

Abstract
Azurin from Pseudomona aeruginosa is a small copper protein with a single tryptophan (Trp) buried in the structure. The Gibbs free energies associated with the folding of holo azurin, calculated monitoring Trp fluorescence and changes in absorbance on the ligand-to-metal band, are different because these techniques probe their local environments, thereby being able to probe different conformational changes. The presence of an intermediate state was observed during the chemical denaturation of the protein. Upon denaturation, a 30-fold increase is observed in the magnitude of the quenching constant of the tryptophan fluorescence by acrylamide, because this residue becomes more accessible to the quencher. Entrapping the protein in sol-gel materials lowers its stability possibly because the solvation properties of the macromolecule are changed. The thermal denaturation of azurin immobilized in a sol-gel monolith is irreversible, which tends to rule out an aggregation mechanism to account for the irreversibility of the denaturation of the protein free in solution. Unlike the Cu(II) ion, the Gd(III) ion accommodates in site B of azurin with high affinity and the folding free energy of Gd-azurin is larger than that of apo azurin.
INTRODUCTION
Azurin from Pseudomona aeruginosa is a small copper protein (128 amino acids) that displays an abnormally large stability. The crystal structure of native azurin as well as that of several mutants, zinc azurin and apo azurin, have been resolved (Baker, 1988; Nar et al., 1991). This protein consists of an α-helix and two β-sheets that form a β-barrel motif. The copper ion is coordinated to the polypeptide chain in five positions and these interactions contribute to the azurin stability (Leckner et al., 1997). The protein displays an intense charge-transfer absorption band with maximum around 625 nm that arises from the bond formed between Cu(II) and Cys-112 (Gilardi et al., 1994). This band probes changes around the metal ion as well as its oxidation state. The presence of the copper ion increases dramatically the folding rate of the macromolecule (Pozdnyakova and Wittung-Stafshede, 2001). Besides the binding site occupied by Cu(II) in native azurin, the presence of another site has been postulated that can accommodate cations such as Hg(II) (Naro et al., 2000). An important contribution to the stability of the macromolecule also comes from a disulfide bond formed between Cys-3 and Cys-26 (Bonander et al., 2000). In fact, the folding energy decreases ∼50% upon replacing Cys-3 and Cys-26 by alanines employing site-directed mutagenesis techniques (Guzzi et al., 1999). Azurin Pae has a single tryptophan (Trp-48) buried in the hydrophobic core of the protein (Hansen et al., 1990). When this environmentally sensitive fluorophore is selectively excited at 295 nm, the maximum of the fluorescence spectrum of native azurin is located around 308 nm, whereas upon denaturation the spectrum red shifts ∼40 nm. The Trp fluorescence yield gauges conformational changes that result upon adding chemical denaturants, or upon changing the pressure, temperature, or pH. In particular, a dramatic increase in the Trp fluorescence quantum yield is observed when the copper ion is removed from the azurin structure, indicating that the copper ion is quenching this residue (Burstein et al., 1977). Although it was thought that Trp was embedded in a rigid environment in the core of the protein, this view changed after several fluorescence decay components were observed (Hutnik and Szabo, 1989a,b).
From the biomedical point of view, azurin Pae has recently attracted intense interest. In particular, Yamada et al. (2002) discovered that azurin enters human cancer cell (melanoma UISO-Mel-2) and induces apoptosis. Particularly strong evidence was provided indicating that the traffic of azurin to the nucleus is mediated by the tumor suppressor protein p53 (Yamada et al., 2002).
To understand the physics that underlies the “anomalously” high thermal stability of azurin Pae, various research approaches have been implemented. Differential scanning calorimetric and electron spin resonance studies indicate that the protein undergoes a reversible denaturation process around 76°C, which could be described using a two-state model, followed by an irreversible process at 82°C (Leckner et al., 1997). Another approach to disentangle the factors that confer the high conformational stability to this family of proteins has been through chemical denaturation protocols. Mei et al. (1999) found, using guanidine hydrochloride (GuHCl), that the denaturation was reversible and measured ΔG0 for the process.
Because numerous spectroscopic tools may be combined to study site-specific changes in azurin, this protein lends itself to investigate local conformational changes that occur upon adding chemical denaturants (or stabilizers), upon thermal denaturation or upon immobilizing the protein in porous materials. The outcome from such studies may be compared with those obtained employing techniques that report on global properties such as differential scanning calorimetry. In this article, we report various experiments aimed at improving the understanding of the factors that contribute to the stability of azurin. In particular, we carried out chemical and thermal denaturation experiments in solution on holo azurin, apo azurin, and gadolinium-azurin and observed in some cases the presence of “intermediate states” between the native and the unfolded forms of azurin. Quenching experiments at different stages of the denaturation using guanidine hydrochloride provided information about the accessibility of the Trp residue. Another aspect of our work was to alter the solvation properties of the protein. To this end, we immobilized azurin in porous sol-gel materials or added glycerol as a chemical stabilizer.
EXPERIMENTAL
Materials
Azurin from Pseudomona aeruginosa (Buchs, Switzerland) was used as received and it was confirmed that the absorbance ratio was A (625 nm) / A (280 nm) ≈ 0.48. Tetraethylorthosilicate (TEOS, 99.99%), GdCl3·7H2O, KCN, guanidine hydrochloride (GuHCl), and the salts to prepare the acetate and phosphate buffers were purchased from Aldrich (Milwaukee, WI). GuHCl was recrystallized following the procedure described by Nozaki (1970). A saturated solution of GuHCl was prepared in water and heated to 40°C to accelerate the dissolution of the solid. Then, the temperature was lowered slowly until the formation of crystals was observed. The crystals were washed with cold water and dissolved in 50 mM phosphate buffer, pH 7. Nozaki reported an equation that relates the concentration of GuHCl with the difference between the refractive index of the GuHCl solution and that of pure water
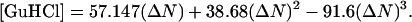 |
Finally, the concentrations of GuHCl were determined from the table of ΔN versus [GuHCl] published by Nozaki (1970). Millipore membranes for ultrafiltration with nominal molecular weight limit (NMWL) of 3000 Da were purchased from Fisher (Cayey, Puerto Rico). All solutions were prepared using fresh deionized water.
