

Abstract
The E462R mutation in the fifth position of the AID (α1 subunit interaction domain) region in the I-II linker is known to significantly accelerate voltage-dependent inactivation (VDI) kinetics of the L-type CaV1.2 channel, suggesting that the AID region could participate in a hinged-lid type inactivation mechanism in these channels. The recently solved crystal structures of the AID-CaVβ regions in L-type CaV1.1 and CaV1.2 channels have shown that in addition to E462, positions occupied by Q458, Q459, E461, K465, L468, D469, and T472 in the rabbit CaV1.2 channel could also potentially contribute to a hinged-lid type mechanism. A mutational analysis of these residues shows that Q458A, Q459A, K465N, L468R, D469A, and T472D did not significantly alter VDI gating. In contrast, mutations of the negatively charged E461, E462, and D463 to neutral or positively charged residues increased VDI gating, suggesting that the cluster of negatively charged residues in the N-terminal end of the AID helix could account for the slower VDI kinetics of CaV1.2. A mutational analysis at position 462 (R, K, A, G, D, N, Q) further confirmed that E462R yielded faster VDI kinetics at +10 mV than any other residue with E462R ≫ E462K ≈ E462A > E462N > wild-type ≈ E462Q ≈ E462G > E462D (from the fastest to the slowest). E462R was also found to increase the VDI gating of the slow CEEE chimera that includes the I-II linker from CaV1.2 into a CaV2.3 background. The fast VDI kinetics of the CaV1.2 E462R and the CEEE + E462R mutants were abolished by the CaVβ2a subunit and reinstated when using the nonpalmitoylated form of CaVβ2a C3S + C4S (CaVβ2a CS), confirming that CaVβ2a and E462R modulate VDI through a common pathway, albeit in opposite directions. Altogether, these results highlight the unique role of E461, E462, and D463 in the I-II linker in the VDI gating of high-voltage activated CaV1.2 channels.
INTRODUCTION
The influx of calcium through voltage-gated Ca2+ channels regulates a wide range of cellular processes, including contraction, activation of Ca2+-dependent enzymes, and gene regulation. To this date, molecular cloning has identified the gene encoding for three distinct families of calcium channel α1 subunits. The CaV1 family encodes the high-voltage activated (HVA) L-type channels; the CaV2 family produces the HVA P/Q-, N-, and R- type channels, whereas CaV3 channels form the low-voltage activated (LVA) T-type channels (Ertel et al., 2000; Lee et al., 1999a; Monteil et al., 2000; Piedras-Renteria and Tsien, 1998). Whereas all voltage-gated Ca2+ channel α1 subunits activate and inactivate in response to membrane depolarization, the HVA CaV1 and CaV2 α1 subunits operate at markedly more positive membrane potentials than LVA CaV3 channel α1 subunits.
Inactivation is a distinctive feature of all voltage-gated ion channels providing a negative feedback response to prolonged depolarizations. Under physiological conditions, inactivation of the L-type CaV1.2 channel proceeds mostly in response to localized elevation of intracellular Ca2+ (deLeon et al., 1995; Bernatchez et al., 1998) through constitutively bound) calmodulin (CaM) (Qin et al., 1999; Zuhlke et al., 1999; Peterson et al., 1999; Lee et al., 1999b). Recent observations suggest that calcium-dependent inactivation (CDI) and VDI could proceed from similar molecular mechanisms since stripping off preassociated CaM (apocalmodulin) from the C-terminal results both in the ablation of CDI and in a striking acceleration of VDI (Liang et al., 2003). CaM preassociation on the C-terminal could thus be a potent determinant of VDI in CaV1 and CaV2 channels (Liang et al., 2003).
