

Abstract
The question of how genetic materials are trafficked in and out of the cell nucleus is a problem of great importance not only for understanding viral infections but also for advancing gene-delivery technology. Here we demonstrate a physical technique that allows gene trafficking to be studied at the single-gene level by combining sensitive fluorescence microscopy with microinjection. As a model system, we investigate the nuclear import of influenza genes, in the form of ribonucleoproteins (vRNPs), by imaging single vRNPs in living cells in real time. Our single-particle trajectories show that vRNPs are transported to the nuclear envelope by diffusion. We have observed heterogeneous interactions between the vRNPs and nuclear pore complexes with dissociation rate constants spanning two orders of magnitude. Our single-particle tracking experiments also provided new insights into the regulation mechanisms for the nuclear import of vRNPs: the influenza M1 protein, a regulatory protein for the import process, downregulates the nuclear import of vRNPs by inhibiting the interactions between vRNPs and nuclear pore complexes but has no significant effect on the transport properties of vRNPs. We expect this single-particle tracking approach to find broad application in investigations of genetic trafficking.
INTRODUCTION
Nuclear trafficking of genetic material is critical to a variety of biological processes. For example, many viruses deliver their genomes to the cell nucleus for replication and expression (Whittaker et al., 2000). Gene therapy relies on the delivery of therapeutic genes into the nuclei of host cells. Only recently have we begun to unravel the molecular mechanisms underlying the nuclear trafficking of viral genes (Whittaker et al., 2000). Even less is known about how synthetic gene-delivery materials help to target foreign genes into the cell nucleus (Li and Huang, 2000). Therefore, efforts to combat viral diseases and to improve gene therapy could both benefit from new experimental techniques for investigating gene trafficking. Here we demonstrate that single-particle tracking in living cells (Byassee et al., 2000; Fusco et al., 2003; Goulian and Simon, 2000; Harms et al., 2001; Kues et al., 2001; Lakadamyali et al., 2003; Seisenberger et al., 2001) is a powerful technique to investigate the nuclear trafficking of genetic material. Specifically, tracking a single genetic carrier particle in a living cell can directly identify interactions between the particle and specific cellular machinery. The physical trajectory of a single genetic particle can provide unambiguous insights into its transport mechanisms. This technique can also resolve interesting dynamics of the nuclear trafficking process that may be obscured in ensemble measurements.
We explore the nuclear trafficking of influenza viral genes. Influenza has been used as a model system for understanding the cellular entry of viruses. Influenza viruses enter cells by receptor-mediated endocytosis and then progresses to late endosomes where fusion between the viral and endosomal membrane leads to the release of viral genes (Klasse et al., 1998; Lamb and Krug, 2001; Martin and Helenius, 1991b; Matlin et al., 1981; Skehel and Wiley, 2000; White et al., 1982; Yoshimura and Ohnishi, 1984). These genes, packaged in the form of ribonucleoproteins (vRNPs), are then imported into the nucleus where they direct viral replication and expression (Herz et al., 1981; Kemler et al., 1994; Krug et al., 1989; Lamb and Krug, 2001; Martin and Helenius, 1991a,b).
The influenza genome consists of eight segmented RNAs, ranging from 890 to 2341 nucleotides in length (Lamb and Krug, 2001; McGeoch et al., 1976). Each single-stranded RNA is wrapped around multiple copies of the influenza nucleoprotein, with approximately one nucleoprotein for every 24 nucleotides of RNA (Compans et al., 1972; Lamb, 1989; McGeoch et al., 1976; Murti et al., 1988; Ortega et al., 2000; Pons et al., 1969). The ends of the RNA are bound to a trimeric polymerase complex that transcribes the RNA both to make mRNA and to replicate the virus (Herz et al., 1981; Hsu et al., 1987; Krug et al., 1989). The protein components of vRNP contain nuclear localization signals to facilitate the nuclear import of vRNP via nuclear pore complexes (NPCs) (Bullido et al., 2000; Davey et al., 1985; Jones et al., 1986; Nieto et al., 1994; O'Neill et al., 1995).
In the early stage of infection, vRNPs are predominantly imported into the nucleus; in the late stage, progeny vRNPs assembled in the nucleus are exported from the nucleus, and the import of vRNPs is inhibited (Bui et al., 1996, 2000; Kemler et al., 1994; Martin and Helenius, 1991a,b; Neumann et al., 2000; O'Neill et al., 1998; Whittaker et al., 1996a,b). The influenza matrix protein M1 is responsible for downregulating the nuclear import of vRNP, whereas both M1 and influenza NS2 play a role in the nuclear export of vRNP (Bui et al., 1996, 2000; Martin and Helenius, 1991a; Neumann et al., 2000; O'Neill et al., 1998; Whittaker et al., 1996a,b).
Despite the above advances in understanding the nuclear import and export of vRNP, several important questions remain open. Among these is the molecular mechanism responsible for the transport of vRNPs in the cell. It was found that actin and microtubule-depolymerizing drugs did not block the import of vRNPs (Martin and Helenius, 1991b); however, it remains unknown whether vRNPs are transported by active mechanisms or by diffusion in cells with an intact cytoskeleton. It is also unclear what molecular mechanism is used by M1 to inhibit the nuclear import of vRNPs.
In this work, we use fluorescence microscopy to track the behavior of single influenza vRNP particles in living cells in real time. This has allowed us to show unambiguously that vRNPs are transported in the cytoplasm and nucleus by diffusion. We have also directly observed the interaction between vRNPs and the nuclear envelope, via NPCs. The binding between vRNPs and the NPCs is highly heterogeneous, with dissociation rate constants ranging from 1 to 100 s. In the late stage of infection when M1 is expressed in the cell, the interactions between the vRNPs and nuclear envelope are inhibited significantly, but the transport properties of the vRNPs are nearly identical to that in the early stage of infection. This suggests that M1 downregulates the nuclear import of vRNP by directly inhibiting its binding to the NPCs.
