsion at distances closer than the dimer state. The reaction coordinate and free energy diagram for each model are shown in Fig. 6.
FIGURE 6.

The definition of each reaction coordinate and the free energy diagrams (Eqs. 5 and 6) are shown for the two model protein systems used in this work. For the spheres, the association/dissociation reaction coordinate, x, is the distance between the sphere centers. For the planes, it is the shortest distance between the planes (which are always parallel). The value x is 0 when the two proteins overlap each other completely.
Calculating the effect of an additive
We compute the free energy along the reaction coordinate in the presence of a solution additive by combining the free energy in the absence of additive (Eqs. 5 and 6) with an appropriate transfer free energy of
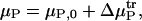 |
(7) |
where μP, 0 is the free energy of a given protein state in the absence of additive, μP is the free energy of the same state when the additive is present, and  is the transfer free energy. The transfer free energy can be computed via the equation
is the transfer free energy. The transfer free energy can be computed via the equation
 |
(8) |
where mX is the molality of the additive and mP is the molality of the protein. The integrand above can be split into a derivative involving properties of only the additive and water, and one that describes binding,
 |
(9) |
where ΓXP, the preferential binding coefficient of additive to the protein in water, is defined as
 |
(10) |
Following our earlier work in all-atom molecular dynamics simulations of preferential binding (Baynes and Trout, 2003), we introduce the relation
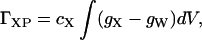 |
(11) |
where cX is the bulk concentration of additive, gX and gW are the additive and water radial distribution functions with respect to the protein, respectively, and the integral is over the solvent volume around the protein. Note that gX and gW typically differ only in the first two solvation shells. For the association/dissociation reactions being modeled here, the protein state is a pair of protein molecules, and dV is a function of x.
The above relation can be substituted into Eq. 9 to yield
 |
(12) |
We now invoke three assumptions that allow significant simplification of the above equation:
The additive free energy (μX) is thermodynamically ideal, or
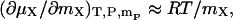 where R is the gas constant. Since the ternary system in question here is dominated by the water and additive, this assumption effectively means that water-additive interactions are ideal. For aqueous solutions of many additives of interest, such as those of NaCl, glycerol, sucrose, and urea, the experimental activities of water-additive mixtures are within 10% of ideality at molalities up to 1 mol/kg (Scatchard et al., 1938; Ninni et al., 2000). For other real systems where experimental thermodynamic activity data on the binary system of water and additive are available, these can be used to more accurately evaluate the partial derivative in question. The assumption used here does not limit the complexity of the additive-protein and water-protein interactions, which ultimately will lead to changes in the transfer free energy via the ΓXP term.
where R is the gas constant. Since the ternary system in question here is dominated by the water and additive, this assumption effectively means that water-additive interactions are ideal. For aqueous solutions of many additives of interest, such as those of NaCl, glycerol, sucrose, and urea, the experimental activities of water-additive mixtures are within 10% of ideality at molalities up to 1 mol/kg (Scatchard et al., 1938; Ninni et al., 2000). For other real systems where experimental thermodynamic activity data on the binary system of water and additive are available, these can be used to more accurately evaluate the partial derivative in question. The assumption used here does not limit the complexity of the additive-protein and water-protein interactions, which ultimately will lead to changes in the transfer free energy via the ΓXP term.The concentrations of protein and additive are sufficiently low such that molar and molal concentrations are equivalent (cX ≈ mX).
The radial distribution functions of water and the additive are independent of the concentration of additive. This is expected to be true at low additive concentrations (mX < 1 m) for additives that interact weakly with the protein (Courtenay et al., 2000; Greene and Pace, 1974; Record et al., 1998).
Applying these approximations to Eq. 12 yields
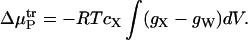 |
(13) |
To enable computation of the transfer free energies, we now require a model for the radial distribution functions of the additive and water around the proteins. In prior studies of all-atom molecular dynamics of proteins in mixed solvents (Baynes and Trout, 2003), we noted that the radial distribution function of water as a function of distance from the protein, gW(r), did not vary whether the protein was RNase A or RNase T1, two proteins with significantly different amino acid sequences. Thus, in this work, we use the water radial distribution function found previously, and assume it to be independent of the protein model and reaction coordinates employed here.
