

Abstract
We report on single-molecule fluorescence measurements performed on the phycobiliprotein allophycocyanin (APC). Our data support the presence of a unidirectional Förster-type energy transfer process involving spectrally different chromophores, α84 (donor) and β84 (acceptor), as well as of energy hopping amongst β84 chromophores. Single-molecule fluorescence spectra recorded from individual immobilized APC proteins indicate the presence of a red-emitting chromophore with emission peaking at 660 nm, which we connect with β84, and a species with the emission peak blue shifted at 630 nm, which we attribute to α84. Polarization data from single APC trimers point to the presence of three consecutive red emitters, suggesting energy hopping amongst β84 chromophores. Based on the single-molecule fluorescence spectra and assuming that emission at the ensemble level in solution comes mainly from the acceptor chromophore, we were able to resolve the individual absorption and emission spectra of the α84 and β84 chromophores in APC.
INTRODUCTION
Room-temperature single-molecule fluorescence detection (SMD) became in the past decade a powerful tool to monitor the dynamic processes in individual biomolecules (Xie, 1996; Lu et al., 1998; Weiss, 2000; Cotlet et al., 2001a; Yang et al., 2003; Zehetmeyer et al., 2003). Probing individual biomolecules via their spectroscopic properties removes the inherent averaging present in ensemble experiments, thus making SMD the method of choice to yield information at the most detailed level. Static and dynamic disorder, that is, the distribution of a molecular parameter within a population of single biomolecules and the time-dependent fluctuation of a parameter for an individual biomolecule within the probed population can now be accessed with SMD. Phenomena not necessarily predictable by ensemble experiments, such as fluctuations in the fluorescence intensity, lifetime, or emission maximum, can be detected by SMD.
Here we report on single-molecule fluorescence experiments performed on allophycocyanin trimers. Cyanobacteria are lower organisms containing phycobilisomes as light-harvesting complexes, i.e., supramolecular aggregates attached on the outer surface of the photosynthetic membrane. They absorb visible light, from 450 to 650 nm and transfer the excitation energy to the reaction center where it is converted to chemical energy via fast electron transfer. Phycobilisomes consist of ordered assemblies of several phycobiliproteins, like phycoerythrins, phycoerythrocyanins, and allophycocyanins, and are organized in rod-like structures with phycoerythrins at the outer ends of the rods, phycoerythrocyanins in the middle, and allophycocyanins in the center, that is, in close proximity to the reaction center. The excitation energy collected upon the absorption of a photon is transferred to the reaction center with an efficiency approaching unity (Van Grondelle, 1985). Allophycocyanin (APC) is a disc-like, trimeric protein with a radius of 11 nm, a thickness of roughly 3 nm and a central channel of 3.5 nm (Brejc et al., 1995). Each monomer contains two phycocyanobilin chromophores, the α84 and the β84 chromophores (Fig. 1 A). Shown in Fig. 1 B is the chemical structure of the phycocyanobilin chromophore.
FIGURE 1.

(A) Schematic structure of APC as it was obtained by means of x-ray crystallography. The chromophores are arranged in three pairs. (B) Chemical structure of the phycocyanobilin chromophore. The individual rings have been named from A to D.
Within a trimer, two chromophores of the neighboring monomers approach each other to a distance of 2.1 nm whereas the distance between chromophores within the same monomer is 5.1 nm (Fig. 1 A). It has been shown that the chromophores are packed rigidly in their respective binding pockets (Brookhaven Protein Data Bank file entry number: 1ALL; Brejc et al., 1995).
There is still an intense debate on the nature of the interaction between close-lying α84 and β84 chromophores in APC which determines the shape of the absorption spectrum (Fig. 2 B, solid line). The absorption spectrum of the APC trimer differs significantly from that of the monomer. Whereas the monomer has a broad and structureless absorption spectrum with a maximum at 615 nm, the APC trimer has a red-shifted and more structured absorption spectrum with a broad shoulder at 632 nm and a sharp peak at 650 nm (MacColl et al., 1981). The change in the absorption spectrum upon going from monomers to trimers is obviously due to interacting chromophore pairs, but the precise nature of this interaction is still a matter of debate. Most of the spectroscopic reports on the APC trimer indicate strong coupling between α84 and β84 chromophores within a pair, hence resulting in a split of the visible absorption spectrum in upper and lower exciton bands. The shape of the circular dichroism (CD) spectrum has been interpreted in terms of the exciton coupling model. However, it is argued that a model where there is a very fast energy transfer from donor to acceptor chromophores can also explain the shape of the CD spectrum (MacColl et al., 1980, Csatorday et al., 1984). Two-color femtosecond pump-probe anisotropy experiments on allophycocyanin trimers have shown a fast femtosecond component, indicating that there initially is a coherently excited chromophore pair. The slower components that were found reflect the dephasing process, which results in a localization of the excitation energy on a chromophore of a weakly coupled chromophore pair. The timescale of the localization was found to be in agreement with a Förster type mechanism (Beck and Sauer, 1992; Edington et al., 1995). Single-molecule experiments on APCs immobilized on glass cover slides have shown three-step photobleaching behavior of fluorescence transients. Each level in the fluorescence intensity was attributed to the emission of a strongly coupled pair (Ying and Xie, 1998). In the assumption of an intermediate-to-strong coupling interaction between α84 and β84 chromophores within a pair, the 654- and 632-nm absorption peaks were assigned to the lower and higher excitonic bands, respectively (Förster, 1965). Chromophores from different pairs would interact in the weak coupling regime that is described by the Förster mechanism. Other reports classify the coupling between α84 and β84 chromophores within a pair as falling in the intermediate to weak coupling range. A femtosecond absorption technique was used to reveal ground state recovery with a time constant of 440 fs. It was concluded that this component could be attributed to fast energy transfer from donor chromophores to acceptor chromophores and that this energy transfer process is in agreement with a Förster type excitation energy transfer mechanism. It was argued that neither a vibrational relaxation process nor an interexciton state relaxation were responsible for the ultrafast component because neither of these two processes would lead to such a fast ground state recovery. The coupled dimer would still be in the excited state (Sharkov et al., 1992). A similar fast component was observed in C-phycocyanin trimers with similar structure as allophycocyanin trimers and interpreted in terms of a Förster-type mechanism (Gillbro et al., 1993). X-ray crystal data reported by Brejc and co-workers (1995) indicate that protein environment, and hence the conformation of the α84 and β84 chromophores, are different in a trimer and, as a result α84 chromophores show a more planar geometry. On the other hand, the Tyrβ90 residue is in close distance and nearly parallel oriented to the D ring of the β84 chromophore (Fig. 1 B). Most probably, Tyrβ90 interacts both sterically and electronically with the D ring, thereby extending the conjugation of the β84 chromophore. In this view, an APC trimer would indeed contain three equivalent donors and three equivalent acceptors and the energy transfer processes should be described in terms of a Förster excitation energy transfer mechanism. The shape of the absorption spectrum is then defined by the sum of the individual absorption spectra of the donor and the acceptor chromophores.