MATERIALS AND METHODS
Reagents
X-31 virus (490715) was purchased from Charles River Laboratories (North Franklin, CT). A monoclonal antibody to influenza nucleoprotein (20302) was purchased from QED Biosciences (San Diego, CA). A monoclonal antibody to nuclear pore o-linked N-acetylglucosamine (ab2734), RL1, was purchased from Novus Biologicals (Littleton, CO). The N2 monoclonal antibody to influenza hemagglutinin was a gift from Dr. Judith White (University of Virginia, Charlottesville, VA). Monoclonal antibody to influenza M1 was a gift from Dr. Adolfo Garcia-Sastre (Mount Sinai School of Medicine, New York). Tetramethylrhodamine goat-anti-mouse (T2762) was purchased from Molecular Probes (Eugene, OR). Wheat germ agglutinin (L-1020) (WGA) was from Vector Laboratories (Burlingame, CA). The BS-C-1 cell line (CCL-26) and cell culture media were from ATCC (Manassas, VA).
Dye labeling of vRNP
Influenza vRNP was purified essentially as described previously (Kemler et al., 1994) with modifications to facilitate dye labeling. Briefly, influenza viruses (1 ml at 2 mg/ml) were pelleted by centrifugation at 35,000 rpm for 40 min at 4°C in a Beckman (Fullerton, CA) SW55 Ti rotor. The pellet was resuspended in disruption buffer of pH 8.1 that contains 100 mM KPO4, 100 mM KCl, 5 mM MgCl2, 5% (w/v) glycerol, 50 mM n-octyl-b-d-glucopyranoside, 10 mg/ml lysolecithin, and 1.5 mM dithiothreitol (DTT) at 31°C for 35 min. Disrupted virus was loaded onto a glycerol step gradient (1 ml 70%, 0.75 ml 50%, 0.375 ml 40%, and 1.8 ml 33% (w/v) glycerol in 100 mM KPO4, 150 mM NaCl, pH 7.8) and centrifuged at 45,000 rpm for 4 h at 4°C; 300-μl fractions were collected and analyzed on an SDS-PAGE gel (data not shown). Fractions 11–13 contain purified vRNPs, consistent with previous results (Kemler et al., 1994). These fractions were pooled, diluted in 10 mM KPO4, 120 mM KCl, pH 8.0, pelleted by centrifugation at 45,000 rpm for 2 h at 4°C, and then resuspended in 50 μl 10 mM KPO4, and 120 mM KCl, pH 8.0. Purified vRNPs were then incubated with amine reactive Cy3 (PA23001; Amersham Biosciences, Piscataway, NJ) in a carbonate buffer (pH 9.3) with occasional mixing for 1 h at room temperature. The reaction was quenched with 10 mM Tris and 120 mM KCl, pH 8.0. Excess dye was removed by three cycles of 1:10 dilution of the vRNP solution into 10 mM Tris and 120 mM Kcl, pH 8.0, followed by concentration with a centricon YM-30 concentrator (42410; Millipore, Chicago, IL). Measurements of the absorption of the dye after each cycle indicated that this procedure was sufficient to remove the free dye and additional cycles of purification do not further reduce the number of free dye molecules in the solution. We note that individual free dye molecules were not detectable under our experimental conditions. The remaining free dyes in the vRNP solution, if any, did not lead to a noticeable increase in the fluorescence background of the cell after the injection of vRNP solutions into the cell. We measured the labeling efficiency by ultraviolet-visible (UV-VIS) absorption spectroscopy and determined that the average number of dyes per nucleoprotein is one. There are on average 70 nucleoproteins in each vRNP complex; so each complex was labeled with ∼70 dye molecules. After labeling and free dye removal, the vRNPs were analyzed by a second glycerol step gradient similar to the one used for purifying vRNPs from the viruses. As determined by SDS-PAGE gel analysis, the dye-labeled vRNPs appeared at identical fractions in the step gradient as the vRNPs before labeling, suggesting that there was no significant degradation or aggregation of vRNP during the labeling process.
Cell culture and drug treatment
BS-C-1 cells were maintained in a 5% CO2 environment in Minimum Eagle Medium (MEM; ATCC) with 10% fetal bovine serum (FBS; ATCC) and passaged every 2–3 days. For fluorescence imaging, BS-C-1 cells were cultured in MEM with 10% FBS in petri dishes with glass coverslips on the bottom. Before fluorescence experiments, cells were washed and incubated in serum-free and phenol red-free MEM for at least 30 min. Cells are viable under these conditions; however, they will not replicate in the absence of FBS. This procedure reduced autofluorescence background due to both phenol red and FBS. In experiments that test the effect of microtubules on the transport properties of vRNPs, the cells were incubated for 30 min before experiments in medium containing 60 μM nocodazole to disrupt microtubules (De Brabander et al., 1976). In experiments that test the effect of actin, cells were incubated for 30 min before experiments in medium containing 20 μM cytochalasin D to disrupt actin filaments (Cooper, 1987). The drugs were maintained in the cell culture media throughout the experiments. Disruption of the actin cytoskeleton by 20 μM cytochalasin D was verified by fixing treated cells and staining with Alexa Fluor 532 phalloidin (A22282; Molecular Probes). Disruption of microtubules by 60 μM nocodazole was verified by immunofluorescence with mouse monoclonal anti-α-tubulin (A11126; Molecular Probes).