In the case of the additive, we wish to introduce a simple, physically based model for the additive-protein radial distribution function, gX. To do this, we relate gX to the potential of mean force between the additive and protein, 〈UXP〉,
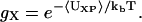 |
(14) |
We then choose the form of the potential of mean force as a standard intermolecular potential function. To select a suitable function, we fit the parameters of standard, physically based intermolecular potentials, such as Lennard-Jones, Kihara, and exponential-6 (Exp-6) to the radial distribution functions of water, urea, and glycerol obtained from all-atom molecular dynamics simulations (Baynes and Trout, 2003). In each case, the intermolecular potential parameters were fit by nonlinear minimization (Marquardt method) of the squared residuals although constraining (Lagrange method) the radial distribution to give the same preferential binding coefficient (ΓXP, via Eq. 11) as the actual additive radial distribution function. Preferential binding coefficients were preserved in the fitting procedure because of their tight relationship (via Eq. 9) to the transfer free energy, the property we ultimately wish to model.
After obtaining the best fits with each potential function, it was observed that the Lennard-Jones potential did not adequately fit the data, and the Kihara potential did not fit the data well at physically meaningful parameter values. Therefore, the three-parameter Exp-6 potential was selected as a model of the additive-protein potential of mean force as
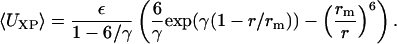 |
(15) |
In the above equation, r is the distance between the solvent molecule and protein, and rm, ε, and γ are the Exp-6 parameters, described below. Results of the fitting process are shown in Fig. 7 and Table 1. Note that the first peak in the radial distribution functions occurs at a value of r smaller than what might be expected. This is because in our case, r is defined as the distance from the center of mass of the solvent or additive to the van der Waals shell of the protein, not to the nucleus of the atoms at the protein surface. This also leads to a value of rm that may be smaller than expected.
FIGURE 7.

Fits of the protein-water and protein-additive radial distribution functions from molecular dynamics simulations for various additives with the protein RNase T1 using the exponential-6 intermolecular potential. Note that r = 0 is at the surface of the protein. The observed data are shown as crosses, the fits as lines. The corresponding parameters are shown in Table 1.
TABLE 1.
Exponential-6 potential parameters for averaged interaction energies of water, urea, and glycerol with RNase A and T1
| Molecule | ε (kcal/mol) | rm (Å) | γ |
|---|---|---|---|
| Water | 0.662 | 0.925 | 3.65 |
| Urea | 0.917 | 1.59 | 3.17 |
| Glycerol | 0.497 | 2.11 | 4.25 |
The parameters were obtained by constrained fitting to radial distribution functions obtained from all-atom molecular dynamics data.
It is undoubtedly possible to obtain a tighter three-parameter fit to the radial distribution functions shown in Fig. 7 by using a broader set of basis functions to model 〈UXP〉. However, we wished to constrain ourselves to standard potential functions whose parameters had some physical meaning. Fits are also shown for water because water-protein radial distribution function data was available; however, the full radial distribution function for water was used for all of the calculations in this work.
The Exp-6 potential combines an exponential repulsive term with an inverse sixth-power attractive term and has a single minimum at U(r = rm) = −ε. The last Exp-6 parameter, γ, is related to the breadth of the minimum near r = rm, and reflects the rigidity and shape of the additive.
In extending the Exp-6 representation to neutral crowders and other additives for which no radial distribution functions are available, rm is used as a measure of additive size; γ is held constant at 3.7, the mean of the observed values for water, glycerol, and urea; and ε is set to yield a desired preferential binding coefficient between the additive and dissociated protein state. For a neutral crowder, ΓXP is set to 0 at x → ∞ (the dissociated state) by the constraint that such an additive should not affect the free energy of isolated protein molecules.
RESULTS
The gap effect can contribute significantly to association and dissociation rate constants
The transfer free energies for placing the model proteins into 1 M solutions of neutral crowders were computed over the entire association/dissociation reaction coordinates (via Eqs. 13–15) and are presented in Fig. 8. The value rm was chosen to have values of 2, 4, 6, or 8 Å to give a range of crowder sizes. In each case, ε was set according to the neutrality condition (ΓXP = 0 for the dissociated state, x → ∞).