FIGURE 2.

(A) Transient absorption spectrum of APC in PBS as reproduced from Su-Ping et al., 2001. (B) Steady-state absorption (solid line) and fluorescence (dashed line with stars, excitation at 632 nm) spectra of APC in a PBS buffer, pH 7.4. Shown in the same panel are the calculated individual absorption (solid line with open circles for α84 and solid line with stars for β84) and fluorescence spectra (dashed line with solid circles for α84 and dashed line with open stars for β84) of the chromophores in APC (see text for details).
It is the aim of this work to contribute to the elucidation of the nature of the interaction between α84 and β84 chromophores in APC trimers. For this purpose, we have performed single-molecule experiments on APC trimers and hence monitored the time course of the fluorescence intensity, lifetime, polarization, and spectra. Our data indicate the presence, within individual APC trimers, of spectrally different species, donor and acceptor chromophores, which interact in the weak coupling regime, therefore suggesting that protein-chromophore interaction is responsible for the particular shape of the APC absorption spectrum.
MATERIALS AND METHODS
Materials
Cross-linked allophycocyanin (APC) was purchased from Molecular Probes (Cat no. A-819, Eugene, OR). Cross-linking allows APC to be used in very dilute solutions without destroying its trimeric structure (Yeh et al., 1987). APC was dissolved in a phosphate-saline buffer (PBS, pH 7.4, Sigma, St. Louis, MO) and extensively dialyzed to remove eventual impurities. For ensemble spectroscopy, we used solutions with a concentration of APC of around 10−7 M to avoid inner filter effects. For single-molecule measurements, APC was diluted to subnanomolar concentrations in a PBS buffered polyvinyl alcohol (PVA) solution (1 wt % PVA) and a drop of the solution was spin-casted on a cleaned coverslip.
Ensemble spectroscopy
Room-temperature absorption and fluorescence spectra of APC in PBS were recorded on a Perkin-Elmer Lambda 40 spectrophotometer (Perkin-Elmer, Wellesley, MA) and a Spex Fluorolog 1500 fluorimeter (Spex Industries, Metuchen, NJ), respectively. Fluorescence decays were recorded by the time-correlated single-photon counting (TCSPC) technique on a setup described previously (Maus et al., 2001c). In brief, linearly polarized laser excitation was provided by a frequency doubled/mode-locked ps Ti:Sapphire laser (Tsunami 1.2-ps pulse, 8.13 MHz repetition rate, Spectra Physics, Mountain View, CA). Fluorescence was collected at magic angle (54.7°), spectrally separated by a monochromator (9030, 10-nm bandwith, Sciencetech, London, ON) and detected with a microchannel plate photomultiplier (E 3059-500, Hamamatsu, Japan). The signal from the microchannel plate photomultiplier was input into a TCSPC PC card (SPC 630, Becker-Hickl, Berlin, Germany). The experimental instrumental response function of the TCSPC setup is 30 ps. Fluorescence decays were collected in 4096 channels with 10,000 photocounts in the peak and analyzed by a weighted iterative reconvolution method based on the Marquardt algorithm (Maus et al., 2001c). We estimated the contribution of the decay times as relative amplitudes according to
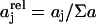 |
(1) |
Single-molecule detection
Room-temperature single-molecule fluorescence spectroscopy of individual APCs embedded in PVA was performed on a scanning stage confocal microscope described previously (Cotlet et al., 2001a). Single-molecule samples were mounted on an Olympus IX 70 inverted fluorescence microscope equipped with a feedback controlled scanning stage (Physics Instruments, Waldbrown, Germany). Optical excitation with either pulsed or continuous wave (CW) laser light occurred by an oil immersion objective (1.4 NA, 60×, Olympus, Tokyo, Japan). Fluorescence was collected by the same objective, passed through a dichroic mirror (DRLP590 or DRLP640, Chroma Technologies, Rockingham, VT), filtered through a notch filter (Kaiser Optics, Ann Arbor, MI) and confocally imaged on a single-photon counting avalanche photodiode (APD, SPCM 15 EG&G, Gaithersburg, MD).