Microinjection
Microinjection was done with a homebuilt injector mounted on an Olympus IX70 inverted microscope (Olympus, Melville, NY) at 37°C in serum-free and phenol red-free MEM. The injector consisted of a Narishigi (hi-7; Bioscience Tools–CB Consulting, San Diego, CA) needle holder mounted on a Newport XYZ (460A-XYZ; Newport, Irvine, CA) translation stage. Regulated, low-pressure compressed air was then applied to back of the injection needle to create a constant flow of the injection solution out of the needle. The air pressure applied to the back of the injection needle was measured with a digital pressure gauge (3834K12; McMaster-Carr, Chicago, IL).
For single vRNP tracking experiments, the stock solution of 100 nM vRNP was typically diluted 1:10 into 10 mM Tris and 120 mM KCl, pH 8.0, before injection into the cell,. As vRNPs can stick to the glass injection needle, it is likely that the concentration of vRNP is significantly less after passing through the injection needle. For ensemble experiments that do not require individual vRNPs to be resolved, the stock vRNP solution was directly injected into the cell. Movies were taken at least 2 min after injection to allow labeled vRNP that had leaked from the needle into the extracellular media to dissipate. Furthermore, as free Cy3 dye is membrane permeable, this allowed time for any free dye that might contribute to background to diffuse out of the injected cell.
Epifluorescence and DIC microscopy
The Olympus IX70 microscope was used for both epifluorescence and differential interference contrast (DIC) illumination. Cy3 dye was excited with a 532 nm diode-pumped Nd:YAG laser (GCL-025-M; Crystalaser, Reno, NV). A 545-nm dichroic (545DCLP; Chroma, Brattleboro, VT) was used to reflect the laser line onto the sample. The fluorescent emission was collected and imaged with a NA = 1.45 60× oil immersion objective (Olympus). A 550-nm longpass emission filter (550LP; Chroma) was used to block scattered laser light and to select for Cy3 emission. A charge-coupled device (CCD) camera (Coolsnap HQ; Roper Scientific, Trenton, NJ) and custom software were used to capture movies at 10 frames per second (fps), 1 fps, or 0.2 fps. In all cases, the total exposure time for each frame was 0.1 s. As the Coolsnap is a progressive scan camera, the time to shift the current frame into the read-out pixels is negligible. The 1-fps and 0.2-fps data were taken by opening a computer-controlled shutter (VMM-D1 and LS6T2; Vincent Associates, Rochester, NY.) in front of the laser, and synchronized with the camera, for 0.1 s every 1 s or 5 s. All experiments were conducted at 37°C.
Single-particle tracking
To follow the motion of the vRNP, each frame in the movie was first processed by convolution with a Gaussian of width 1.4 pixels and mean equal to zero. This removed diffuse, low-frequency background signals and helped to reduce noise in the image. Peaks in the fluorescence image due to labeled vRNP were detected using an algorithm that first searched each image for pixels of an intensity that was greater than that of their immediate neighbors. Then, starting at each local maximal pixel, recursion was used to identify all the pixels belonging to a single peak using the criteria that the next pixel along the progress direction is of lower intensity then the pixel under consideration. The intensity of all pixels in the peak was summed to determine the peak brightness. If the peak brightness was above a threshold, the local maximum was marked as a potential vRNP peak.
The trajectories of mobile vRNP were then reconstructed in a semiautomated fashion using the previously identified peak locations. To study the transport properties of the vRNP, we analyze only those vRNPs that could be tracked for at least 20 frames before they disappeared or overlapped with a second vRNP. After we identified the first frame from which the vRNP can be tracked, an automated program was used to determine the peak in the successive frame that is nearest to the peak location in the current frame to construct the physical trajectory of the vRNP. Mistracking was manually corrected. A vRNP particle was characterized as mobile if it did not remain within 0.18 μm (the pixel size) of its current position for more then two frames.
The trajectories of vRNP bound to the nuclear envelope were determined in a manner similar to that described above. A vRNP was identified as being bound to the nuclear envelope if it met the following criteria: i), the total distance traveled in five frames is <0.18 mm (one pixel); ii), it was no more than 0.5 μm outside or 1.5 μm inside the location of nuclear envelope as determined by a DIC image of the cell taken immediately before the start of the fluorescence movies. We note that binding of vRNP in the vicinity of the nuclear envelope was dramatically reduced in the presence of anti-NPC antibodies, indicating that these binding events are indeed due to association of vRNPs with NPCs.
The microscope did not drift by more than 0.2 μm on the timescale of the longest experiments performed (16 min). In the time course of 16 min, ∼60% of the time, the cell morphology would change. It was however still possible to tell the location of the nuclear envelope to within 1–2 μm. This is in part due to the discernable differences in the background fluorescence between the cytoplasm and the nucleus and in part because the vRNP particles that were fixed in the cell change their positions along with the morphology change of the cell; thus the positions of these particles allow us to determine the new position of the nuclear envelope.
Analysis of the motion of fixed vRNPs indicated that the error in our measurement of the vRNP position was 0.11 μm. As the vRNPs contain on average 70 dye molecules and thus are relatively bright, cell autofluorescence does not contribute significantly to the error in the position measurements. At the illumination powers used, the cytoplasm of uninjected cells would give a fluorescence signal comparable to 5–10% of that of a vRNP and fluorescence from the nucleus was negligible.
Simulation
The simulated vRNP trajectories in two dimensions were generated from a series of random numbers with a normal distribution. These numbers were multiplied by a constant factor that was chosen to match the average diffusion coefficient of a real data set. Individual trajectory lengths were randomly chosen from a uniform distribution with a mean equal to the average data set length and a width equal to twice the difference between the average data set length and the shortest trajectory in the data set. The trajectories were rounded to the nearest pixel to more exactly match the real data.