FIGURE 8.

Transfer free energies for pairs of protein molecules transferred into 1 M additive solution as a function of position along the association reaction coordinate, x, are shown. The sizes of the additives are varied although keeping the second virial coefficients constant. The curves are labeled with the additive sizes (rm in Å) to which they correspond. The left-hand figure is for the association of two spherical proteins 20 Å in radius, and the right-hand figure is for the association of two pseudo-infinite planar proteins with an area of 400 πÅ2 on each face.
It is readily apparent that at constant ΓXP, the gap effect on the transfer free energy increases proportionately with increasing additive size, rm. For the additive sizes illustrated, the effect on transfer free energy ranges from 0 to almost 6 kcal/mol. At the same additive size and change in surface area of reaction, the planes exhibit approximately double the gap effect of the spheres. This is because the lack of curvature of the planes necessitates that their gap effect is concentrated over a narrower region of the reaction coordinate.
These transfer free energy effects can be superimposed onto a free energy diagram by simple addition (Eq. 7). The final free energy diagrams are shown in Fig. 9. In the case of the spherical model, the transition state in the original free energy surface (μP, 0(x)) is near the maxima in the transfer free energies, so the transfer free energy effects make significant changes to the activation free energy of the association and dissociation reactions in an additive solution at all values of rm. For the planar model, the location of the maximum in the transfer free energy depends on rm. Consequently, the transfer free energy maximum for the planar model does not always build on the existing free energy barrier in μP, 0(x). In fact at higher rm, the transition state for association and dissociation results completely from the gap effect. The original transition state is “drowned out.”
FIGURE 9.

The protein free energies along the reaction coordinate for association/dissociation in the presence of neutral crowders at 1 M concentration are shown. This combines μP, 0 (Eqs. 5 and 6) with the transfer free energies shown in Fig. 8. The curves are labeled with the additive sizes (rm in Å) to which they correspond.
Using Eq. 4 to estimate the resulting changes in reaction rate, the relative reaction rates (k/k0) can be determined as a function of additive size (rm) at constant ΓXP, as shown in Figs. 10 and 11. Increasing ΓXP can also be seen to decelerate association and accelerate dissociation. This is a well-known result consistent with the fact that denaturants are used to slow protein association reactions. The magnitude of this change depends on whether the free surface area of the transition state is more similar to that of the monomer or to that of the dimer. If the transition state is similar to the dimer, as in the case of the planar proteins, there is a strong effect on ka and almost no effect on kd. For the spherical protein geometry, the transition state is closer to the monomer, and the effect of ΓXP is larger on kd than on ka.
FIGURE 10.

The change in association and dissociation rates for 20 Å spherical proteins caused by a 1 M additive is shown as a function of the additive size (x axis) and additive-protein preferential binding coefficient, ΓXP (labels).
FIGURE 11.

The change in association and dissociation rates for planar proteins (with 400 πÅ2 area on a face) caused by a 1 M additive is shown as a function of the additive size (x axis) and additive-protein preferential binding coefficient, ΓXP (labels). For the dissociation rate, the ΓXP dependence is negligible.
We also see that increasing additive size at constant ΓXP decreases both the association and dissociation rates, consistent with the gap effect hypothesis. An approximately two-orders-of-magnitude drop in the association rate constant can be seen over the range of additive sizes shown (2–8 Å). In the case of two associating planes, this effect does not appear at moderate additive sizes (2.5 < rm < 5.5 Å) because, although the maximum in transfer free energy keeps increasing, it moves away from the original transition state on the reaction coordinate.
Designing additives for the control of aggregation
It may be possible to exploit the gap effect in designing solvent additives for the prevention of protein aggregation. Prevalent additives that work via a pure free surface effect, such as guanidinium and urea, have apparent radii (rm) of ∼2–3 Å. These have the disadvantage, however, that they can also enhance the unfolding or partial unfolding of proteins because of their positive preferential binding coefficients. The results of the preceding section suggest that if the size of these additives could be increased to ∼8 Å although maintaining their preferential binding coefficient with isolated protein molecules, the gap effect can potentially contribute another 1–2 order-of-magnitude depression in the association rate.