Instrumentation
Absorption spectra were collected in a Varian Cary 1 spectrophotometer (Palo Alto, CA). The emission and excitation spectra were recorded using an ISS spectrofluorometer (PC1) (Champaign, IL). The fluorescence lifetime measurements were performed in a PTI Time Master spectroflourometer (Photon Technology International, Lawrenceville, NJ). The light source of this instrument was an N2 laser pumped-dye laser (rhodamine 590), equipped with a frequency doubling crystal to generate light in the 280–295 nm spectral range. The time resolution of the instrument was 20–25 ps/channel.
Preparation of sol-gel monoliths
Sol-gel silica monoliths were prepared following the procedure described by Brenan and co-workers (Zheng et al., 1997). To initiate the hydrolysis of TEOS an acidified aqueous solution was added, but the formation of a new phase was observed. The resulting biphasic sample was sonicated for 1 h to obtain a monophasic sol, which was stored for 7 days at −20°C to obtain TEOS completely hydrolyzed. To form the monoliths, equal volumes of the sol and phosphate buffer solution, pH 7.2 were mixed and transferred to disposable plastic cassettes with a width of 1 mm (Invitrogen, Atascadero, CA). The resulting solution solidifies within 2 min. After rinsing the monoliths with deionized water (without removing them from the cassettes), buffer solution was added to store them for 24 h at 4°C. At the end of this initial aging period, the monoliths were once again rinsed with water and after adding buffer solution again, they were allowed to age for 10 additional days at 4°C. Following this procedure we obtained monoliths with excellent optical and mechanical properties. The aged monoliths were removed from the cassette and cut to fit inside 1-cm quartz cuvettes. Monoliths doped with azurin were prepared adding the protein to the buffer solution to be mixed with the sol.
Preparation of apo azurin
The Cu(II) ion was removed from the protein structure titrating azurin with a solution 0.1 M KCN. Both the azurin and the KCN solutions were prepared in 200 mM phosphate buffer solution, pH 7.4. The KCN solution was added drop by drop until the blue color disappeared. The excess KCN was removed by ultrafiltration employing a chamber with a capacity of 10 mL to which a 3000 NMWL membrane was installed. The concentration of apo azurin was determined from the absorption spectrum and the known absorption extinction coefficient 5930 M−1 cm−1 (Naro et al., 2000). Initially, we followed the dialysis protocol reported by Naro et al. (2000) to remove the metal, but we obtained identical results employing the procedure previously described.
Preparation of Gd-azurin
Gd-azurin was prepared adding a solution of GdCl3 to a solution of apo azurin. The solution of apo azurin was prepared in a 50 mM acetate buffer, pH 6.5. The formation of Gd-azurin was monitored from the increase in the absorbance at 260 nm (Naro et al., 2000). After the titration was completed, the excess Gd3+ was removed by ultrafiltration using a 3000 NMWL membrane.
Chemical denaturation of azurin
Samples of holo and apo azurin were prepared in 50 mM phosphate buffer pH 7.2 and placed in 10-mm quartz cuvettes for spectroscopic measurements. Gd-azurin solution was prepared in acetate buffer pH 6.5. Aliquots of 8.5 M GuHCl in the same buffer were added to the azurin solution and the fluorescence spectra of the sample were collected after each addition of denaturant.
RESULTS AND DISCUSSION
Chemical denaturation
Fig. 1 displays fluorescence spectra of azurin for several concentrations of the chemical denaturating agent GuHCl. The protein concentration was 1.0 × 10−6 M and the pH was adjusted to 7.2 using the phosphate buffer. The sample was excited at 295 nm to observe fluorescence selectively from Trp. As curve a of the inset of Fig. 1 shows, the emission intensity at 308 nm decreases until [GuHCl] ≈ 3.2 M, and at higher concentration of GuHCl the fluorescence signal remains constant. In contrast, the fluorescence intensity at 348 nm (curve b) is zero below [GuHCl] ≈ 3.2 M, but shows an abrupt increase above [GuHCl] = 3.2 M. This means that native azurin never coexists with its denatured form, indicating the presence of an “intermediate state”. Thus, the chemical denaturation of azurin occurs through a two-step mechanism: a), N ↔ I; and b), I ↔ D, where N, I, and D denote the native, “intermediate,” and denaturated states of the protein, respectively.
FIGURE 1.

Fluorescence spectra of azurin free in solution obtained during the chemical denaturation adding GuHCl. The curves correspond to the following molar concentrations of denaturant: (a) 0, (b) 1.42, (c) 2.43, (d) 3.20, (e) 3.60, (f) 3.78, (g) 3.95, and (h) 4.5 M. Several other curves were not presented for clarity. The inset shows denaturation plots for azurin in solution following the (a) 308 nm band and (b) 348 nm bands.