Immunofluorescence
Immunofluorescence was performed essentially as described previously (Martin and Helenius, 1991b). Cells were fixed in phosphate buffered saline (PBS) with 3% formaldehyde for 15 min, neutralized for 15 min in PBS with 50 mM NH4Cl, permeabilized for 6 min in PBS with 0.1% triton, and blocked for 20 min in PBS with 10% FBS (PBS-FBS). Then they were incubated for 30 min with the primary antibody in PBS-FBS. After washing twice using PBS-FBS, they were incubated for another 30 min with the fluorescent secondary antibody in PBS-FBS. Finally, cells were washed twice with PBS-FBS before imaging.
RESULTS
To allow imaging of individual influenza vRNP particles in living cells, we labeled the vRNPs with Cy3 dyes at the level of one dye per nucleoprotein. This corresponds to 70 dye molecules per vRNP on average. Labeled vRNPs were injected into the cytoplasm of BS-C-1 cells at 37°C. After 30 min, the vRNPs were essentially all imported into the nucleus (Fig. 1, A and B). Both previous data (Kemler et al., 1994) and ours (not shown) have demonstrated that microinjected vRNPs are competent for viral protein expression. We compared the import kinetics of the Cy3-labeled and unlabeled vRNPs by injecting cells with the labeled and unlabeled vRNPs, respectively, and then fixing the cells with 3% formaldehyde at varying times after injection. The intracellular vRNP distribution of cells injected with labeled vRNPs was visualized directly by the Cy3 fluorescence, whereas that of cells injected with the unlabeled vRNPs was visualized by immunofluorescence with an antinucleoprotein antibody. The import kinetics of Cy3-labeled vRNPs was essentially identical to that of unlabeled vRNPs (Fig. 1 C), indicating that dye labeling does not change the kinetics of the nuclear import process for vRNPs.
FIGURE 1.

Nuclear import of dye-labeled vRNP. (A) A DIC image of an injected cell. (B) A fluorescence image of the same cell, taken 30 min after vRNP injection, showing nuclear import of the labeled vRNPs. Scale bars: 10 μm. (C) Nuclear import kinetics of labeled and unlabeled vRNPs. The amount of vRNPs in the cytoplasm and nucleus were quantified using Cy3 fluorescence and antinucleoprotein immunofluorescence for the labeled and unlabeled vRNPs, respectively. Fin and Fout indicate, respectively, the average fluorescence intensities inside and outside the nucleus after subtraction of a background term determined from areas outside the injected cells. The t = 0 point indicates the time of injection. The value of Fin/(Fin + Fout) at t = 0 was determined by coinjecting WGA (5mg/ml) with labeled vRNP to block import. This value is not zero because vRNPs that are outside but directly above or below the nucleus contribute to Fin.
It has been shown previously that vRNPs are imported into the nucleus via NPCs on the nuclear envelope (Kemler et al., 1994; Martin and Helenius, 1991b). To confirm that the dye-labeled vRNPs used the same pathway to enter the nucleus, we coinjected labeled vRNPs with 5 mg/ml WGA or 2 mg/ml anti-NPC, both of which are known to block nuclear import via NPCs (Featherstone et al., 1988; Finlay et al., 1987). In both cases, the nuclear import of labeled vRNPs was blocked (data shown later), indicating that labeled vRNPs indeed enter the nucleus via the NPCs.
Next, we explored the transport mechanism of vRNP by tracking single vRNP particles in living BS-C-1 cells. Low concentrations of labeled vRNPs were injected into the cells so that the instantaneous positions of individual vRNP particles could be tracked (Fig. 2 A and Supplementary Movie S1). The flat morphology of this cell type helps tracking of individual vRNPs by keeping them in or near the focal plane of the microscope. Under typical experimental conditions, ∼60% of the vRNP particles were mobile, and individual vRNPs could be tracked for ∼5 s (50 frames).
FIGURE 2.

Tracking the movement of single vRNPs. (A) A DIC image of part of a cell with the trajectories of two example vRNP particles overlaid as black lines. Scale bar: 10 μm. (B) The measured mean-square displacement (〈Δr2〉) versus time (Δt) for four example vRNPs in the cytoplasm of a cell. Lines are the best fit to 〈Δr2〉 = 4DΔt, with D being the diffusion coefficient. (C) 〈Δr2〉 versus Δt for four example vRNPs in the nucleus of a cell. The vRNP trajectories were determined 15 min after injection; this allowed the majority of the vRNPs to import into the nucleus. (D) The average mean-square displacement for all the vRNP in the cytoplasm (□) or in the nucleus (○). The cytoplasm data include 108 vRNP trajectories from six different cells, and the nucleus data include 37 vRNP trajectories from six different cells. The average trajectory was 113 frames long in the cytoplasm and 54 frames long in the nucleus.
To test whether vRNPs are transported by diffusion or by directed, active transport mechanisms, we determined the relation between the mean-square distance (〈Δr2〉) traveled by each mobile vRNP particle and the traveling time (Δt). For vRNPs both in the cytoplasm and in the nucleus 〈Δr2〉 increases linearly with Δt (Fig. 2, B and C), and the velocity autocorrelation calculated from all the vRNP trajectories decays to zero in a single step (Fig. 3, A and B). We averaged the 〈Δr2〉 versus Δt traces of all mobile vRNP particles in either the cytoplasm or the nucleus to more carefully check for anomalous diffusion of the vRNPs. As shown in Fig. 2 D, the average 〈Δr2〉 is also linear with Δt, further indicating that the vRNP particles move by diffusion. The slope of the best-fit line gives an average diffusion coefficient of 0.7 μm2/s in the cytoplasm.
FIGURE 3.

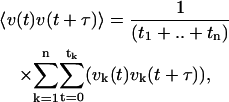 |
Electron microscopy indicates that the eight distinct vRNPs are ∼20 nm in diameter and 30–100 nm long (Compans et al., 1972; Pons et al., 1969). A spherical particle of diameter 30–60 nm, an object similar in size to a vRNP, would have a diffusion coefficient of 7 to 15 μm2/s in water. In the cytoplasm, such a particle would have a diffusion coefficient ∼10–20 times smaller than that in water (Luby-Phelps, 2000). This means that the expected diffusion coefficient would be ∼0.3–1.5 μm2/s, very close to what we observed for vRNP particles.