Voltage-dependent inactivation (VDI) has been traditionally investigated in the presence of Ba2+ as the charge carrier. Fast and slow VDI mechanisms have been proposed in CaV1.2 channels based on the kinetics of Ba2+-dependent inactivation. The analysis of gating currents has further shown that the fast VDI component (<1 s depolarization) involves charge immobilization similar to voltage-gated Na+ and K+ channels (Ferreira et al., 2003), suggesting that cationic selective voltage-gated channels share similar structural mechanisms of VDI. As in voltage-gated K+ channels (Liu et al., 1996), mutations in the pore region (IIS6, IIIS6, and IVS6) of CaV1.2 have been shown to slow VDI gating (Hering et al., 1996, 1998; Stotz et al., 2000; Stotz and Zamponi, 2001; Berjukow and Hering, 2001; Shi and Soldatov, 2002). In addition to C-type inactivation, a hinged-lid type mechanism could contribute to the fast VDI gating in HVA CaV1 and CaV2 channels (see for review, Stotz et al., 2004). Molecular studies have rapidly converged toward the high-affinity CaVβ subunit binding site AID (α1 subunit interaction domain) in the I-II linker of HVA CaV channels (Page et al., 1997; Herlitze et al., 1997; Cens et al., 1999; Stotz et al., 2000; Bernatchez et al., 2001a,b; Berrou et al., 2001). The AID region displays a high degree of identity between the HVA CaV1 and CaV2 families with 10 out of 18 residues being strictly conserved (see Fig. 2 A). We have shown that introducing negatively charged residues at the fifth position of the AID region significantly decreased the VDI kinetics and voltage dependence of CaV2.3, whereas the combined mutations of other nonconserved residues had little impact on VDI gating (Berrou et al., 2001). These observations have led to the attractive suggestion that the AID region forms a blocking particle contributing to a hinged-lid type inactivation mechanism in HVA CaV2 channels (Stotz et al., 2004; Kim et al., 2004). The presence of a negatively charged residue at the equivalent position in L-type CaV1.2 is believed to account for the slower VDI kinetics in this channel, although in that case, the data have long been limited to the single E462R mutation (Herlitze et al., 1997; Berrou et al., 2001).
FIGURE 2.

E461A accelerates VDI kinetics in CaV1.2. (A) CaVβ subunit binding site on the α1 subunit (AID) is located within 22 residues of the IS6 transmembrane segment. The primary sequence for the AID helix in CaV2.3 and CaV1.2 channels is shown with the residues putatively accessible for protein interaction highlighted in bold letters. The residues specific to CaV2.3 are shown in italic. (B) Whole-cell current traces are shown from left to right for CaV1.2 (XhoI) wt, E461A, K465N, and T472D in 10 mM Ba2+. Unless specified otherwise, mutants were expressed in Xenopus oocytes in the presence of CaVα2bδ and CaVβ3 subunits, and currents were recorded using the two-electrode voltage-clamp technique in the presence of 10 mM Ba2+. Holding potential was −80 mV throughout. Oocytes were pulsed from −40 mV to +60 mV using 10 mV steps for 450 ms. Capacitive transients were erased for the first millisecond after the voltage step. All mutants tested expressed significant whole-cell currents with similar activation properties (Table 1). (C) r300 values (the fraction of whole-cell currents remaining at the end of a 300 ms pulse) are shown as mean ± SE for the mutated hydrophilic residues from 0 to +20 mV for CaV1.2 (XhoI) wt; E461A, E462R, K465N, L468R, D469A, T472D, and Q473K (from left to right). The r300 ratios varied from 0.73 ± 0.01 at 0 mV to 0.67 ± 0.02 at +20 mV (23) for CaV1.2 (XhoI) wt; 0.53 ± 0.02 at 0 mV to 0.44 ± 0.03 at +20 mV (11) for E461A; 0.38 ± 0.02 at 0 mV to 0.29 ± 0.01 at +20 mV (20) for E462R; 0.87 ± 0.02 at 0 mV to 0.82 ± 0.01 at +20 mV (10) for K465N; 0.82 ± 0.01 at 0 mV to 0.76 ± 0.02 at +20 mV (7) for L468R; 0.69 ± 0.02 at 0 mV to 0.63 ± 0.03 at +20 mV (11) for D469A; 0.85 ± 0.01 at 0 mV to 0.77 ± 0.01 at +20 mV (16) for T472D; and 0.81 ± 0.01 