Using fluorescence data presented in the inset of Fig. 1 we calculated the fraction of the protein converted to the “intermediate state”, fI = [I]/[N]. Because fN + fI = 1, the equilibrium constant is given by Keq = fI / fN for a two-state equilibrium, N ↔ I, as a function of [GuHCl]. In this case, fI is measured at 308 nm as a function of [GuHCl]. Likewise, the constant that governs the I ↔ D equilibrium is obtained measuring fD = [D]/[I] at 348 nm as a function of [GuHCl]. Because an equilibrium condition is established after each addition of GuHCl, one can calculate ΔG0 for different [GuHCl]. However, the quantity of interest, which is the Gibbs free energy for the folding process in pure water (denoted 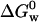 ), is obtained from the intercept of a plot of ΔG0 versus [GuHCl]. Following this procedure we calculated
), is obtained from the intercept of a plot of ΔG0 versus [GuHCl]. Following this procedure we calculated 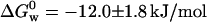 for the N ↔ I equilibrium, and
for the N ↔ I equilibrium, and 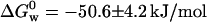 for the I ↔ D equilibrium. Leckner et al. reported the free energy measured at 348 nm to be
for the I ↔ D equilibrium. Leckner et al. reported the free energy measured at 348 nm to be 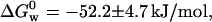 which is in good agreement with our value (Leckner et al., 1997). However, we argue that the Gibbs free energy for the overall folding process is the sum of the free energies for the two consecutive steps,
which is in good agreement with our value (Leckner et al., 1997). However, we argue that the Gibbs free energy for the overall folding process is the sum of the free energies for the two consecutive steps, 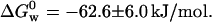 This line of reasoning is supported by the close agreement between our value of
This line of reasoning is supported by the close agreement between our value of 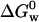 and the one measured by La Rosa and co-workers employing the microcalorimetry technique,
and the one measured by La Rosa and co-workers employing the microcalorimetry technique, 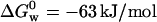 (La Rosa et al., 1995). Our findings indicate that at low concentrations of GuHCl (i.e., [GuHCl < 3.2 M), Trp probes a conformational change that occurs without exposing the core of the protein to the solvent. The electronic energy of Trp is efficiently quenched at the end of the first stage of the denaturation process, namely, when the N ↔ I equilibrium has been shifted to the right. In contrast, during the second stage of the denaturation, the core of the macromolecule is being exposed to the solvent (water containing GuHCl and the buffer salts) and this is confirmed by the red shift of ≈40 nm on the maximum of the Trp fluorescence band. The observation that above [GuHCl] ≈ 3.2 M the Trp fluorescence at 348 nm increases encompasses two opposite effects. The fluorescence yield of Trp should decrease as the fraction of this residue exposed to the solvent increases, but should increase if the interaction that quenches its energy in the first stage of the denaturation disappears (i.e., [GuHCl] < 3.2 M). Clearly, the latter effect is the dominant.
(La Rosa et al., 1995). Our findings indicate that at low concentrations of GuHCl (i.e., [GuHCl < 3.2 M), Trp probes a conformational change that occurs without exposing the core of the protein to the solvent. The electronic energy of Trp is efficiently quenched at the end of the first stage of the denaturation process, namely, when the N ↔ I equilibrium has been shifted to the right. In contrast, during the second stage of the denaturation, the core of the macromolecule is being exposed to the solvent (water containing GuHCl and the buffer salts) and this is confirmed by the red shift of ≈40 nm on the maximum of the Trp fluorescence band. The observation that above [GuHCl] ≈ 3.2 M the Trp fluorescence at 348 nm increases encompasses two opposite effects. The fluorescence yield of Trp should decrease as the fraction of this residue exposed to the solvent increases, but should increase if the interaction that quenches its energy in the first stage of the denaturation disappears (i.e., [GuHCl] < 3.2 M). Clearly, the latter effect is the dominant.
We also measured the folding Gibbs free energy for azurin monitoring the disappearance of the charge-transfer absorption at 625 nm as a function of [GuHCl] to be 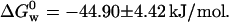 That we obtain different values of
That we obtain different values of 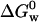 depending on whether the denaturation of azurin is measured monitoring fluorescence from Trp or absorbance on the charge-transfer band calls for an explanation. An important spectroscopic observation is that when the fluorescence intensity at 308 nm has reached its minimum, the absorption band at 625 nm is still present, meaning that these two measurements are not correlated. These observations stress the fact that the Trp relative fluorescence yield and the absorbance spectra on the ligand-to-metal transition gauge different local conformational changes.
depending on whether the denaturation of azurin is measured monitoring fluorescence from Trp or absorbance on the charge-transfer band calls for an explanation. An important spectroscopic observation is that when the fluorescence intensity at 308 nm has reached its minimum, the absorption band at 625 nm is still present, meaning that these two measurements are not correlated. These observations stress the fact that the Trp relative fluorescence yield and the absorbance spectra on the ligand-to-metal transition gauge different local conformational changes.
We measured 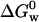 for apo azurin to be −27.9 ± 4.2 kJ/mol. Our value of
for apo azurin to be −27.9 ± 4.2 kJ/mol. Our value of 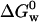 for apo (Table 1) is similar to that obtained by Pozdnyakova et al.,
for apo (Table 1) is similar to that obtained by Pozdnyakova et al., 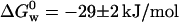 (Pozdnyakova et al., 2001). Note that the stability is ∼50% smaller than that of holo azurin. As opposed to holo azurin, when apo azurin is examined the band at 348 nm never grows up.
(Pozdnyakova et al., 2001). Note that the stability is ∼50% smaller than that of holo azurin. As opposed to holo azurin, when apo azurin is examined the band at 348 nm never grows up.
TABLE 1.