For lifetime experiments we used the 590-nm output from the ps laser system used in ensemble TCSPC experiments. Here, fluorescence from single APC trimers was registered by a TCSPC PC card (SPC 630, Becker-Hickl, Berlin, Germany) using the FIFO (first-in, first-out) mode. In FIFO mode, for each detected photon it is possible to register the time lag with respect to the excitation pulse (tn microtime, when using pulsed excitation) and the time lag with respect to the previously detected photon (Tn macrotime or chronological time). Such a single-molecule fluorescence trajectory (FIFO data set) allows us to construct the fluorescence intensity trajectory with a bin time as short as 100 ns and fluorescence decays using bins of a certain amount of photons. We estimated fluorescence lifetimes from bins of 1000 photons over a single-molecule fluorescence trajectory. Lifetimes were estimated by maximum likelihood estimator (MLE) fitting and reiterative convolution of the instrumental response function of the setup (0.38 ns) with an exponential model function (M),
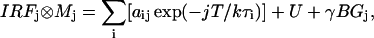 |
(2) |
with j = 1,k with k the number of channels over which the photons of a decay are spread and i the number of exponential terms. Here T is the time window of the experiment, a and 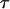 are amplitude and decay time, U is a constant accounting for noncorrelated background and γ is a scaling factor correcting for the presence of correlated background (BG), i.e., scattered Rayleigh and/or Raman photons (Maus et al., 2001a).
are amplitude and decay time, U is a constant accounting for noncorrelated background and γ is a scaling factor correcting for the presence of correlated background (BG), i.e., scattered Rayleigh and/or Raman photons (Maus et al., 2001a).
For isotropic and polarization experiments, we used the 632-nm linearly polarized light from a He-Ne laser (Melles-Griot, Carlsbad, CA). For isotropic experiments, the fluorescence collected from a single APC trimer was focused on one APD. For polarization experiments the fluorescence was split by a polarizing beam-splitter cube and focused onto two APDs to detect the parallel (Ipar) and perpendicular (Iperp) components of the fluorescence intensity and to construct the time course of the polarization degree according to
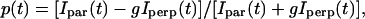 |
(3) |
with g a correction factor accounting for the difference in polarization sensitivity of the two APDs.
For the recording of the single-molecule fluorescence spectra, optical excitation occurred with the 568-nm laser light from an Ar-Kr ion laser (Stabilite 2018, Spectra Physics). Fluorescence was spectrally resolved by a polychromator (SP 150, Acton Research, Acton, MA) and detected with a liquid-nitrogen cooled CCD (LNCCD, Roper, Trenton, NJ).
RESULTS
Ensemble spectroscopy
APC features an absorption spectrum with a main and sharp peak at 654 nm and a broad shoulder at 632 nm (Fig. 2 B, solid line). The protein has a molar extinction coefficient in the absorption maximum of 700,000 M−1 cm−1. Fluorescence from APC, emitted with a sharp main peak at 660 nm and a shoulder at 720 nm (Fig. 2 B, dashed line with open stars, 632-nm excitation) has a quantum yield of fluorescence of 0.68 (on 654-nm excitation). Scanning the excitation wavelength over the absorption spectral range of the protein does not lead to changes in shape or in peak position of the fluorescence spectrum. Moreover, fluorescence and absorption spectra have different shapes, especially at the low- and high-energy sides, respectively. The fluorescence excitation spectrum resembles the absorption spectrum and does not feature any change either in shape or in peak position when monitoring the fluorescence at 660 and 720 nm.
On 590-nm pulsed excitation, the fluorescence of APC detected at 660 nm decays biexponentially with decay times of 1.6 ns (90% weight) and 0.8 ns (10%). Similar values were reported earlier by Maxson and Sauer (1988). The contribution of these components stays similar if fluorescence is detected at 720 nm. Similar decay times with similar contributions are recovered on 640-nm excitation, independent of the detection wavelength, i.e., either 660 or 720 nm.
Single-molecule detection
Fluorescence intensity trajectories
Shown in Fig. 3, A–D, are examples of fluorescence intensity trajectories we detected from single APC trimers immobilized in PVA on 632-nm excitation (excitation power at the sample of 0.5 kW/cm2). Although recorded with a short dwell time (100 ns), trajectories were binned to 60 ms to clearly show the presence of different intensity levels during the survival time of the single APC trimers. They feature different intensity levels, stepwise changes, a behavior characteristic for multichromophoric systems (Ying and Xie, 1998; Hofkens et al., 2000; Cotlet et al., 2001b; Bopp et al., 1997) and hence different from immobilized, single-chromophoric, single-dye molecules which usually show a single intensity level followed by one-step photobleaching (Xie, 1996; Ambrose et al., 1994). Within the probed population of single APC trimers (70 molecules), 76% feature six levels of intensity. Fig. 3 A is an example of a single APC trimer featuring six intensity levels. Six levels in the fluorescence intensity might account for the presence of six absorbers in a single APC trimer. The rest of the single APC trimers show from five to three levels of intensity (Fig. 3, B–D), most probably due to the bleaching of one or more chromophores during the process of imaging of the sample. In fact, most of the APC trimers show the highest intensity level at the beginning of the trajectory (Fig. 3). Intensity in single APC trimers switches from high to dim and back to high levels. For most of the molecules, before complete photobleaching occurs, fluorescence is detected as a long-lasting dim intensity level (Fig. 3, B–D). While in such dim levels, some of the single APC trimers switch off their fluorescence for periods lasting up to tens of seconds.
FIGURE 3.

Isotropic fluorescence intensity trajectories (60-ms dwell time) from single APC trimers in PVA recorded at 632-nm CW linear polarized excitation showing (A) six, (B) five, (C) four, and (D) three levels of intensity. For each panel the inset is a zoom (10-ms dwell time) of the first part of the trajectory and the graph at right represents the frequency histograms, in logarithmic scale (10-ms dwell time), of the data shown in the main panels.