As the size of an additive is increased, its preferential binding coefficient will tend to decrease as the third power of radius. This is because increasing additive size increases the excluded volume of additive and protein, which decreases the preferential binding coefficient. To compensate for this excluded volume difference and return ΓXP to its original value, an additional additive-protein attraction must be introduced into the molecule.
If it is not possible to increase the additive-protein attraction in some way as size is increased, the additive will have a large, negative preferential binding coefficient, and a gap effect will not appear. A gap effect arises for neutral crowders because there is a region of solvent that is inaccessible to the additive around encounter complexes but not around isolated protein molecules. In the case of an “excluded crowder” with a large, negative preferential binding coefficient, the volume of exclusion is actually larger in the dissociated state than in the encounter complex or associated state. Thus, in stark contrast to neutral crowders, excluded crowders like sugars, polyols, and large, hydrophilic polymers favor association (Linder and Ralston, 1995; Kosk-Kosicka et al., 1995; Nichol et al., 1981).
CONCLUSIONS
In this work, a simple framework for modeling protein association and dissociation reactions in the presence of solution additives was developed and analyzed. Our model extends prior work in binding theory by considering various geometric models of the protein surface, the protein-protein association/dissociation transition states, and solvent radial distribution functions obtained from all-atom molecular dynamics simulations (Baynes and Trout, 2003).
Our analysis of the model supports the hypothesis that a “gap effect,” analogous to osmotic stress, will occur in association reactions when large solution additives with sufficient protein affinity are present. This gap effect affects the free energy of protein-protein encounter complexes, such as the association transition state, and has only a small effect on the end states. Thus, we have demonstrated how it is possible for an additive to exert a purely kinetic effect on protein association/dissociation. We call additives which have these properties “neutral crowders”: they are neutral in that they do not significantly shift the free energy of isolated protein molecules, but they decrease the rate of protein association and dissociation by being excluded from the interprotein gap in protein-protein encounter complexes for steric reasons.
For an optimal effect, the maximum in the transfer free energy induced by the gap effect must be near the original association free energy barrier. When this is not the case, the gap effect will be strongest when the original barrier is small (<1–2 kcal/mol) or nonexistent, such as in diffusion-controlled reactions.
As the size of a neutral crowder is increased, the gap effect becomes proportionately larger, but maintaining neutrality is difficult as size increases. At a constant protein-additive interaction energy, increasing additive size would decrease the protein-additive preferential binding coefficient as the third power of additive size due to an excluded volume effect. Thus, to make a large neutral crowder, additive-protein interactions must become significantly more attractive as size is increased. If this cannot be achieved, the gap effect will diminish and ultimately disappear.
Today, the best known additives for suppressing protein association reactions are small denaturants such as urea and guanidinium chloride. Our gap effect model predicts that if a significantly larger additive, perhaps 4–5-times the size of these small additives, can be developed, and it were a “neutral crowder,” it would depress association rates by a factor of 100–1000-times more than guanidinium or urea at the same molar concentration.
Acknowledgments
We thank Prof. D. I. C. Wang for inspiration for this study and very helpful advice.
The authors thank the National Institutes of Health Biotechnology Training Program, the Singapore-Massachusetts Institute of Technology Alliance, and the National University of Singapore for funding.
APPENDIX: RELATION TO VIRIAL COEFFICIENTS
Given the approximations used in this work, the transfer free energy can be related to the additive-protein and water-protein second virial coefficients. This follows directly from the McMillan-Mayer formula for the second virial coefficient (McMillan and Mayer, 1945) of
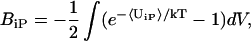 |
(16) |
where BiP is the second virial coefficient and 〈UiP〉 is the potential of mean force between a solvent component i (additive or water) and the protein. In terms of the radial distribution functions, this is
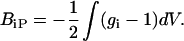 |
(17) |
The preceding equation can then be substituted into the integrand of Eq. 13 for the water and additive to yield
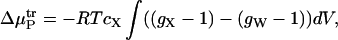 |
(18) |
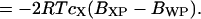 |
(19) |
Thus, the additive protein and water protein second virial coefficients are related to the radial distribution functions (Eq. 17) and to the transfer free energy (Eq. 19).