Folding Gibbs free energies for azurin and derivatives
| ΔG (308 nm) kJ/mol | ΔG (348 nm) kJ/mol | ΔG total kJ/mol | ||
|---|---|---|---|---|
| Azurin (native) | −12.0 ± 1.8 | −50.6 ± 4.2 | −62.2 ± 6.0 | This work |
| – | −52 ± 4.7 | −52 ± 4.7 | * | |
| Apo azurin | – | −27.9 ± 4.2 | – | This work |
| – | −29 ± 2.3 | – | * | |
| Azurin in sol-gel | −5.6 ± 0.3 | −36.4 ± 0.3 | −42.0 ± 0.6 | This work |
| Azurin/15% glycerol | −8.1 ± 1.4 | −55.8 ± 1.0 | −71 ± 15. | This work |
| −63.2 ± 14.4 | ||||
| Gd-azurin | −54.3 ± 2.6 | – | – | This work |
We carried out denaturation experiments on the protein immobilized in silica sol-gels similar to those previously described for the free protein. In the absence of GuHCl, the fluorescence spectrum displayed by the entrapped protein was identical to that of the native protein free in solution. As can be appreciated from Fig. 2 B, the GuHCl-induced unfolding occurs at a lower concentration (fD = 0.5 at [GuHCl] = 3.0 M) than for the protein free in solution, meaning that its chemical stability has decreased. As for the protein free in solution, the intensity of the fluorescence band with maximum at 308 nm decreases to an “intermediate state”, then shifts to 348 nm, and finally the fluorescence band at 348 nm begins to grow with [GuHCl]. The free-energy change associated with the overall folding process, 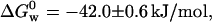 was obtained adding the free energies measured at 308, ΔG0 = −5.6 ± 0.3 and at 348 nm, ΔG0 = −36.4 ± 0.3 kJ/mol. Thus, the immobilized protein is 33% less stable than azurin free in solution. The decrease in the stability may arise from changes in the solvation shell of the protein. It is also feasible that the entrapment may favor conformation states different to those populated when the protein is free in solution.
was obtained adding the free energies measured at 308, ΔG0 = −5.6 ± 0.3 and at 348 nm, ΔG0 = −36.4 ± 0.3 kJ/mol. Thus, the immobilized protein is 33% less stable than azurin free in solution. The decrease in the stability may arise from changes in the solvation shell of the protein. It is also feasible that the entrapment may favor conformation states different to those populated when the protein is free in solution.
FIGURE 2.

Denaturation of azurin adding GuHCl monitoring the relative fluorescence yield of tryptophan: (A) the curve of azurin free in solution monitored at 308 different probes the N ↔ I (○), whereas the curve at 348 nm probes the I ↔ D; (B) denaturation of azurin immobilized in a sol-gel monolith followed at 308 and 348 nm. The denaturation followed at 348 nm occurs at lower [GuHCl] than free in solution indicating a decrease in the stability of protein; (C) in the presence of glycerol the curve at 348 nm shows an increase in the stability of the macromolecule, whereas the curve at 308 nm shows a plateau, indicating the presence of an intermediate conformation; and (D) denaturation curve for gadolinium azurin followed at 308 nm.
According to the work of Friedman and co-workers it is possible to mechanically alter the distribution of vibrational modes accessible to proteins immobilized in sol-gel materials prepared using protocols that involve the addition of glycerol (Gottfried et al., 1999; Juszczak and Friedman, 1999; Khan et al., 2000; Samuni et al., 2000). In particular, we employed one of Friedman's protocols, which involves the addition of 15% glycerol. Using this procedure we observed a marked increased in the stability of the protein (data not shown) (fD = 0.5 at [GuHCl] ≈ 4 M) as compared to the protein immobilized using Brenan's protocol (Fig. 2 B) (Zheng et al., 1997). However, the stability of the protein immobilized using Friedman's method is comparable to that of the protein free in solution, 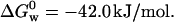
As a control experiment, we carried out a denaturation experiment for the protein free in solution, in the presence of 15% glycerol. It is clear from Fig. 2 C (refer to the curve that corresponds to the measurement at 348 nm) that azurin is more stable in the presence of glycerol than free in solution. On the other hand, a plateau is observed monitoring the process at 308 nm in the 1.5–2.8 M GuHCl concentration range, which indicates the presence of an intermediate state not observed in the absence of glycerol. We determined the free energies for the process that occurs in the 0–2.5 M GuHCl concentration range, 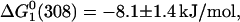 and that for the process that occurs beyond [GuHCl] = 2.5 M,
and that for the process that occurs beyond [GuHCl] = 2.5 M, 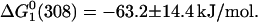 Thus, the free energy for the overall folding process is −71.3 ± 15.8 kJ/mol, which represents a 15% increase in the stability with respect to the protein free in solution. This is the anticipated result because glycerol stabilizes the folded state of proteins (Schellman, 2003; Tanford, 1970) whereas GuHCl stabilizes the unfolded states (Winkler et al., 1997). On the other hand, from the increase of the band at 348 nm we obtained,
Thus, the free energy for the overall folding process is −71.3 ± 15.8 kJ/mol, which represents a 15% increase in the stability with respect to the protein free in solution. This is the anticipated result because glycerol stabilizes the folded state of proteins (Schellman, 2003; Tanford, 1970) whereas GuHCl stabilizes the unfolded states (Winkler et al., 1997). On the other hand, from the increase of the band at 348 nm we obtained, 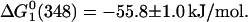
Our results should be put in the context of previous work in this area. Samuni et al. observed that myoglobin (Mb), at low pH and free in solution, tends to loose the heme group, whereas the immobilized protein did not (Samuni et al., 2000). Even though these authors provided strong evidence indicating that the individual helices do unfold, they attributed the retention of the heme group to the spatial constraint that the sol-gel matrix imposes on the protein. This group also observed several intermediate states during the denaturation of the immobilized protein, which they attributed to the combination of effects such as the slowing down of the unfolding kinetics, decrease in the pKa of unfolding, and changes in the kinetics at which the solvation shell around the protein may be changed. On the other hand, Eggers and Valentine presented evidence that apomyoglobin was partially denaturated upon immobilization (Eggers and Valentine, 2001). The circular dichroism spectra recorded by these authors lead them to conclude that the conformational changes presented by apomyoglobin upon encapsulation are due to the unique properties of water confined inside the interstices of the silica sol-gel. Since we performed fluorimetric studies on azurin, we were not able to detect changes in the secondary structure upon encapsulation. This would have been possible using the Raman technique as in Samuni et al. (2000) experiments or circular dichroism spectroscopy as in the work of Eggers and Valentine (2001). However, the decrease in the Trp fluorescence that we observed upon adding GuHCl should be due to changes in the secondary structure of azurin. The fact that we observed a decrease in the stability of azurin immobilized in a sol-gel as compared to the protein free in solution, suggests the possibility that azurin may be undergoing conformational changes when it is immobilized in sol-gel monoliths.