Polarization trajectories
For a single immobilized molecule changes in the polarization degree of fluorescence, Δp, are directly related to changes in the orientation of the transition dipole moment of the emitting chromophore, Δθ, according to
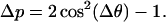 |
(4) |
For a single dye molecule, if reorientations in the matrix are excluded, the degree of polarization of fluorescence p will show no fluctuations during the measurement and hence during the survival time. Such a behavior can be regarded as a fingerprint of the presence of a single emitter. From the single APC trimers probed using polarization-sensitive detection, we selected for the following discussion only the ones displaying six levels in the isotropic fluorescence intensity trajectory and hence accounting for the presence of six absorbers. Shown in Fig. 4, A and C, are trajectories of the polarization degree from two individual APC trimers reconstructed according to Eq. 3. The polarization data shown in Fig. 4, A and B account for a single-molecule with three levels in the p trajectory, on average 0.08, 0.48, and −0.44. The switch from one level to the next one happens suddenly, as can be seen in Fig. 4 A. The histogram from Fig. 4 B clearly accounts for the presence of a three-modal distribution of the polarization degree. We previously showed that single proteins of smaller size than APC are rigidly immobilized in PVA (Cotlet et al., 2001b) and hence eventual reorientations of a whole single APC trimer can be excluded during the measurement. The chromophores in APC are known to be rigidly bound in their pockets. Consequently, the sudden and relatively large jumps we detect in the time course of the polarization degree of fluorescence must relate to the presence of different emitters, here three, i.e., each level in the p trajectory will relate to a single emitter. The data shown in Fig. 4, C and D, account for a single APC trimer displaying four levels in the p trajectory, on average −0.68, −0.21, 0.09, and 0.30. Because some of the levels are relatively short and hence contribute little to the histogram shown in Fig. 4 D, more than four levels of polarization might be present here. Switching from one level to another happens also suddenly and hence for this single molecule we claim the presence of more than three consecutive emitters. Within the population of single APC molecules we probed (96 molecules), 16% showed less than three levels, 59% showed three levels, and the rest showed more than three levels. The observation of polarization trajectories with more than three levels strongly supports the absence of strong coupling between the chromophores within a pair and hence of the excitonic splitting of the absorption of APC (vide infra).
FIGURE 4.

(A and C) Time courses of the polarization degree (20 ms/bin) of single APC on 632-nm CW linear polarized excitation. (B and D) Histograms, in logarithmic scale, and Gauss fits of the polarization data from panels A and C, respectively. Also shown are the main values resulting from the fit.
Single-molecule fluorescence lifetimes
Shown in Fig. 5 A is the isotropic fluorescence intensity of a single APC trimer recorded with 590-nm pulsed excitation (excitation power at the sample of 0.5 kW/cm2). The maximum count rate detected from single APC trimers as well as the survival time under pulsed excitation is by far lower than under CW excitation, an observation reported previously by Ying and Xie and related to S1-S1 annihilation (Ying and Xie, 1998). For the specific single APC trimer accounting for Fig. 5, the total fluorescence decay constructed by binning all the detected photons shows a biexponential profile with lifetimes of 1.6 and 0.8 ns (Fig. 5 C), similar to the ensemble decay of APC in PBS. Multiexponential single-molecule fluorescence decays have been reported previously for biological systems and were related to fluctuating lifetimes during the measurement (Edman et al., 1996; Eggeling et al., 1998; Yang et al., 2003), in contrast to single exponential decays of single-chromophoric single-dye molecules (Xie and Dunn, 1994). To reveal lifetime fluctuations, we next estimate the lifetimes by MLE fitting every 1000 photons over the single-molecule fluorescence trajectory with a single exponential model (Fig. 5 B). MLE lifetimes are indeed found to fluctuate during the survival time of the single APC trimer we probe. Two representative single-exponential decays, each of 1000 total photons, are shown in Fig. 5, D and E, and account for lifetimes of 1.62 and 0.78 ns, respectively. By histogramming the lifetimes from Fig. 5 B we recover, as mean values, the lifetimes found in the total decay shown in Fig. 5 C. For the single APC molecule accounting for Fig. 5, the lifetimes correlate with the isotropic intensity (Fig. 5 G), suggesting that intensity fluctuations are mainly due to lifetime changes. However, some of the individual APC trimers we probed do not show lifetimes correlated with the isotropic fluorescence intensity (data not shown).
FIGURE 5.

Representative time-resolved data accounting for a single APC trimer in PVA. (A) Isotropic fluorescence intensity and (B) fluorescence lifetime trajectories. Lifetimes were estimated by MLE fitting each 1000 photons over the single-molecule fluorescence trajectory. (C) Total fluorescence decay constructed from the single-molecule fluorescence trajectory. (D) Unquenched and (E) quenched single-molecule fluorescence decays (1000 photons each) observed during the measurement of the single APC trimer. (F) Histogram of lifetimes from panel B and Gauss fits. (G) Intensity versus lifetime correlation constructed from panels A and B.
A histogram containing MLE lifetimes estimated from all the single APC trimers we probed is shown in Fig. 6. Each lifetime is an estimate from a single-molecule decay of 1000 photons using a single exponential function. Gauss fits to the histogram result in mean values of 1.71 and 1.08 ns, contributing 83 and 17%, respectively. The MLE histogram from Fig. 6 is in good agreement with the averaged ensemble isotropic fluorescence decay of APC in PBS, both in value and contribution.
FIGURE 6.

Overall histogram containing lifetimes estimated from decays of 1000 photons from a population of 70 single APC trimers. Also shown is a bimodal Gauss fit (lines with circles).
Single-molecule fluorescence spectra
We recorded fluorescence spectra from single APC trimers on 568-nm CW excitation with an integration time of 1 s by spectrally resolving all the photons collected from single APC trimers. Most of the single APC trimers show solely emission peaking at 660 nm until photobleaching occurs, as previously reported by Ying and Xie (1998). However, in some cases, apart from the 660-nm maximum, we detected a blue-shifted band with a maximum around 630 nm. An example related to such a molecule is shown in Fig. 7, A and B. In the beginning of the emission, we detect a fluorescence spectrum peaking at 660 nm with an “ensemble”-like shape (Fig. 7 C). The spectrum stays the same, both in peak position and shape, experiencing only a drop in intensity, most probably due to sequential bleaching of the chromophores. After 28 s the spectrum suddenly shifts its maximum to shorter wavelengths, that is, around 630 nm, and broadens considerably (Fig. 7 D). During the next seconds we detect a spectrum containing both 660- and 630-nm peaks (Fig. 7 E). After 40 s from the beginning of the experiment the spectrum shifts its maximum to longer wavelengths, i.e., around 650 nm. Our data clearly show that two spectrally different emitting species are present in APC. Such dynamics in the single-molecule fluorescence spectrum of APC was not observed previously. We suggest that the 660- and 630-nm peaks relate to emission from acceptor and donor chromophores, respectively, in a single APC trimer (vide infra). The spectrum carrying both the 660- and 630-nm peaks is a combination of the emission of the donor and acceptor chromophores.