at 0 mV to 0.71 ± 0.01 at +20 mV (15) for Q473K. As compared with CaV1.2 (XhoI) wt, the r300 were significantly smaller at p < 10−16 for E462R and at p < 10−6 for E461A when measured at 0 mV, whereas they were significantly larger for K465N, L468R, T472D, and Q473K at 0.001 < p < 0.01 under the same conditions. In contrast, r300 were not significantly different between CaV1.2 (XhoI) wt and D469A. (D) Voltage dependence of inactivation was estimated from isochronal inactivation data points measured after 5 s conditioning pulses applied between −100 and +30 mV from a holding potential of −100 mV. The fraction of the noninactivating current was recorded at the end of the pulse and data were fitted to Boltzmann Eq. 1. The voltage dependence of inactivation was similar for all constructs. The final fraction of noninactivating Ba2+ current decreased from 0.46 ± 0.03 (9) for T472D and 0.46 ± 0.05 (10) for K465N; to 0.38 ± 0.04 (7) for L468R and 0.30 ± 0.03 (15) for Q473K; to 0.20 ± 0.02 (19) for CaV1.2 (XhoI) wt; to 0.14 ± 0.04 (10) for E461A, 0.13 ± 0.02 (5) for D469A; and 0.09 ± 0.01 (19) for E462R at + 10 mV. Complete set of fit values are shown in Table 1.
In the recently solved crystal structures of the AID-CaVβ regions in L-type CaV1.1 and CaV1.2 channels (Van Petegem et al., 2004; Chen et al., 2004; Opatowsky et al., 2004), the AID region adopts an α-helical structure upon binding to the CaVβ subunit with hydrophilic residues lined up exclusively on the face of the helix opposite to CaVβ (Fig. 1). The hydrophilic E462 was thus shown to be correctly positioned to interact with other proteins and/or regions of the channel as postulated in a hinged-lid type inactivation mechanism. According to the published three-dimensional structures, the side chains of Q458, Q459, E461, K465, D469, and T472 are equally available and poised to interact with other proteins/regions of the channel. In this regard, Q458, Q459, and E461 residues are strictly conserved between CaV1 and CaV2 families and were consequently believed to participate to CaVβ binding. Their role in VDI gating was never investigated before.
FIGURE 1.

(A) Three-dimensional representation of the AID helix of the rabbit CaV1.2 obtained with INSIGHT II using the human Cav1.2 crystal structure cocrystallized with CaVβ2a (Protein Data Bank: 1T0J.pdb) as a template (Van Petegem et al., 2004). The core of the AID helix appears in white. Side chains are color-coded: E461 (orange), E462 (yellow), D463 (turquoise), D469 (green), and Q473 (red). The hydrophobic face of the AID helix interacts with the CaVβ2a subunit shown in blue. (B) Two-dimensional helical wheel representation of the AID region between Q458 and E475 as predicted in the rabbit CaV1.2. Helical wheel projections were carried out with ANTHEPROT. As seen, the hydrophobic (circles) and hydrophilic (diamonds) residues line up on opposite sides of the helix. Filled black symbols highlight the residues that were shown to interact strongly with CaVβ2a or CaVβ3 in every crystal structure published to this date (Van Petegem et al., 2004; Chen et al., 2004; Opatowsky et al., 2004); the empty symbols represent residues that were shown to be clearly noninteracting with either CaVβ2a or CaVβ3; and the shaded symbols show residues for which some degree of interaction was found in either crystal structure.
Given that the I-II linker has been proposed as a universal gating particle in both HVA CaV1 (Erickson et al., 2003) and CaV2 channels, we undertook a detailed mutational analysis of the structural determinants underlying VDI within the I-II linker of CaV1.2. Point mutations Q458A, Q459A, K465N, L468R, D469A, T472D, and Q473K did not significantly increase VDI gating. In contrast, mutations of the negatively charged E461, E462, and D463 to neutral or positively charged residues increased VDI gating, suggesting that the cluster of negatively charged residues in the N-terminal end of the AID helix could account for the slower VDI kinetics of L-type CaV1.2 channels.