Fluorescence lifetimes
We measured the fluorescence lifetime of azurin at different stages of the chemical denaturation with GuHCl. Even though the fluorescence lifetime of native azurin has been reported in the literature, we measured it again to check our procedures. Table 2 lists fluorescence lifetimes of azurin at different stages of the denaturation process, as well as that of apoazurin and Gd-azurin free in solution. Fig. 3 shows how the data and the fits of the decays look like for denaturated azurin. The first two entries of Table 2 compare our results for native holo azurin with the corresponding one reported in the literature (Hutnik and Szabo, 1989b; Hansen et al., 1990). The decay curves for native and denaturated holo azurin were fitted to three exponential decays, whereas apo azurin required only one component. The fluorescence decay time of denaturated azurin is shorter than that of the native form. This decrease in the fluorescence lifetime may be ascribed to the increased accessibility of Trp to the solvent as well as to the increased mobility of the fluorophore. The lifetime of the denatured azurin, 2.25 ± 0.15 ns, is close to the value obtained for tryptophan free in solution, 2.8 ± 0.2 ns.
TABLE 2.
Fluorescence lifetimes for azurin and derivatives in solution
| α1 | τ1 (ns) | α2 | τ2(ns) | α3 | τ3(ns) | χ2 | ||
|---|---|---|---|---|---|---|---|---|
| Azurin native | 0.11 | 4.86 ± 0.33 | 0.21 | 0.61 ± 0.21 | 0.68 | 0.16 ± 0.1 | 0.98 | This work |
| Azurin native | 0.02 | 4.80 | 0.05 | 0.36 | 0.92 | 0.097 | – | * |
| Azurin denatured | 0.18 | 2.25 ± 0.15 | 0.22 | 0.54 ± 0.07 | 0.60 | 0.06 ± 0.02 | 0.95 | This work |
| Apoazurin | 1 | 5.08 | – | – | – | – | – | * |
| Apoazurin | 0.61 | 4.71 | 0.12 | 1.31 | 0.16 | 0.13 | – | † |
| Gd-azurin | 0.87 | 4.29 ± 0.06 | 0.17 | 0.06 ± 0.02 | – | – | 1.1 | This work |
| Hg-azurin | 0.05 | 3.20 | 0.95 | 0.38 | – | – | – | ‡ |
| Co-azurin | 0.26 | 4.70 | 0.74 | 0.85 | – | – | – | ‡ |
| Ni-azurin | 0.21 | 4.56 | 0.79 | 0.104 | – | – | – | ‡ |
| Zn-Azurin | 1.0 | 4.34 | – | – | – | – | – | ‡ |
FIGURE 3.

Fluorescence decay of azurin chemically denatured free in solution. The curve was fitted to three exponential decay components, τ1 = 2.25 ns, τ2 = 0.54 ns, and τ3 = 0.06 ns.
The lifetime of Gd-azurin was fitted to two exponential decay components. The magnitude of the longer lifetime is in the 4.3–4.7-ns range, which is shorter than that of Cu(II)-azurin. The decays of Ni(II)-azurin and the rest of the divalent metals listed at the end of Table 2 also consists of two decay components (see Table 2). These results tend to indicate that Gd(III) is occupying the same site as these ions.
Fluorescence quenching at different stages of the chemical denaturation process
To obtain information about the accessibility of the single Trp of azurin to the bulk solution, we performed steady-state and real-time fluorescence experiments on the native form, on the “intermediate state” and on the unfolded form. Stern-Volmer plots were constructed employing acrylamide as a fluorescence quencher. Fig. 4, line a, corresponds to the experiment carried out on the native form of azurin (i.e., no guanidine present in the solution). The slope of the SV plot equals KSV = 0.40 M−1. For the native protein the real-time fluorescence decay was adjusted to three exponential decays and the longer decay component, τ1 = 4.86 ns (see Table 2), was combined with KSV to obtain the quenching constant, kQ = 8.2 × 107 M−1 s−1. We choose the long-lived component because it is the only one involved in diffusion controlled processes.
FIGURE 4.

Stern-Volmer plots obtained adding acrylamide as a fluorescence quencher to: (a) native azurin, (b) the intermediate state, and (c) chemically denatured azurin. The different slopes reflect the different accessibilities of Trp to the quencher.