FIGURE 7.

(A) Fluorescence spectral run (1 s integration/spectrum) detected from a single APC trimer on 568-nm CW excitation. (B) Time course of the emission maximum of the spectra from panel A. Single-molecule fluorescence spectra accounting for (C) single β84, (D) single α84, and (E) mixture of single β84 and α84 chromophores.
DISCUSSION
The ensemble and especially the single-molecule results reported here strongly suggest the presence, within APC trimers, of spectrally different chromophores and hence of an intermediate-to-weak coupling interaction between chromophores within a pair which is governed by a Förster-type excitation energy transfer mechanism (vide infra). With this picture in mind, within a pair of nearby chromophores, Förster-type resonance energy transfer (FRET) will take place from α84 (donor, D) to β84 (acceptor, A). Femtosecond ground-state recovery experiments on the APC trimer reported by Sharkov and co-workers (1992) were indicative of the presence of a subpicosecond component that the authors attributed to unidirectional FRET from α84 to β84 chromophores within a pair. Assuming a Förster radius of 5 nm as reported by Brejc and co-workers (1995), for a center-to-center distance of 2.1 nm, FRET from α84 to β84 within a pair should occur with an efficiency
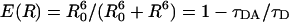 |
(5) |
of 99.45%, assuming that direct optical excitation of the acceptor is excluded. Here τDA and τD are lifetimes of the donor in the presence and absence of the acceptor, respectively. For an efficiency of 99.45% and assuming an unquenched donor lifetime τD = 1.5 ns (Sauer et al., 1987), FRET should then occur as fast as 8 ps, a value approaching the earlier reported FRET related time constant by Sharkov and co-workers (1992). With a FRET efficiency of 99.45%, we expect only the acceptor to contribute to the fluorescence emission of the APC trimer. If so, the ensemble fluorescence spectrum of APC (Fig. 2 B, dashed line with open stars) relates only to the acceptor chromophore (β84). Based on these assumptions and on the single-molecule fluorescence spectra we report in Fig. 7, C and D, it is possible to accurately identify/calculate the individual absorption and emission spectra of the α84 and β84 chromophores for the APC trimer (vide infra).
We next assign the 654-nm peak and the 632-nm shoulder from the absorption spectrum of APC (Fig. 2 B) to the acceptor (β84) and the donor (α84) chromophores, respectively. Assuming mirror symmetry between the absorption and fluorescence spectra of the acceptor, we can decompose the absorption spectrum of APC into individual spectra of the donor and acceptor chromophores. Mirror symmetry has been observed previously for other complex biomolecules with tightly bound chromophores (Tsien, 1998). Fig. 2 B shows the calculated acceptor (solid line with solid stars) and donor (solid line with open circles) ensemble absorption spectra. Image mirroring should be valid for the donor as well. At the single-molecule level we detect donor emission with a peak at 630 nm (Fig. 7 D). Hence, we next build the ensemble fluorescence spectrum of the donor as a mirror image of the calculated absorption spectrum from Fig. 2 B with the main peak at 630 nm. Interestingly, the donor calculated ensemble fluorescence spectrum (Fig. 2 B, dashed line with solid circles) highly resembles in shape the single-molecule fluorescence spectrum we attribute to the donor (Fig. 7 D). This is an important observation that supports the methodology we use here to calculate individual spectra of α84 and β84 chromophores and hence the assumption that emission from APC relates mainly to the acceptor, that is, the β84 chromophore. Absorption and emission spectra of α84 and β84 chromophores differ considerably in shape, with the latter having a more defined vibronic structure. According to the x-ray crystal data, α84 and β84 chromophores probe different environments in the pockets to which they are bound and, as a result, they have different spectral properties. The red shift in absorption/emission of β84 as compared to α84 is the result of the interaction between Tyrβ90 residue and the D ring of the β84 chromophore (Brejc et al., 1995).