MATERIAL AND METHODS
Recombinant DNA techniques
cDNAs coding for wild-type (wt) rabbit CaV1.2 (GenBank X15539), rat CaVβ3 (GenBank M88751) (Castellano et al., 1993), and rat CaVβ2a (GenBank M80545) were kindly donated by Dr. E. Perez-Reyes (University of Virginia). The wild-type human CaV2.3 (GenBank L27745) was a gift from Dr. T. Schneider (University of Köln). The rat brain CaVα2bδ subunit provided by Dr. T. P. Snutch (University of British Columbia) is > 90% similar to GenBank NM_000722 (Williams et al., 1992).
Point mutations in CaV1.2, CEEE, and CaVβ2a were obtained with the Quick-Change XL-mutagenesis kit (Stratagene, La Jolla, CA) using 39 bp primers. The CEEE chimera was constructed using the CaV1.2 (XhoI) channel as described previously (Bernatchez et al., 2001a). CaV1.2 and CEEE mutations were performed by cassette cloning using the naturally occurring SacI (956) site and the XhoI site that was engineered at position 1530 nt in the I-II linker of CaV1.2 (42 residues before IIS1) (Berrou et al., 2001; Bernatchez et al., 2001a). This is a nonsilent mutation creating a Gly to Arg mutation (G511R). The resulting CaV1.2 (XhoI) channel (that will be referred to herein as CaV1.2 (XhoI) wt) displayed inactivation and activation kinetics similar to the wild-type CaV1.2 (Berrou et al., 2001; Bernatchez et al., 2001a) (Fig. 2). Constructs were verified by restriction mapping after relegation of the mutated fragment into the SacI/XhoI sites of the wild-type CaV1.2 and the CEEE chimera. Recombinant clones were screened by double-stranded sequence analysis of the entire ligated cassette. cDNA constructs for the wild-type and mutated CaVα1 subunits were linearized at the 3′ end by HindIII digestion, whereas the rat brain CaVβ3 and CaVβ2a subunits were digested by NotI. Run-off transcripts were prepared using methylated cap analog m7G (5′)ppp(5′)G and T7 RNA polymerase with the mMessage mMachine transcription kit (Ambion, Austin, TX). The final cRNA products were resuspended in diethylpyrocarbonate-treated H2O and stored at −20°C. The integrity of the final product and the absence of degraded RNA were determined by a denaturing agarose gel stained with ethidium bromide.
Functional expression of wild-type and mutants channels
Oocytes were obtained from female Xenopus laevis clawed frog (Nasco, Fort Atkinson, WI) as described previously (Berrou et al., 2002; Parent et al., 1997, 1995). Individual oocytes free of follicular cells were obtained after 30–40 min incubation in a calcium-free solution (in mM: 82.5 NaCl; 2.5 KCl; 1 MgCl2; 5 HEPES; pH 7.6) containing 2 mg/ml collagenase (Gibco, Burlington, Ontario, Canada). A solution of 46 nl containing between 35 and 50 ng of cRNA coding for the wild-type or mutated α1 subunit was injected 16 h later into stage V and VI oocytes. cRNA coding for rat brain CaVα2bδ and rat brain CaVβ3 were coinjected with the α1 subunit at a 3:1:2 weight ratio. In some cases, the wild-type CaVβ2a or the CaVβ2a C3S + C4S (referred to as CaVβ2a CS) mutant replaced the CaVβ3 subunit. Oocytes were incubated at 19°C in a Barth's solution (in mM): 100 NaCl; 2 KCl; 1.8 CaCl2; 1 MgCl2; 5 HEPES; 2.5 pyruvic acid; 100 units/ml of penicillin; 50 μg/ml gentamicin (pH 7.6). The inactivation properties of each mutant channel herein described was studied in a minimum of three different oocyte batches. Furthermore, the wild-type channel was always measured under the same experimental conditions with every new mutant, thus ensuring that the inactivation properties of the channels would be recorded under the same level of endogenous CaVβ subunits (Lacerda et al., 1994; Tareilus et al., 1997).