References
- Arakawa, T., and S. N. Timasheff. 1985. The stabilization of proteins by osmolytes. Biophys. J. 47:411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa, T., and S. N. Timasheff. 1982. Stabilization of protein structure by sugars. Biochemistry. 21:6536–6544. [DOI] [PubMed] [Google Scholar]
- Arakawa, T., and K. Tsumoto. 2003. The effects of arginine on refolding of aggregated proteins: not facilitate refolding, but suppress aggregation. Biochem. Biophys. Res. Commun. 304:148–152. [DOI] [PubMed] [Google Scholar]
- Armstrong, N., A. de Lencastre, and E. Gouaux. 1999. A new protein folding screen: application to the ligand binding domains of a glutamate and kainite receptor and to lysozyme and carbonic anhydrase. Protein Sci. 8:1475–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora, D., and N. Khanna. 1996. Method for increasing the yield of properly folded recombinant human γ-interferon from inclusion bodies. J. Biotechnol. 52:127–133. [DOI] [PubMed] [Google Scholar]
- Baynes, B. M., and B. L. Trout. 2003. Proteins in mixed solvents: a molecular-level perspective. J. Phys. Chem. B. 107:14058–14067. [Google Scholar]
- Buchner, J., and R. Rudolph. 1991. Renaturation, purification and characterization of recombinant fab-fragments produced in Escherichia coli. Biotechnology. 9:157–162. [DOI] [PubMed] [Google Scholar]
- Cleland, J. L. 1991. Mechanisms of protein aggregation and refolding. Ph.D. thesis, Massachusetts Institute of Technology, Cambridge, MA.
- Cleland, J. L., C. Hedgepeth, and D. I. C. Wang. 1992. Polyethylene glycol enhanced refolding of bovine carbonic anhydrase B. J. Biol. Chem. 267:13327–13334. [PubMed] [Google Scholar]
- Cleland, J. L., M. F. Powell, and S. J. Shire. 1993. The development of stable protein formulations: a close look at protein aggregation, deamidation, and oxidation. Crit. Rev. Ther. Drug Carrier Syst. 10:307–377. [PubMed] [Google Scholar]
- Courtenay, E. S., M. W. Capp, C. F. Anderson, and M. T. Record, Jr. 2000. Vapor pressure osmometry studies of osmolyte-protein interactions: implications for the action of osmoprotectants in vivo and for the interpretation of “osmotic stress” experiments in vitro. Biochemistry. 39:4455–4471. [DOI] [PubMed] [Google Scholar]
- Dill, K. A. 1990. Dominant forces in protein folding. Biochemistry. 29:7133–7155. [DOI] [PubMed] [Google Scholar]
- Greene, Jr., R. F., and D. N. Pace. 1974. Urea and guanidine hydrochloride denaturation of ribonuclease, lysozyme, α-chymotrypsin, and β-lactoglobulin. J. Biol. Chem. 249:5388–5393. [PubMed] [Google Scholar]
- Kendrick, B. S., J. F. Carpenter, J. L. Cleland, and T. W. Randolph. 1998. A transient expansion of the native state precedes aggregation of recombinant human interferon-γ. Proc. Natl. Acad. Sci. USA. 95:14142–14146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefhaber, T., R. Rudolph, H.-H. Kohler, and J. Buchner. 1991. Protein aggregation in vitro and in vivo: a quantitative model of the kinetic competition between folding and aggregation. BioTechnology. 9:825–829. [DOI] [PubMed] [Google Scholar]
- Kosk-Kosicka, D., M. M. Lopez, I. Fomitcheva, and V. L. Lew. 1995. Self-association of plasma membrane Ca2+-ATPase by volume exclusion. FEBS Lett. 371:57–60. [DOI] [PubMed] [Google Scholar]
- Lee, J. C., and S. N. Timasheff. 1981. The stabilization of proteins by sucrose. J. Biol. Chem. 256:7193–7201. [PubMed] [Google Scholar]