Fig. 4, line b, corresponds to the case when [GuHCl] = 3.2 M, which is the concentration at which the emission at 308 nm no longer decreases, and the native form of azurin has been converted into the “intermediate state”. From this plot we obtained KSV = 4.4 M−1. Due to the smallness of the signal/noise ratio at this point of the titration, we were unable to measure reliable fluorescence decay times to report the bimolecular quenching constant of the “intermediate state”. Fig. 4, line c, displays a quenching plot in the presence of [GuHCl] = 4.0 M, which is the minimum concentration necessary to form exclusively the unfolded form of the protein. At this point, Trp emits at 348 nm, indicating that this fluorophore is exposed to the solvent, and has reached its maximum intensity. Following the procedure outlined for the native form, we calculated the quenching constant for the unfolded protein to be kQ = 3.1 × 109 M−1 s−1, combining KSV = 7.0 M−1 and τ1 = 2.25 ns. The fact that the magnitude of the quenching constant increases a factor of 30 upon denaturating the protein further confirms that Trp is more accessible to the quencher as expected for a denaturated protein. It is instructive to compare the magnitude of kQ for the unfolded protein with that of Trp free in solution. The literature value for the quenching constant of free Trp is kQ = 5.9 × 109 M−1 s−1, which corresponds to an efficiency close to unity. Even though Trp in the unfolded protein is fully exposed to the bulk solution, the magnitude of kQ is 1.9 times smaller than that of Trp free in solution because its diffusion coefficient is larger.
Thermal denaturation
In this section we examine the temperature behavior of fD for holo and apo azurin monitoring fluorescence from Trp and absorption on the charge-transfer transition at 625 nm. For holo azurin, the Trp emission at 308 nm decreases upon increasing the temperature from 20 to 45°C. Above 45°C, the maximum of the band red shifts to 348 nm and the intensity begins to increase (data not shown). Fig. 5 depicts the fraction of the denaturated protein, monitored at 348 nm, as a function of temperature. The curve is sigmoid and half of the protein is denaturated (i.e., fD = 0.5) at 83.8°C, but the process is irreversible. On the other hand, measuring fD after the absorption of the charge-transfer transition as a function of temperature we obtained Td = 76°C. The phase change monitored after the charge-transfer (CT) absorption is completed at 80°C, which coincides with the temperature at which the fluorescence intensity at 348 nm begins to increase. Td obtained after the absorption at 625 nm is the same value reported by la Rosa and co-workers obtained using the microcalorimetry technique (La Rosa et al., 1995). These authors demonstrated, using electron paramagnetic resonance and microcalorimetry techniques, that >76°C the process is irreversible. Thus, we did not attempt to derive thermodynamic data from this experiment. On the other hand, the denaturation of apo azurin followed at 348 nm (Fig. 5) shows a hump around 65°C. This behavior may be due to a transition in the secondary structure of the protein. The experiment with apo azurin clearly demonstrates that the metal confers considerable stability to the protein because Td is much lower. In addition, the metal prevents changes in the secondary structure as the denaturation curve monitored at 348 nm is sigmoid (i.e., it does not show humps).
FIGURE 5.

Thermal denaturation curves for holo azurin monitored by fluorescence at 348 nm (Holo Az) and by the absorption band at 625 nm (Holo CT band) and for apo azurin. The temperature was increased at a rate of 0.5°C/min. The denaturation temperature measured for holo azurin monitoring fluorescence from Trp is larger than that obtained measuring the decrease in the CT band at 628 nm, indicating that each technique probes different local conformational changes. The apo azurin denaturation curve presents a hump in the 50–70°C temperature interval and the slope where the unfolding occurs is more abrupt than for holo azurin.
Properties of gadolinium-azurin
The inclusion of different metals in the copper active site of azurin has been studied to understand the interactions of the protein with metals. Foreign metal such as Ag(I), Co(II), Hg(II), and Zn(II) have been incorporated into the structure (Naro et al., 2000) of the protein. Those studies suggested the presence of a second, smaller metal binding site. The observation that the fluorescence yield of Trp increases when the Cu(II) ion is removed from the structure indicates that the metal is quenching Trp. This suggests that the Cu(II) ion may interact with electronically excited Trp partially quenching its fluorescence.
The fluorescence maximum of native Gd-azurin lies at 308 nm, as for holo and apo azurins, as Fig. 2 D displays. Following this band we carried out a denaturation experiment similar to those previously described for azurin and apo azurin. This data allowed us to calculate 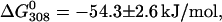 which means that Gd-azurin is 15% less stable than holo azurin, but ∼50% more stable than apo azurin. Even though the fluorescence band at 308 nm decreases upon adding GuHCl, it never shifts to 348 nm, and at the end of the denaturation experiment the fluorescence is almost completely quenched. We studied the possibility that Gd(III) may be quenching Trp in the protein. To this end, we monitored fluorescence from free Trp as function [Gd(III)], but within the experimental error, no quenching was observed. This indicates that in the protein the metal is orchestrating the interaction between Trp and other residues, through which the electronic energy is quenched. In contrast, when Cu(II)-azurin is denaturated the emission shifts to 348 nm and the protein shows a fluorescence yield larger than that of the native form (Mei et al., 1999). For Gd-azurin the plot of fD against [GuHCl] begins to increase from [GuHCl] = 1.0 M and keeps increasing monotonically up to ∼3.5 M.
which means that Gd-azurin is 15% less stable than holo azurin, but ∼50% more stable than apo azurin. Even though the fluorescence band at 308 nm decreases upon adding GuHCl, it never shifts to 348 nm, and at the end of the denaturation experiment the fluorescence is almost completely quenched. We studied the possibility that Gd(III) may be quenching Trp in the protein. To this end, we monitored fluorescence from free Trp as function [Gd(III)], but within the experimental error, no quenching was observed. This indicates that in the protein the metal is orchestrating the interaction between Trp and other residues, through which the electronic energy is quenched. In contrast, when Cu(II)-azurin is denaturated the emission shifts to 348 nm and the protein shows a fluorescence yield larger than that of the native form (Mei et al., 1999). For Gd-azurin the plot of fD against [GuHCl] begins to increase from [GuHCl] = 1.0 M and keeps increasing monotonically up to ∼3.5 M.