Electrophysiological recordings in oocytes
Wild-type and mutant channels were screened at room temperature for macroscopic barium current 4–6 days after RNA injection using a two-electrode voltage-clamp amplifier (OC-725C, Warner Instruments, Hamden, CT) as described earlier (Berrou et al., 2001; Bernatchez et al., 2001a; Parent et al., 1997). Voltage and current electrodes were filled with 3 M KCl; 1 mM EGTA; 10 mM HEPES (pH 7.4). Whole-cell currents were measured in a 10 Ba2+ solution (in mM; 10 Ba(OH)2; 110 NaOH; 1 KOH; 20 HEPES titrated to pH 7.3 with methane sulfonic acid (MeS)) or exceptionally a 10 Ca2+ solution where Ca(OH)2 replaced Ba(OH)2. To minimize kinetic contamination by the endogenous Ca2+ activated Cl− current, oocytes were injected with 18.4 nl of a 50 mM EGTA (ethylene glycol- bis(b-aminoethyl ether)-N,N,N′,N′-tetraacetic acid) (Sigma, St. Louis, MO) 0.5–2 h before the experiments. Oocytes were superfused by gravity flow at a rate of 2 ml/min that was fast enough to allow complete chamber fluid exchange within 30 s. Experiments were performed at room temperature (20–22°C).
Data acquisition and analysis
PClamp software 6.02 (Axon Instruments, Foster City, CA) was used for on-line data acquisition and analysis. Unless stated otherwise, data were sampled at 10 kHz and low pass filtered at 5 kHz using the amplifier built-in filter. For all recordings, a series of 450-ms voltage pulses were applied from a holding potential of −80 mV at a frequency of 0.2 Hz from −40 to +60 mV. Isochronal inactivation data (h 5000) were obtained from normalized currents measured at 0 or +10 mV after a series of 5 s prepulses that varied from −100 to +30 mV (Berrou et al., 2001; Bernatchez et al., 2001a). For the isochronal inactivation figures, data points represent the mean of n ≥ 5 and were fitted to the Boltzmann Eq. 1:
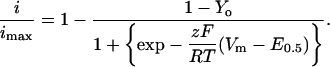 |
(1) |
Pooled data points (mean ± SE) were fitted to Eq. 1 using user-defined functions and the fitting algorithms provided by Origin 6.1 (Microcal Software, Northampton, MA) analysis software. Equation 1 accounts for the fraction of noninactivating current with E0.5, midpoint potential; z, slope parameter; Y0, fraction of noninactivating current; Vm, the prepulse potential; and RT/F with their usual meanings. The fitting process generated values estimating errors on the given fit values.
Activation parameters were estimated from the mean I/V curves obtained for each channel combination. The I/V relationships were normalized to the maximum amplitude and were fitted to the Boltzmann Eq. 2:
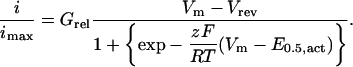 |
(2) |
E0.5 is the potential for 50% activation; Grel is the normalized conductance; z, slope parameter; Vm, the test potential; and RT/F with their usual meanings. The fitting process generated values estimating errors on the given fit values.
Inactivation kinetics were quantified using r300 values, that is the ratio of the whole-cell current remaining at the end of a 300 ms pulse. Capacitive transients were erased for clarity in the final figures. Statistical analyses were performed using the Student's t-test for two independent populations fitting routines provided by Origin 6.1 (Microcal Software).