The isotropic fluorescence intensity trajectories of most of the single APC trimers we probed on CW excitation display six levels, which is a clear indication that within an APC trimer six absorbers are present. On the other hand, polarization data indicate, in most of the cases, that three emitters are present in a single APC trimer, and, moreover, that each of them consecutively acts as an emissive site as a function of time (Fig. 4, A–B). This is possible only if we assume unidirectional transfer of the excitation energy between the spectrally different chromophores, i.e., FRET from α84 to β84. According to the calculated spectra of individual chromophores in APC (Fig. 2 B), there is almost perfect overlap between the emission of α84 and the absorption of β84. The α84 and β84 chromophores are bound in the protein pockets such that they are oriented toward each other with an angle of 60° and are situated, within a pair, at a center-to-center distance of 2.1 nm. These are conditions for a highly efficient unidirectional FRET from α84 to β84 chromophores within a pair. Based on the calculated spectra of the donor and acceptor chromophores and on the x-ray crystal data (Brejc et al., 1995), we estimate according to Eq. 5 a Förster radius of 6.8 nm and hence a FRET-related time constant in the order of 1.3 ps, a value that is in better agreement with the earlier results of Sharkov and co-workers (1992). If so, at the excitation wavelength at which fluorescence intensity and polarization trajectories are recorded, both types of chromophores can undergo optical excitation, such that, within an APC trimer, six chromophores can absorb. Upon optical excitation, the α84 chromophores transfer their excitation energy via unidirectional FRET to the β84 chromophores located within the same pair such that the final emission relates to β84 chromophores, here three. Because most of the single APC trimers show three levels in the polarization trajectory, it might be possible that, within a pair, the donor chromophore bleaches first. If so, bleaching of the donor might take place from the singlet excited state since triplet formation is rather unlikely in the presence of a highly efficient unidirectional FRET. Bleaching from the singlet excited state has been observed previously in single peryleneimide-terrylenediimide molecules undergoing highly efficient unidirectional FRET (Gronheid et al., 2002). The assumption of photo-unstable donors might explain why single APC trimers show the highest intensity of fluorescence just in the beginning of the emission and for a short time and why some of the trimers show less than six intensity levels. Another reason for the observation of mainly three p levels might be that it is impossible to distinguish between the p values of nearly parallel chromophores from different subunits. Förster-type excitation energy transfer (hopping) is also involved between β84 chromophores (vide infra). Single-molecule spectroscopic studies on immobilized multichromophoric systems containing identical chromophores have shown that, if favorable conditions for energy hopping are present, fluorescence can be emitted from a specific chromophore at a given time, namely the one acting as an emissive trap toward its neighbors (Hofkens et al., 2000, 2001). These neighbors are situated at center-to-center distances within the Förster radius for energy hopping. Bleaching of the emitting chromophore in a multichromophoric system can be seen as a sudden change in the transition dipole moment and hence as a sudden change in the value of polarization degree of fluorescence, as we observed for single APC trimers. In our case, β84 chromophores are displaced at a center-to-center distance of 3.1 nm and are oriented toward each other with an angle of 120°. According to the spectra we calculated here for the β84 chromophore (Fig. 2 B), there is a large spectral overlap between the absorption and fluorescence. Therefore, in an APC trimer, conditions for energy hopping amongst β84 chromophores are present. On the basis of the x-ray crystal data reported by Brejc and co-workers (1995) and the spectral data reported here, we estimate a Förster-radius for energy hopping of 4.5 nm and hence we expect a depolarization of the fluorescence because of the energy hopping of 25 ps. We tried to measure the time-resolved fluorescence anisotropy decay of APC (590-nm excitation/660-nm detection) to find evidence for an anisotropy decay time connected with energy hopping (Maus et al., 2001b). Unfortunately, fluorescence from APC depolarizes too fast to be resolved in TCSPC experiments (data not shown) and hence the anisotropy decay time connected with energy hopping falls within the response time of the instrument, here 30 ps.
Single-molecule fluorescence spectra of APC trimers that we report here (Fig. 7 A) show that bleaching of the 660-nm emitter is followed by donor emission (630 nm). After donor bleaching, red fluorescence recovers but is slightly blue shifted, peaking at around 650 nm. At the ensemble level in solution bleaching does not pose a problem and hence by no means can the donor be spectrally identified. Due to a highly efficient unidirectional FRET, here of 99.45%, emission in solution will always come from the most spectrally red-shifted species, here the acceptor emitting at 660 nm. Interestingly, the spectral data from Fig. 7 show that, within a single APC trimer, even the β84 chromophores are spectrally different. Indeed, acceptor emission is seen in the beginning peaking at 660 nm whereas, after the bleaching of the donor (peak at 630 nm), the acceptor shows up again but peaking around 650 nm and with a slightly broader shape.
The isotropic fluorescence intensity trajectories detected from single APC trimers feature the highest count rate/level in the beginning, just after optical excitation is applied. Single APC trimers are found to reversibly switch their isotropic emission between high and dim intensity levels. Such behavior was also observed earlier by Ying and Xie (1998) in single-molecule experiments and attributed to the formation of temporary and long-lived radical cation traps. Recently, Su-Ping and co-workers (2001) reported the detection of a transient intermediate assigned as a radical cation of APC and with a lifetime of tens of μs. Shown in Fig. 2 A, top panel, is the spectrum of the radical cation of APC as reproduced from literature (Su-Ping et al., 2001). With an absorption peaking at 690 nm and overlapping considerably with the emission of the acceptor chromophore, the radical cation can therefore act as a nonradiative trap for the acceptor emission in APC. Being a temporary trap, once it disappears, the emission from the acceptor restores. Recalling the single-molecule lifetime data we report here (Fig. 5, B–F), the subnanosecond lifetimes associated with dim isotropic intensity levels (Fig. 5 A) might account for quenched fluorescence from a single APC trimer due to radical cation formation. The radical cation is most likely formed from the emitting chromophore since that one is already the lowest in energy and it already acts as an emissive trap. It is not merely a coincidence that the single-molecule lifetime data match remarkably with the ensemble TCSPC data, both in value and in contribution (vide supra). Therefore we attribute the 1.6- and the 0.8-ns lifetimes of APC to unquenched and quenched fluorescence of the acceptor chromophores, respectively, the latter quenched by the radical cation. According to the report of Su-ping et al. (2001), the formation of the APC radical cation in a polar solution occurs through a monophotonic process and with a quantum yield of formation of 0.17, meaning APC can be easily ionized at the ensemble level.
However, some of the quenched lifetimes we detect at the single-molecule level might relate to emission from a donor chromophore within a pair having the acceptor already bleached (vide infra). According to the center-to-center distances predicted by the x-ray crystal data and for a Förster radius of 6.8 nm, FRET from an α84 donor to a β84 acceptor located in a different pair (at a center-to-center distance of 5.2 nm) will take place with an efficiency of ∼83% and hence a time constant of 270 ps. If the acceptor in a specific pair bleaches and simultaneously a radical cation is formed in another pair, donor emission might be seen alone. This is because the quenching of donor fluorescence by the radical cation will happen with an even lower efficiency due to a smaller spectral overlap between donor emission and radical cation absorption (Fig. 2, A and B). If detected at the single-molecule level, donor emission should lead to the observation of more than three levels in the polarization degree trajectory of a single APC trimer (Fig. 4, C and D).
The observations we present here, which are mainly based on single-molecule data, can be compiled in a photophysical model as shown in Fig. 8. According to this model, an APC trimer will contain three pairs of chromophores, each pair composed of two spectrally different α84 and β84 chromophores. Within a pair, an α84 (donor) transfers its excitation to the β84 (acceptor) via unidirectional FRET whereas energy hopping is present among β84 chromophores from different pairs. The radical cation, if formed, will quench the emission of the whole APC trimer.