We prepared azurin substituted with the Gd(III) ion, which is a paramagnetic metal, adding GdCl3 to a solution of apo azurin. The pH was adjusted to 6.5 using the acetate buffer. As Fig. 6 shows, the absorbance below 290 nm increases upon adding the Gd(III) ion. This behavior is characteristic of metals that occupy site B of the protein (Naro et al., 2000). The inset of Fig. 6 displays the absorbance measured at 260 nm versus [GuHCl]. The curve reaches a plateau meaning that all the apo azurin is converted into Gd-azurin. Monitoring changes in absorbance at 280 nm we calculated the binding constant of gadolinium to apo azurin, Gd(III) + apo azurin = Gd-azurin. A plot of log{(A − Ao)/(AGd − A)} against log[Gd3+], gives a straight line that corresponds to log Keq (El Baraka et al., 1994). The equilibrium constant was estimated to be Keq = 3.31 × 103 M−1, which in turn allowed us to estimate ΔG0 ≈ −20 kJ/mol, indicating that the metal gives stability to the structure. Because we found that the folding energies of Gd-azurin and apo azurin are −53.4 ± 2.6 and 27.9 ± 4.2 kJ/mol, within the experimental error, the free energy estimated for the Gd binding reaction accounts for the difference in energy between Gd-azurin and apo azurin.
FIGURE 6.

Formation of gadolinium azurin upon adding Gd(III) to apo azurin in solution. The increase in absorbance below 290 nm is characteristic of ions that occupy site B in the protein. The inset displays the dependence of the absorbance at 260 nm with gadolinium concentration.
Further evidence of the Gd(III) binding comes from fluorescence lifetime measurement. Unlike the equilibrium Cu(II) + apo azurin ↔ Cu(II)-azurin, the equilibrium between apo azurin and Gd(III) is reached very fast and for this reason we were able to measure Keq. Even though the absorption spectrum of azurin changes upon adding Gd(III), neither changes are observed in the shape of the fluorescence spectra nor in the fluorescence yield. In contrast, the fluorescence yield of apo azurin is reduced 20-fold upon adding Cu(II), which indicates that the Cu(II) ion quenches the Trp fluorescence.
CONCLUDING REMARKS
The Gibbs free energies associated to the folding of holo azurin, calculated through chemical denaturation experiments monitoring Trp fluorescence and changes in absorbance on the ligand-to-metal band are different, the reason being that these techniques probe their local environment and report on different conformational changes. Close examination to the chemical denaturation process monitored using fluorescence techniques reveals that the denaturation occurs via an “intermediate” state. The agreement between the data obtained in our laboratory employing spectroscopic techniques and the calorimetric data available from the literature further supports the presence of an intermediate state. The immobilization of the protein in sol-gel materials lowers its stability, possibly by altering the solvation properties of the protein. Upon denaturation, the magnitude of the quenching constant of the single tryptophan of azurin by acrylamide increases by a factor of 30, which reflects that this residue is more accessible to the quencher, as anticipated. Thermal denaturation experiments performed increasing the temperature at 0.5°C/min monitoring fluorescence from Trp and absorbance on the charge-transfer band display different denaturation temperatures, again indicating that different conformational changes occur at different stages of the denaturation. The thermal denaturation of azurin immobilized in a sol-gel is irreversible as is also the case for the free protein. It follows that this experiment rules out the aggregation of the protein to account for the irreversibility of the process. The thermal denaturation curve for apo azurin is not sigmoid as that of holo azurin. Instead, the curve exhibits a hump, once again reflecting the important role played by the Cu(II) ion in providing stability of the macromolecule. The Gd(III) ion accommodates in site B of azurin with high affinity, but this paramagnetic ion does not quench the Trp fluorescence. The folding free energy of Gd-azurin is higher than that of apo azurin indicating that this metal ion increases the stability of the protein, although not as much as the Cu(II) ion.
Acknowledgments
The authors acknowledge the generous support provided by the National Institutes of Health SCORE/RISE Program.
References
- Baker, E. N. 1988. Structure of azurin from Alcaligenes denitrificans refinement at 1.8 A resolution and comparison of the two crystallographically independent molecules. J. Mol. Biol. 203:1071–1095. [DOI] [PubMed] [Google Scholar]
- Bonander, N., J. Leckner, H. Guo, B. G. Karlsson, and L. Sjölin. 2000. Crystal structure of the disulfite bond-deficient azurin mutant C3A/C26A. How important is the S-S bond for folding and stability? Eur. J. Biochem. 267:4511–4519. [DOI] [PubMed] [Google Scholar]
- Burstein, E. A., E. A. Permyakov, V. A. Yashin, S. A. Burkhanov, and A. Finazzi-Agro. 1977. The fine structure of luminescence spectra of azurin. Biochim. Biophys. Acta. 491:155–159. [DOI] [PubMed] [Google Scholar]