To further characterize the diffusion properties of vRNPs in the cytoplasm, we compare our measured distribution of diffusion coefficients with a simulated distribution (Fig. 3 C). The simulated distribution was derived from a set of 1000 simulated vRNP diffusion trajectories generated using the average diffusion coefficient obtained from experiments as the diffusion coefficient. The length of the trajectories in time was randomly chosen from a range that mimics the experimental trajectory lengths (see Material and Methods). As seen in Fig. 3 C, the distribution of diffusion coefficients is moderately broader then expected from the simulation. This extra width may be due to the size of distribution of vRNP particles: the influenza genome includes eight segmented RNA genes, ranging from 890 to 2341 nucleotides in length. Another possible reason for the additional width may arise from the local differences in the microenvironment of the cell (Goulian and Simon, 2000; Kues et al., 2001). We note that the only the vRNPs that can be tracked for more than 20 frames (2 s) are analyzed, and such analysis may bias the distribution toward the more slowly diffusing vRNPs.
In the early stage of infection, incoming vRNPs are imported and retained in the nucleus. By observing the trajectories of vRNPs after import into the nucleus, we found that the movement of vRNP was also diffusive with a moderately lower diffusion coefficient (Figs. 2, C and D, and 3 D), indicating that the vRNP are not retained in the nucleus by interactions with fixed nuclear structures.
To further explore the transport properties of vRNPs in cells, we carried out two control experiments and measured the transport properties of vRNPs in cells treated with either a microtubule-disrupting drug (nocodazole) or an actin-disrupting drug (cytochalasin D). Although these drugs disrupt the microtubules (De Brabander et al., 1976) or the actin filaments (Cooper, 1987; Cooper et al., 1987) and block the actin- or microtubule-dependent transport of influenza viruses inside cells (Lakadamyali et al., 2003), we found that the transport properties of vRNPs in treated cells was similar to that in untreated cells and that the nuclear import of vRNPs was not perturbed by these two drugs (Martin and Helenius, 1991b). These observations further support the conclusion that the vRNP are transported primarily by diffusion in the cytoplasm.
Next, we investigate the interaction of vRNPs with NPCs. After labeled vRNPs were injected into a cell, a fluorescent ring was observed around the nucleus before significant import, indicating the preferential binding of the vRNPs to the nuclear envelope (Fig. 4 A). In a control experiment, a monoclonal antibody against nuclear pore o-linked N-acetylglucosamine (RL1) that is known to block nuclear import (Featherstone et al., 1988) was coinjected with the vRNPs. Western blots have shown that this antibody binds to a subset of the protein components of purified nuclear pore complexes (Snow et al., 1987). In addition, immunofluorescence and immunogold electron microscopy experiments have demonstrated that this antibody specifically targets the nuclear pore complex in fixed cells (Snow et al., 1987). The import of vRNPs was indeed blocked by the presence of this antibody, and no fluorescent ring was observed around the nucleus (Fig. 4 B), suggesting that the binding of vRNPs to the nuclear envelope was mediated by the NPCs. By contrast, when WGA, a molecule that is known to only inhibit translocation across the NPCs but does not block binding sites on the NPC (Newmeyer and Forbes, 1988), was coinjected with the vRNPs, import of vRNPs was again blocked, but the accumulation of vRNPs at the nuclear envelope was more pronounced (Fig. 4 C). The above results indicate that the observed association of the vRNPs with the nuclear envelope was due to their binding to the NPCs, probably through karyopherin molecules, the nuclear import factors that mediate the binding of vRNP to the NPC (O'Neill et al., 1995).
FIGURE 4.

The interaction of the vRNP with the nuclear envelope. (A) A fluorescence image of a cell taken 2 min after injection of labeled vRNP. The ring at the nuclear envelope indicates association of vRNPs with the nuclear envelope. (B) A fluorescence image of a cell that was coinjected with labeled vRNP and anti-NPC (RL1). (C) A fluorescence image of a cell that was coinjected with labeled vRNP and WGA. (D) Same as A but with a lower concentration of labeled vRNP to allow the detection of individual vRNP particles. The gray line indicates the location of the nuclear envelope, determined using DIC microscopy. The image was convolved with a Gaussian filter to reduce noise. Scale bars: 10 μm.
To characterize this interaction quantitatively, we measured the time that each vRNP molecule spent bound to the nuclear envelope. Fig. 4 D and Supplementary Movie S1 show that preferential binding of the vRNP at the nuclear envelope can be observed at the single-vRNP level when a lower concentration of vRNPs is injected into the cell. The time distribution of these binding events shows that vRNP dissociates from the nuclear envelope with dramatically different time constants. We therefore performed single-particle tracking experiments at three different time resolutions: 0.1 s, 1 s, and 5 s. Although the high time resolution data allow us to determine more accurately the smaller dissociation time constants, laser excitation is much lower in the lower time resolution experiments and thus allows us to track the vRNP particles for a longer time, leading to a more accurate determination of the larger time constants. At each time resolution, the total number of frames was kept to 200, corresponding to 20s, 200s, and 1000s data acquisition times for the three different time resolutions, respectively (see Materials and Methods for details). Photobleaching is negligible in 200 frames. At a certain time resolution, only the dissociation time constants that are much smaller than the observation time window are trusted with confidence. At all but the lowest time resolution, the time distribution of these vRNP binding events cannot be fit well by single exponential decays but requires at least two time constants to fit the experimental data. The combination of all three sets of data appears to indicate the existence of three discrete dissociation constants: 1 s, 10 s, and 90 s (Fig. 5, A–C). We do not formally preclude the possibility that vRNPs bind to the nuclear envelope with a continuous distribution of time constants. The above results indicate the presence of multiple types of interactions between the vRNP and the NPC. The occurrence frequency of the binding events is higher than that of the nuclear import events of vRNPs, as estimated from their import kinetics. This result suggests that a vRNP particle binds to NPCs multiple times before its translocation through the NPCs. We note that it is difficult to determine directly whether a binding event leads to nuclear entry by tracking the vRNP position because a vRNP above or below the nucleus may appear to be inside the nucleus due to the depth of focus of the wide-field microscope.