RESULTS
Accessible residues in the AID helix contributes to VDI in CaV1.2
The AID region of the rabbit CaV1.2 channel QQLEEDLKGYLDWITQAE, with conserved residues shown in bold letters, forms an α-helix upon binding to CaVβ subunits (Van Petegem et al., 2004; Chen et al., 2004; Opatowsky et al., 2004). In the three-dimensional representation shown in Fig. 1 B, Y467, W470, and I471 are seen to be buried in the CaVβ subunit fold and unavailable to interact with other proteins. In contrast, Q458, Q459, E461, E462, K465, L468, D469, T472, and Q473 line the face of the helix opposite to the CaVβ subunit (Fig. 1 B) and appear to remain fairly accessible even in the presence of CaVβ. In fact, no interaction could be detected for Q458, Q459, E461, E462, K465, D469, or T472 and the CaVβ subunit in any of the three crystal structures of the AID region (Van Petegem et al., 2004; Chen et al., 2004; Opatowsky et al., 2004). To investigate the role of these free residues in a hinged-lid type inactivation mechanism, alanine residues were introduced at the positions occupied by conserved residues (Q458A, Q459A, and E461A) whereas nonconserved residues were mutated to their equivalent in CaV2.3 channels (E462R, K465N, L468R, D469A, T472D, and Q473K) (Fig. 2 A). Except for Q458A and Q459A, all point mutations involve a change in the net charge carried by the residue. Mutant channels were coexpressed with CaVβ3 that emphasizes closed-state inactivation (Patil et al., 1998). Robust Ba2+ currents were measured for all mutants (Fig. 2 B) (see Table 1 for details). As shown in the r300 analysis of Fig. 2 C, E461A inactivated significantly faster than the parent CaV1.2 (XhoI) wt channel (p < 0.001), although it remained slightly slower than E462R (p < 0.01). In contrast, Q458A (not shown), Q459A (not shown), and D469A were similar to CaV1.2, whereas K465N, L468R, T472D, and Q473K mutants were significantly slower than the wt channel (p < 0.01).
TABLE 1.
Biophysical properties of CaV1.2 mutants
| With α2bδ/β3 in 10 Ba2+ | Inactivation (5 s)
|
Activation
|
||
|---|---|---|---|---|
| E0.5 (mV) | Fractional currents | E0.5 (mV) | Peak IBa (μA) | |
| Wt | −23 ± 1 (21) z = 2.5 | 0.17 ± 0.02 (21) | −8 ± 2 (23) z = 3.7 ± 0.4 | −2.5 ± 0.3 (43) |
| XhoI | −24 ± 1 (19) z = 2.3 | 0.20 ± 0.02 (19) | −5 ± 1 (16) z = 3.8 ± 0.4 | −1.9 ± 0.3 (23) |
| Q458A | −16 ± 1 (10) z = 1.7 | 0.23 ± 0.04 (10) | −7 ± 1 (14) z = 4.0 ± 0.2 | −1.3 ± 0.3 (14) |
| Q459A | −22 ± 1 (10) z = 2.4 | 0.21 ± 0.02 (10) | −7 ± 1 (13) z = 3.7± 0.3 | −1.9 ± 0.3 (13) |
| E461A | −25 ± 2 (8) z = 2.3 | 0.14 ± 0.02 (10) | −10 ± 1 (17) z = 4.3 ± 0.7 | −0.9 ± 0.2 (9) |
| E462R | −24 ± 1 (19) z = 2.9 | 0.09 ± 0.01 (19) | −1 ± 2 (4) z = 3.5 ± 0.6 | −1.7± 0.7 (17) |
| E462K | −22 ± 1 (14) z = 1.9 | 0.22 ± 0.02 (14) | −4 ± 1 (7) z = 4.2 ± 0.4 | −0.7 ± 0.2 (4) |
| E462A | −20 ± 1 (6) z = 2.7 | 0.09 ± 0.02 (6) | −0.3 ± 0.4 (13) z = 3.3 ± 0.1 | −1.0 ± 0.1 (7) |
| E462G | −19 ± 1 (7) z = 1.9 | 0.27 ± 0.01 (7) | 0.3 ± 0.5 (11) z = 3.5 ± 0.3 | −0.9 ± 0.1 (11) |
| E462N | −20 ± 2 (4) z = 2.7 | 0.26 ± 0.06 (4) | −4 ± 1 (6) z = 3.7 ± 0.4 | −1.1 ± 0.1 (6) |