- Lilie, H., E. Schwarz, and R. Rudolph. 1998. Advances in refolding of proteins produced in E. coli. Curr. Opin. Biotechnol. 9:497–501. [DOI] [PubMed] [Google Scholar]
- Linder, R., and G. Ralston. 1995. Effects of dextran on the self-association of human spectrin. Biophys. Chem. 57:15–25. [DOI] [PubMed] [Google Scholar]
- Lumry, R., and H. Eyring. 1954. Conformation changes of proteins. J. Phys. Chem. 58:110–120. [Google Scholar]
- Mahadevan, H., and C. K. Hall. 1990. Statistical-mechanical model of protein precipitation by nonionic polymer. AIChE J. 36:1517–1528. [Google Scholar]
- McMillan, W. G., and J. E. Mayer. 1945. The statistical thermodynamics of multicomponent systems. J. Chem. Phys. 13:276–305. [Google Scholar]
- Nichol, L. W., A. G. Ogston, and P. R. Willis. 1981. Effect of inert polymers on protein self-association. FEBS Lett. 126:18–20. [DOI] [PubMed] [Google Scholar]
- Ninni, L., M. S. Camargo, and A. J. A. Meirelles. 2000. Water activity in polyol systems. J. Chem. Eng. Data. 45:654–660. [Google Scholar]
- Rand, R. P. 1992. Raising water to new heights. Science. 256:618. [DOI] [PubMed] [Google Scholar]
- Record, Jr., M. Thomas, W. Zhang, and C. F. Anderson. 1998. Analysis of effects of salts and uncharged solutes on protein and nucleic acid equilibria and processes: a practical guide to recognizing and interpreting polyelectrolyte effects, Höfmeister effects, and osmotic effects of salts. Adv. Protein Chem. 51:281–353. [DOI] [PubMed] [Google Scholar]
- Rinas, U., B. Risse, R. Jaenicke, K.-J. Abel, and G. Zettlmeissl. 1990. Denaturation-renaturation of the fibrin-stabilizing factor XIII a-chain isolated from human placenta. Biol. Chem. Hoppe-Seyler. 371:49–56. [DOI] [PubMed] [Google Scholar]
- Rudolph, R., S. Fischer, and R. Mattes. 1985. Process for the Activation of tPA after Expression in Prokaryotic Cells. Patent DE3537708.
- Scatchard, G., W. J. Hamer, and S. E. Wood. 1938. Isotonic solutions. I. The chemical potential of water in aqueous solutions of sodium chloride, potassium chloride, sulfuric acid, sucrose, urea and glycerol at 25°. J. Am. Chem. Soc. 60:3061–3070. [Google Scholar]
- Schreiber, G. 2002. Kinetic studies of protein-protein interactions. Curr. Opin. Struct. Biol. 12:41–47. [DOI] [PubMed] [Google Scholar]
- Selzer, T., and G. Schreiber. 2001. New insights into the mechanism of protein-protein association. Proteins. 45:190–198. [DOI] [PubMed] [Google Scholar]
- Shiraki, K., M. Kudou, S. Fujiwara, T. Imanaka, and M. Takagi. 2002. Biophysical effect of amino acids on the prevention of protein aggregation. J. Biochem. 132:591–595. [DOI] [PubMed] [Google Scholar]
- Taneja, S., and F. Ahmad. 1994. Increased thermal stability of proteins in the presence of amino acids. Biochem. J. 303:147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. 1999. Instability, stabilization, and formulation of liquid protein pharmaceuticals. Int. J. Pharm. 185:129–188. [DOI] [PubMed] [Google Scholar]
- Webb, J. N., S. D. Webb, J. L. Cleland, J. F. Carpenter, and T. W. Randolph. 2001. Partial molar volume, surface area, and hydration changes for equilibrium unfolding and formation of aggregate transition state: high-pressure and cosolute studies on recombinant human IFN-γ. Proc. Natl. Acad. Sci. USA. 98:7259–7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zettlmeissl, G., R. Rudolph, and R. Jaenicke. 1979. Reconstitution of lactic dehydrogenase. Noncovalent aggregation vs. reactivation. I. Physical properties and kinetics of aggregation. Biochemistry. 18:5567–5571. [DOI] [PubMed] [Google Scholar]