FIGURE 5.

Integrated time histograms of the duration of binding events of individual vRNPs to the nuclear envelope. (A) The number of vRNP binding events with disassociation times shorter than the time indicated on the horizontal axis. The binding times were determined from movies taken at 10 fps (0.1-s time resolution) on 13 cells. The solid line is the best-fit single exponential decay with a time constant τ = 2.2 s and the shaded line is the best-fit double exponential decay with two time constants τ1 = 1.1 s and τ2 = 5.8 s. The second time constant is probably shorter than the actual dissociation time constant due to the finite observation window (20 s). (B) Integrated time histograms of the binding events of vRNPs to the nuclear envelope determined from movies taken at 1 fps (1-s time resolution) on six cells. The solid line is the best-fit single exponential decay (τ = 24 s), and the shaded line is the best-fit double exponential decay (τ1 = 10.4 s and τ2 = 113 s). Again the second time constant is probably not accurately determined due to the finite observation window (200 s). (C) Integrated time histograms of the binding events determined from movies taken at 0.2 fps (5-s time resolution) on eight cells. The solid line is the best-fit single exponential decay (τ = 89 s). These three experiments combined suggest the presence of three dissociation time constants, 1 s, 10 s, and 89 s.
Late in infection, the nuclear import of vRNPs is inhibited to prevent progeny vRNPs from reentering the nucleus, thus facilitating virus packaging. The protein responsible for this downregulation is influenza M1 (Bui et al., 1996, 2000; Whittaker et al., 1996a). How M1 inhibits the nuclear import of vRNP is, however, unclear. Several different mechanisms may be possible: M1 could inhibit the nuclear import of vRNPs by promoting their interactions with cellular matrices, thus keeping them away from the nuclear envelope; by blocking the binding of vRNPs to the NPCs; or alternatively by preventing the vRNPs bound to NPCs from translocating through the pore.
To distinguish the above mechanisms, we injected labeled vRNP into BS-C-1 cells that were preinfected with influenza viruses for 6 h. Under this condition, M1 proteins are expressed in cells, as determined by immunofluorescence (data not shown), and the nuclear import of vRNPs was blocked (Fig. 6 A). Tracking of single vRNPs revealed that they still moved diffusively, as indicated by the linear dependence of 〈Δr2〉 on Δt (not shown) and the velocity autocorrelation function (Fig. 6 B). The diffusion coefficients were slightly larger than those in the absence of M1 (Fig. 6 C). This shows that M1 does not block import of the vRNP by promoting the interaction of vRNP with cellular matrices or inhibiting the mobility of vRNPs.
FIGURE 6.

vRNP transport and vRNP-NPC interaction during the late stage of infection. (A) A fluorescence image of a cell taken 10 min after injection with labeled vRNPs. The cell was infected with influenza viruses 6 h before injection. No fluorescent ring is observed at the nuclear envelope in contrast to Fig. 3 A. (B) The velocity autocorrelation of all the vRNP trajectories in the cytoplasm of preinfected cells (163 trajectories and 17 cells). (C) The distribution of diffusion coefficients, D, in the cytoplasm of infected cells (gray) compared to that of uninfected cells (white).
When labeled vRNPs were injected into cells preinfected with influenza, preferential association of vRNPs with the nuclear envelope was not observed (Fig. 6 A), whereas such a behavior (the fluorescent ring at the nuclear envelope) was observed when the same concentration of vRNPs were injected into uninfected cells (Fig. 4, A and C). This suggests that preinfection significantly inhibits the binding interaction between the vRNP and the nuclear envelope. In addition, we performed experiments at the single-vRNP level by injecting a lower concentration of vRNPs. We found that in movies taken at 10 fps there were on average 1.3 binding events per preinfected cell (15 cells total) versus 14 binding events per uninfected cell (13 cells total). After accounting for the apparent differences in vRNP concentration in the cell by counting the average number of vRNP visible in the infected versus uninfected cells, we determined that the binding frequency of vRNP to the nuclear envelope is reduced by fourfold.
These results indicate that in the late stage of infection the binding of the vRNPs to the nuclear envelope is significantly inhibited. We propose that M1 proteins block the nuclear localization signals on the vRNP from interacting with karyopherin molecules, the nuclear import factors that mediate the binding of vRNP to the NPC (O'Neill et al., 1995). This effectively blocks the interaction of vRNPs with NPCs and thus inhibits the nuclear import of the vRNPs.
DISCUSSION
In the course of an influenza infection, the vRNP enter the cytoplasm after viral fusion with a late endosomal compartment. The sites where this fusion occurs are often at some distance from the nucleus, and thus the mechanism by which the vRNP are transported in the cytosol is important for the nuclear import of vRNP. Our single-vRNP trajectories directly show that influenza genes are transported by diffusion, both in the cytoplasm and in the nucleus. This experiment allowed the transport mechanisms of vRNPs to be determined without the use of depolymerizing drugs that perturb normal cellular functions. The time trajectories of single vRNPs have also revealed binding between the vRNPs and the nuclear envelope, with dissociation rate constants ranging from 0.01 to 1 s−1. Experiments in the presence of anti-NPC and WGA indicate that these binding events are due to vRNP-NPC interactions, likely mediated by nuclear import factors. We found that the binding of vRNPs to NPCs in the late stage of infection was inhibited in comparison to the early infection stage, but that the transport properties of vRNPs are almost identical in both stages. These results suggest that M1, expressed in the late stage of infection, inhibits the nuclear import of vRNPs by directly blocking their interactions with NPCs. M1 may block the vRNP-NPC interaction by blocking the nuclear localization signals on the vRNP, preventing their interaction with the nuclear import factors that mediate the binding of vRNP to the NPC (O'Neill et al., 1995).