FIGURE 8.

Photophysical scheme accounting for the processes observed in APC. D, donor; A, acceptor; RC*, radical cation; kabs, k(D)fl, k(A)fl, and kFRET are rate constants for absorption, donor fluorescence, acceptor fluorescence, and Förster energy transfer. kNR1 and kNR2 are rate constants for nonradiative deactivation.
CONCLUSION
We have performed single-molecule fluorescence measurements on the phycobiliprotein allophycocyanin (APC) in order to understand the mechanism of interaction between the phycocyanobilin α84 and β84 chromophores. Our data strongly support the presence of a unidirectional Förster-type energy transfer process involving spectrally different α84 (donor) and β84 (acceptor) chromophores as well as of energy hopping amongst β84 chromophores. The isotropic fluorescence intensity detected from single APC trimers shows predominantly six levels, which definitely accounts for the presence of six absorbers and hence weak interaction amongst the chromophores within a trimer. Single-molecule fluorescence spectra we report here substantiate the presence of spectrally different emitters, that is, a red-emitting chromophore (peak at 660 nm) that we connect with β84 (acceptor), and a blue-shifted species (peak at 630 nm) that we connect with α84 (donor). Polarization data from single APC trimers point to the presence of mainly three consecutive red emitters, strongly suggesting energy hopping amongst β84 chromophores. Based on the single-molecule fluorescence spectra, and assuming that emission at the ensemble level in solution comes mainly from the acceptor chromophore, we were able to resolve the individual absorption and emission spectra of the α84 and β84 chromophores in APC. A scheme accounting for the photophysical processes present in APC is proposed and supported by the ensemble and single-molecule data reported here as well as by earlier work on APC.
Acknowledgments
Dr. Th. Gensch is acknowledged for discussion at the early stages of this project.
D.L. thanks the Instituut voor Innovatie door Wetenschap en Technologie and Katholieke Universiteit Leuven (KUL) for financial support. M.C. thanks Onderzoeksfonds of KUL for a postdoctoral fellowship. S.H. thanks the Japan Society for the Promotion of Science for a postdoctoral fellowship. The federal science policy through IAP/V/03 and the KUL through GOA 01/2 are thanked.
Davey Loos and Mircea Cotlet contributed equally to this work.
References
- Ambrose, W. P., P. M. Goodwin, J. C. Martin, and R. A. Keller. 1994. Alterations of single molecule fluorescence lifetimes in near-field optical microscopy. Science. 265:364–367. [DOI] [PubMed] [Google Scholar]
- Beck, W. F., and K. Sauer. 1992. Energy-transfer and exciton-state relaxation processes in allophycocyanin. J. Phys. Chem. 96:4658–4666. [Google Scholar]
- Bopp, M. A., Y. Jia, L. Li, R. J. Cogdell, and R. M. Hochstrasser. 1997. Fluorescence and photobleaching dynamics of single light-harvesting complexes. Proc. Natl. Acad. Sci. USA. 94:10630–10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brejc, K., R. Ficner, R. Huber, and S. Steinbacher. 1995. Isolation, crystallization, crystal structure analysis and refinement of allophycocyanin from the cyanobacterium Spirulina platensis at 2.3 Å resolution. J. Mol. Biol. 249:424–440. [DOI] [PubMed] [Google Scholar]
- Cotlet, M., J. Hofkens, S. Habuchi, G. Dirix, M. Van Guyse, J. Michiels, J. Vanderleyden, and F. C. De Schryver. 2001a. Identification of different emitting species in the red fluorescent protein DsRed by means of ensemble and single-molecule spectroscopy. Proc. Natl. Acad. Sci. USA. 98:14398–14403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotlet, M., J. Hofkens, F. Köhn, J. Michiels, G. Dirix, M. Van Guyse, J. Vanderleyden, and F. C. De Schryver. 2001b. Collective effects in individual oligomers of the red fluorescent coral protein DsRed. Chem. Phys. Lett. 336:415–423. [Google Scholar]
- Csatorday, K., R. MacColl, V. Csizmadia, J. Grabowski, and C. Bagyinka. 1984. Exciton interaction in allophycocyanin. Biochemistry. 23:6466–6470. [DOI] [PubMed] [Google Scholar]
- Edington, M. D., R. E. Riter, and W. F. Beck. 1995. Evidence for coherent energy transfer in allophycocyanin trimers. J. Phys. Chem. 99:15699–15704. [Google Scholar]
- Edman, L., Ü. Metz, and R. Rigler. 1996. Conformational transitions monitored for single molecules in solution. Proc. Natl. Acad. Sci. USA. 93:6710–6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggeling, C., J. R. Fries, L. Brand, R. Günther, and C. A. M. Seidel. 1998. Monitoring conformational dynamics of a single molecule by selective fluorescence spectroscopy. Proc. Natl. Acad. Sci. USA. 95:1556–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förster, Th. 1965. Delocalized excitation and excitation transfer. In Modern Quantum Chemistry. O. Sinanoglu, editor. Academic Press, New York. 93–137.