- Eggers, D. K., and J. S. Valentine. 2001. Molecular confinement influences protein structure and enhances thermal protein stability. Protein Sci. 10:250–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Baraka, M., R. García, and E. Quiñones. 1994. A study of the inclusion complexes of beta-cyclodextrin with three electronic states of 4-(N,N-dimethylamino)benzonitrile. J. Photochem. Photobiol. A-Chem. 79:181–187. [Google Scholar]
- Gilardi, G., G. Mei, N. Rosato, G. W. Canters, and A. Finazzi-Agro. 1994. Unique environment of Trp48 in Pseudomonas aeruginosa azurin as probed by site-directed mutagenesis and dynamic fluorescence spectroscopy. Biochemistry. 33:1425–1432. [DOI] [PubMed] [Google Scholar]
- Gottfried, D. S., A. Kagan, B. M. Hoffman, and J. M. Friedman. 1999. Impeded rotation of a protein in a sol-gel matrix. J. Phys. Chem. B. 103:2803–2807. [Google Scholar]
- Guzzi, R., L. Sportelli, C. La Rosa, D. Milardi, D. Grasso, M. P. Verbeet, and G. W. Canters. 1999. A spectroscopic and calorimetric investigation on the thermal stability of the Cys3Ala/Cys26Ala azurin mutant. Biophys. J. 77:1052–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, J. E., J. W. Longworth, and G. R. Fleming. 1990. Photophysics of metalloazurins. Biochemistry. 29:7329–7338. [DOI] [PubMed] [Google Scholar]
- Hutnik, C. M., and A. G. Szabo. 1989a. Confirmation that multiexponential fluorescence decay behavior of holoazurin originates from conformational heterogeneity. Biochemistry. 28:3923–3934. [DOI] [PubMed] [Google Scholar]
- Hutnik, C. M., and A. G. Szabo. 1989b. A time-resolved fluorescence study of azurin and metalloazurin derivatives. Biochemistry. 28:3935–3939. [DOI] [PubMed] [Google Scholar]
- Juszczak, L. J., and J. M. Friedman. 1999. UV resonance raman spectra of ligand binding intermediates of sol-gel encapsulated hemoglobin. J. Biol. Chem. 274:30357–30360. [DOI] [PubMed] [Google Scholar]
- Khan, I., C. F. Shannon, D. Dantsker, A. J. Friedman, J. Perez-Gonzalez-de-Apodaca, and J. M. Friedman. 2000. Sol-gel trapping of functional intermediates of hemoglobin: geminate and bimolecular recombination studies. Biochemistry. 39:16099–16109. [DOI] [PubMed] [Google Scholar]
- Kroes, S. J., G. W. Canters, G. Gilardi, A. van Hoek, and A. J. W. G. Visser. 1998. Time-resolved fluorescence study of azurin variants: conformational hetorogeneity and tryptophan mobility. Biophys. J. 75:2441–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rosa, C., D. Milardi, D. Grasso, R. Guzzi, and L. Sportelli. 1995. Thermodynamics of the thermal unfolding of azurin. J. Phys. Chem. A. 99:14864–14870. [Google Scholar]
- Leckner, J., N. Bonander, P. Wittung-Stafshede, B. G. Malmstrom, and B. G. Karlsson. 1997. The effect of the metal ion on the folding energetics of azurin: a comparison of the native, zinc and apoprotein. Biochem. Biophys. Acta. 1342:19–27. [DOI] [PubMed] [Google Scholar]
- Mei, G., A. Di Venre, F.F. Malvezzi, G. Gilardi, N. Rosato, F. De Matteis, and A. Finnazzi-Agro. 1999. The effect of pressure and guanidine hydrochloride on azurins mutated in the hydrophobic core. Eur. J. Biochem. 265:619–626. [DOI] [PubMed] [Google Scholar]
- Nar, H., A. Messerschmidt, R. Huber, M. van de Kamp, and G. W. Canters. 1991. Crystal structure analysis of oxidized Pseudomonas aeruginosa azurin at pH 5.5 and pH 9.0. A pH-induced conformational transition involves a peptide bond flip. J. Mol. Biol. 221:765–772. [DOI] [PubMed] [Google Scholar]
- Naro, F., M. G. Tordi, G. M. Giacometti, F. Tomei, A. M. Timerio, and L. Zolla. 2000. Metal binding to Pseudomonas aeruginosa azurin: a kinetic investigation. Z. Naturforsch. 55:347–354. [DOI] [PubMed] [Google Scholar]
- Nozaki, Y. 1970. The preparation of guanidine hydrochloride. Methods Enzymol. 26:43–50. [DOI] [PubMed] [Google Scholar]
- Pozdnyakova, I., J. Guidry, and P. Wittung-Stafshede. 2001. Copper stabilizes azurin by decreasing the unfolding rate. Arch. Biochem. Biophys. 390:146–148. [DOI] [PubMed] [Google Scholar]
- Pozdnyakova, I., and P. Wittung-Stafshede. 2001. Biological relevance of metal binding before protein folding. J. Am. Chem. Soc. 123:10135–10136. [DOI] [PubMed] [Google Scholar]
- Samuni, U., M. S. Navati, L. J. Juszczak, D. Dantsker, M. Yang, and J. M. Friedman. 2000. Unfolding and refolding of sol-gel encapsulated carbonmonoxymyoglobin: an orchestrated spectroscopic study of intermediates and kinetics? J. Phys. Chem. B. 104:10802–10813. [Google Scholar]
- Schellman, J. A. 2003. Protein stability in mixed solvents: a balance of contact interaction and exclude volume. Biophys. J. 85:108–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanford, C. 1970. Protein denaturation. C. Theoretical models for the mechanism of denaturation. Adv. Protein Chem. 24:1–95. [PubMed] [Google Scholar]
- Winkler, J. R., P. Wittung-Stafshede, J. Leckner, B. G. Malmstrom, and H. B. Gray. 1997. Effects of folding on metalloprotein active sites. Proc. Natl. Acad. Sci. USA. 94:4246–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada, T., M. Goto, V. Punj, O. Zaborina, M. L. Chen, K. Kimbara, D. Majumdar, E. Cunningham, T. K. Das Gupta, and A. M. Chakrabarty. 2002. Bacterial redox protein azurin, tumor suppressor protein p53, and regression of cancer. Proc. Natl. Acad. Sci. U.S.A. 99:14098–14103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, L., W. R. Reid, and J. D. Brenan. 1997. Measurement of fluorescence from tryptophan to protein doped sol-gel-derived glass monoliths. Anal. Chem. 69:3940–3949. [DOI] [PubMed] [Google Scholar]