Our experiments have shown that the behavior of single viral genes can be tracked in living cells. We expect this single-particle tracking approach to have many applications in investigations of gene trafficking in viral infection and gene delivery technologies. The physical and time trajectories of single genetic particles, the direct visualization of interactions between these particles and cellular machinery, and the transient dynamic information revealed by the time trajectories can provide critical insights, complementary to ensemble measurement results, into the molecular mechanisms underlying the trafficking of genetic materials in cells.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
We thank Dr. Matt Michael for the loan of a microinjector at an early stage of the experiment. We thank Dr. Judy White for the N2 antibody and Dr. Adolfo Garcia-Sastre for the anti-M1 antibody.
This work was supported in part by a Searle Scholar award, a Beckman Young Investigator award, the Office of Naval research, and the National Science Foundation (to X.Z.).
References
- Bui, M., G. Whittaker, and A. Helenius. 1996. Effect of M1 protein and low pH on nuclear transport of influenza virus ribonucleoproteins. J. Virol. 70:8391–8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui, M., E. G. Wills, A. Helenius, and G. Whittaker. 2000. Role of the influenza virus M1 protein in nuclear export of viral ribonucleoproteins. J. Virol. 74:1781–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullido, R., P. Gomez-Puertas, C. Albo, and A. Portela. 2000. Several protein regions contribute to determine the nuclear and cytoplasmic localization of the influenza A virus nucleoprotein. J. Gen. Virol. 81:135–142. [DOI] [PubMed] [Google Scholar]
- Byassee, T. A., W. C. Chan, and S. Nie. 2000. Probing single molecules in living cells. Anal. Chem. 72:5606–5611. [DOI] [PubMed] [Google Scholar]
- Compans, R. W., J. Content, and P. H. Duesberg. 1972. Structure of the ribonucleoprotein of influenza virus. J. Virol. 10:795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, J. A. 1987. Effects of cytochalasin and phalloidin on actin. J. Cell Biol. 105:1473–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper J. A., J. Bryan, B. Schwab III, C. Frieden, D. J. Loftus, and E. L. Elson. 1987. Microinjection of gelsolin into living cells. J. Cell Biol. 104:491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey, J., N. J. Dimmock, and A. Colman. 1985. Identification of the sequence responsible for the nuclear accumulation of the influenza virus nucleoprotein in xenopus oocytes. Cell. 40:667–675. [DOI] [PubMed] [Google Scholar]
- De Brabander, M. J., R. M. Van de Veire, F. E. Aerts, M. Borgers, and P. A. Janssen. 1976. The effects of methyl (5-(2-thienylcarbonyl)-1H-benzimidazol-2-yl) carbamate, (R-17934; NSC-238159), a new synthetic antitumoral drug interfering with microtubules, on mammalian-cells culture in vitro. Cancer Res. 36:905–916. [PubMed] [Google Scholar]
- Featherstone, C., M. K. Darby, and L. Gerace. 1988. A monoclonal-antibody against the nuclear-pore complex inhibits nucleocytoplasmic transport of protein and RNA in vivo. J. Cell Biol. 107:1289–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay, D. R., D. D. Newmeyer, T. M. Price, and D. J. Forbes. 1987. Inhibition of in-vitro nuclear transport by a lectin that binds to nuclear pores. J. Cell Biol. 104:189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco, D., N. Accornero, B. Lavoie, S. M. Shenoy, J. M. Blanchard, R. H. Singer, and E. Bertrand. 2003. Single mRNA molecules demonstrate probabilistic movement in living mammalian cells. Curr. Biol. 13:161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulian, M., and S. M. Simon. 2000. Tracking single proteins within cells. Biophys. J. 79:2188–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms, G. S., L. Cognet, P. H. M. Lommerse, G. A. Blab, H. Kahr, R. Gamsjager, H. P. Spaink, N. M. Soldatov, C. Romanin, and T. Schmidt. 2001. Single-molecule imaging of L-type Ca2+ channels in live cells. Biophys. J. 81:2639–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz, C., E. Stavnezer, R. M. Krug, and T. Gurney. 1981. Influenza-virus, an RNA virus, synthesizes its messenger-RNA in the nucleus of infected-cells. Cell. 26:391–400. [DOI] [PubMed] [Google Scholar]
- Hsu, M.-T., J. D. Parvin, S. Gupta, M. Krystal, and P. Palese. 1987. Genomic RNAs of influenza viruses are held in a circular conformation in virions and in infected cells by a terminal panhandle. Proc. Natl. Acad. Sci. USA. 84:8140–8144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, I. M., P. A. Reay, and K. L. Philpott. 1986. Nuclear localization of all three influenza polymerase proteins and a nuclear signal in polymerase PB2. EMBO J. 5:2371–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemler, I., G. Whittaker, and A. Helenius. 1994. Nuclear import of microinjected influenza virus ribonucleoproteins. Virology. 202:1028–1033. [DOI] [PubMed] [Google Scholar]
- Klasse, P. J., R. Bron, and M. Marsh. 1998. Mechanisms of enveloped virus entry into animal cells. Adv. Drug. Deliv. Rev. 34:65–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug, R. M., and F. V. Alonso-Carlen, I. Julkunen, and M. G. Katze. 1989. Expression and replication of the influenza virus genome. In The Influenza Viruses. R. M. Krug, editor. Plenum Press, New York. 89–152.