- Gillbro, T., A. V. Sharkov, I. V. Kryukov, E. V. Khoroshilov, P. G. Kryukov, R. Fischer, and H. Scheer. 1993. Förster energy transfer between neighbouring chromophores in C-phycocyanin trimers. Biochim. Biophys. Acta. 1140:321–326. [Google Scholar]
- Gronheid, R., J. Hofkens, F. Kohn, T. Weil, E. Reuther, K. Mullen, and F. C. De Schryver. 2002. Intramolecular Förster energy transfer in a dendritic system at the single molecule level. J. Am. Chem. Soc. 124:2418–2419. [DOI] [PubMed] [Google Scholar]
- Hofkens, J., M. Maus, T. Gensch, T. Vosch, M. Cotlet, F. Köhn, A. Herrmann, K. Müllen, and F. De Schryver. 2000. Probing photophysical processes in individual multichromophoric dendrimers by single-molecule spectroscopy. J. Am. Chem. Soc. 122:9278–9288. [Google Scholar]
- Hofkens, J., W. Schroeyers, D. Loos, M. Cotlet, F. Köhn, T. Vosch, M. Maus, A. Herrmann, K. Müllen, T. Gensch, and F. C. De Schryver. 2001. Triplet states as non-radiative traps in multichromophoric entities: single molecule spectroscopy of an artificial and natural antenna system. Spectrochim. Acta. 57:2093–2107. [DOI] [PubMed] [Google Scholar]
- Liu, R., M. W. Holman, L. Zang, and D. M. Adams. 2003. Single-molecule spectroscopy of intramolecular electron transfer in donor-bridge-acceptor systems. J. Phys. Chem. A. 107:6522–6526. [Google Scholar]
- Lu, H. P., L. Xun, and X. S. Xie. 1998. Single molecule enzymatic dynamics. Science. 282:1877–1882. [DOI] [PubMed] [Google Scholar]
- MacColl, R., K. Csatorday, D. S. Berns, and E. Traeger. 1980. Chromophore interactions in allophycocyanin. Biochemistry. 19: 2817–2820 [DOI] [PubMed] [Google Scholar]
- MacColl, R., K. Csatorday, D. S. Berns, and E. Traeger. 1981. The relationship of the quaternary structure of allophycocyanin to its spectrum. Arch. Biochem. Biophys. 208:42–48. [DOI] [PubMed] [Google Scholar]
- Maus, M., M. Cotlet, J. Hofkens, T. Gensch, F. C. De Schryver, J. Schaffer, and C. A. M. Seidel. 2001a. An experimental comparison of the maximum likelihood estimation and nonlinear least-squares fluorescence lifetime analysis of single molecules. Anal. Chem. 73:2078–2086. [DOI] [PubMed] [Google Scholar]
- Maus, M., R. De, M. Lor, T. Weil, S. Mitra, U.-M. Wiesler, A. Herrmann, J. Hofkens, T. Vosch, K. Müllen, and F. C. De Schryver. 2001b. Intramolecular energy hopping and energy trapping in polyphenylene dendrimers with multiple peryleneimide donor chromophores and a terryleneimide acceptor trap chromophore. J. Am. Chem. Soc. 123:7668–7676. [DOI] [PubMed] [Google Scholar]
- Maus. M., E. Rousseau, M. Cotlet, J. Hofkens, G. Schweitzer, M. Van der Aueraer, F. C. De Schryver, and A. Kruger, 2001c. New picosecond laser system for easy tunability over the whole ultraviolet/visible/near infrared wavelength range based on flexible harmonic generation and optical parametric oscillation. Rev. Sci. Instr. 72:36–40. [Google Scholar]
- Maxson, P., and K. Sauer. 1988. Fluorescence spectroscopy off allophycocyanin complexes from Synechococcus 6301 strain AN112. In Photosynthetic Light-Harvesting Systems. H. Scheer and S. Schneider, editors. Walter de Gruyter, New York. 439–449.
- Sauer, K., H. Scheer, and P. Sauer. 1987. Förster transfer calculations based on crystal structure data from Agmenellum quadruplicatum C-phycocyanin. Photochem. Photobiol. 46:427–440. [Google Scholar]
- Sharkov, A. V., I. V. Kryukov, E. V. Khoroshilov, P. G. Kryukov, R. Fischer, H. Scheer, and T. Gillbro. 1992. Femtosecond energy transfer between chromophores in allophycocyanin trimers. Chem. Phys. Lett. 191:633–638. [Google Scholar]
- Su-Ping, Z., P. Jing-Xi, H. Zhen-Hui, Z. Jing-Quan, Y. Si-De, and J. Li-Jin. 2001. Generation and identification of the transient intermediates of allophycocyanin by laser photolytic and pulse radiolytic techniques. Int. J. Radiat. Biol. 77:637–642. [DOI] [PubMed] [Google Scholar]
- Tsien, R. Y. 1998. The green fluorescent protein. Annu. Rev. Biochem. 67:509–543. [DOI] [PubMed] [Google Scholar]
- Van Grondelle, R. 1985. Excitation energy transfer, trapping and annihilation in photosynthetic systems. Biochim. Biophys. Acta. 811:147–195. [Google Scholar]
- Weiss, S. 2000. Measuring conformational dynamics of biomolecules by single molecule fluorescence spectroscopy. Nat. Struct. Biol. 7:724–729. [DOI] [PubMed] [Google Scholar]
- Xie, X. S. 1996. Single-molecule spectroscopy and dynamics at room temperature. Acc. Chem. Res. 29:598–606. [Google Scholar]
- Xie, X. S., and R. C. Dunn. 1994. Probing single molecule dynamics. Science. 265:361–364. [DOI] [PubMed] [Google Scholar]
- Yang, H., G. Luo, P. Karnchanapanurach, T.-M. Louie, I. Rech, S. Cova, L. Xun, and X. S. Xie. 2003. Protein conformational dynamics probed by single molecule electron transfer. Science. 302:262–266. [DOI] [PubMed] [Google Scholar]
- Yeh, S. W., L. J. Ong, J. H. Clark, and A. N. Glazer. 1987. Fluorescence properties of allophycocyanin and a crosslinked allophycocyanin trimer. Cytometry. 8:91–95. [DOI] [PubMed] [Google Scholar]
- Ying, L., and X. S. Xie. 1998. Fluorescence spectroscopy, exciton dynamics, and photochemistry of single allophycocyanin trimers. J. Phys. Chem. 102:10399–10409. [Google Scholar]
- Zehetmayer, P., T. Hellerer, A. Parbel, H. Scheer, and A. Zumbusch. 2003. Spectroscopy of single phycoerythrocyanin monomers: dark state identification and observation of energy transfer heterogeneities. Biophys. J. 83:407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]