- Kues, T., A. Dickmanns, R. Luhrmann, R. Peters, and U. Kubitscheck. 2001. High intranuclear mobility and dynamic clustering of the splicing factor U1 snRNP observed by single particle tracking. Proc. Natl. Acad. Sci. USA. 98:12021–12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakadamyali, M., M. J. Rust, H. P. Babcock, and X. Zhuang. 2003. Visualizing infection of individual influenza viruses. Proc. Natl. Acad. Sci. USA. 100:9280–9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb, R. A. 1989. Genes and proteins of the influenza viruses. In The Influenza Viruses. R. M. Krug, editor. Plenum Press, New York. 1–87.
- Lamb, R. A., and R. M. Krug. 2001. Orthomyxoviridae: the viruses and their replication. In Fields Virology. D. M. Knipe and P. M. Howley, editors. Lippincott Williams and Wilkins, Philadelphia. 1487–1531.
- Li, S., and L. Huang. 2000. Nonviral gene therapy: promises and challenges. Gene Ther. 7:31–34. [DOI] [PubMed] [Google Scholar]
- Luby-Phelps, K. 2000. Cytoarchitecture and physical properties of cytoplasm: volume, viscosity, diffusion, intracellular surface area. Int. Rev. Cytol. 192:189–221. [DOI] [PubMed] [Google Scholar]
- Martin, K., and A. Helenius. 1991a. Nuclear transport of influenza virus ribonucleoproteins: the viral matrix protein (M1) promotes export and inhibits import. Cell. 67:117–130. [DOI] [PubMed] [Google Scholar]
- Martin, K., and A. Helenius. 1991b. Transport of incoming influenza virus nucleocapsids into the nucleus. J. Virol. 65:232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlin, K. S., H. Reggio, A. Helenius, and K. Simons. 1981. Infectious entry pathway of influenza-virus in a canine kidney-cell line. J. Cell Biol. 91:601–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeoch, D., P. Fellner, and C. Newton. 1976. Influenza virus genome consists of eight distinct RNA species. Proc. Natl. Acad. Sci. USA. 73:3045–3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murti, K. G., R. G. Webster, and I. M. Jones. 1988. Localization of RNA-polymerases of influenza virus ribonucleoproteins by immunogold labeling. Virology. 164:562–566. [DOI] [PubMed] [Google Scholar]
- Neumann, G., M. T. Hughes, and Y. Kawaoka. 2000. Influenza A virus NS2 protein mediates vRNP nuclear export through NES-independent interaction with hCRM1. EMBO J. 19:6751–6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newmeyer, D. D., and D. J. Forbes. 1988. Nuclear import can be separated into distinct steps in vitro: nuclear pore binding and translocation. Cell. 52:641–653. [DOI] [PubMed] [Google Scholar]
- Nieto, A., S. Delaluna, J. Barcena, A. Portela, and J. Ortin. 1994. Complex structure of the nuclear translocation signal of influenza-virus polymerase PA subunit. J. Gen. Virol. 75:29–36. [DOI] [PubMed] [Google Scholar]
- O'Neill, R. E., R. Jaskunas, G. Blobel, P. Palese, and J. Moroianu. 1995. Nuclear import of influenza-virus RNA can be mediated by viral nucleoprotein and transport factors required for protein import. J. Biol. Chem. 270:22701–22704. [DOI] [PubMed] [Google Scholar]
- O'Neill, R. E., J. Talon, and P. Palese. 1998. The influenza virus NEP (NS2 protein) mediates the nuclear export of viral ribonucleoproteins. EMBO J. 17:288–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega, J., J. Martin-Benito, T. Zurcher, J. M. Valpuesta, J. L. Carrascosa, and J. Ortin. 2000. Ultrastructural and functional analyses of recombinant influenza virus ribonucleoproteins suggest dimerization of nucleoprotein during virus amplification. J. Virol. 74:156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons, M. W., I. T. Schulze, G. K. Hirst, and R. Hauser. 1969. Isolation and characterization of ribonucleoprotein of influenza virus. Virology. 39:250–259. [DOI] [PubMed] [Google Scholar]
- Seisenberger, G., M. U. Ried, T. Endreβ, H. Buning, M. Hallek, and C. Brauchle. 2001. Real-time single-molecule imaging of the infection pathway of an Adeno-associated virus. Science. 294:1929–1932. [DOI] [PubMed] [Google Scholar]
- Skehel, J. J., and D. C. Wiley. 2000. Receptor binding and membrane fusion in viral entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69:531–569. [DOI] [PubMed] [Google Scholar]
- Snow, C. M., A. Senior, and L. Gerace. 1987. Monoclonal antibodies identify a group of nuclear pore complex glycoproteins. J. Cell Biol. 104:1143–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, J., A. Helenius, and M.-J. Gething. 1982. Hemagglutinin of influenza-virus expressed from a cloned gene promote membrane-fusion. Nature. 300:658–659. [DOI] [PubMed] [Google Scholar]
- Whittaker, G., M. Bui, and A. Helenius. 1996a. Nuclear trafficking of influenza virus ribonucleoproteins in heterokaryons. J. Virol. 70:2743–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker, G., M. Bui, and A. Helenius. 1996b. The role of nuclear import and export in influenza virus infection. Trends Cell Biol. 6:67–71. [DOI] [PubMed] [Google Scholar]
- Whittaker, G. R., M. Kann, and A. Helenius. 2000. Viral entry into the nucleus. Annu. Rev. Cell Dev. Biol. 16:627–651. [DOI] [PubMed] [Google Scholar]
- Yoshimura, A., and S. Ohnishi. 1984. Uncoating of influenza-virus in endosomes. J. Virol. 51:497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.