| E462Q | −22 ± 2 (11) z = 1.9 | 0.21 ± 0.02 (11) | −1.4 ± 0.3 (16) z = 3.4± 0.2 | −1.1 ± 0.1 (16) |
| E462D | −19 ± 2 (12) z = 2.6 | 0.36 ± 0.03 (12) | −2 ± 1 (19) z = 3.5 ± 0.2 | −1.0 ± 0.1 (19) |
| D463A | −21 ± 1 (8) z = 2.5 | 0.07 ± 0.01 (8) | −12 ± 1 (5) z = 5.3 ± 0.5 | −0.73 ± 0.05 (4) |
| D463R | −22 ± 1 (6) z = 3.5 | 0.11 ± 0.01 (6) | −6 ± 1 (12) z = 3.7 ± 0.3 | −2.8 ± 0.5 (5) |
| L464A | −21 ± 0.8 (5) z = 2.4 | 0.31 ± 0.05 (5) | −7.1 ± 0.6 (11) z = 3.4 ± 0.6 | −1.6 ± 0.3 (11) |
| K465N | −17 ± 1 (10) z = 1.5 | 0.46 ± 0.05 (10) | −6 ± 1 (7) z = 3.9 ± 0.2 | −1.4 ± 0.1 (12) |
| L468R | −22 ± 1 (7) z = 2.5 | 0.38 ± 0.04 (7) | −15 ± 1 (12) z = 5.6 ± 0.4 | −2.0 ± 0.3 (7) |
| D469A | −25 ± 1 (13) z = 3.6 | 0.13 ± 0.02 (5) | −3 ± 1 (14) z = 3.8 ± 0.2 | −7 ± 2 (12) |
| D469R | −20 ± 1 (16) z = 1.9 | 0.49 ± 0.03 (16) | −10 ± 1 (16) z = 4.1 ± 0.3 | −1.6 ± 0.2 (14) |
| T472D | −11 ± 1 (9) z = 1.6 | 0.46 ± 0.03 (9) | −8 ± 1 (15) z = 4.2 ± 0.3 | −2.7 ± 0.4 (16) |
| Q473K | −18 ± 1 (15) z = 2.4 | 0.30 ± 0.03 (15) | −3 ± 0.4 (8) z = 3.6 ± 0.4 | −1.7 ± 0.4 (15) |
| Q473R | −27 ± 2 (8) z = 1.6 | 0.51 ± 0.07 (8) | −6 ± 1 (13) z = 3.7 ± 0.2 | −1.0 ± 0.2 (8) |
Biophysical parameters of CaV1.2 wild-type and mutant channels expressed in Xenopus oocytes in the presence of CaVα2bδ and CaVβ3 subunits. The background channel used for the mutations was the CaV1.2 (XhoI) with a unique XhoI site at G511R. Whole-cell currents were measured in 10 mM Ba2+ throughout. The voltage dependence of inactivation was determined from the peak currents measured at 0 mV after 5 s pulses from −100 to +50 mV. Relative currents were fitted to Boltzmann Eq. 1. The fractional currents represent the fraction of whole-cell currents remaining at the end of a 5 s conditioning pulse to + 10 mV. Activation data were estimated from the mean I/V relationships and fitted to Boltzmann Eq. 2. Peak IBa was determined from I/V relationships for the corresponding experiments. The data are shown with the mean ± SE and the number n of samples appears in parentheses.
The voltage dependence was not significantly altered, although the fraction of the noninactivating current remaining after the 5-s prepulses varied significantly among mutants (Fig. 2 D). The least inactivated mutants include T472D and K465N with fractional residual currents as high as 0.46 ± 0.03 (9) and 0.46 ± 0.05 (10), respectively, at +10 mV. L468R and Q473K form a second group with fractional residual currents of 0.38 ± 0.04 (7) and 0.30 ± 0.03 (15). Under the same experimental conditions, inactivation was almost complete for E461A, E462R, and D469A with fractional currents of 0.14 ± 0.02 (10), 0.09 ± 0.01 (19), and 0.11 ± 0.03 (5), respectively, as compared with 0.20 ± 0.02 (19) for the parent channel CaV1.2 (XhoI) wt. These data show that mutations of two consecutive residues E461 and E462 in the N-terminal end of the AID helix accelerate VDI gating, whereas any mutation in the C-terminal region tends to decrease VDI gating.
Alanine scan of the N-terminal end of the AID helix
To further assess the role of the N-terminal end of the AID region, the VDI gating of four consecutive residues from 461 to 464 were analyzed with alanine mutants (Fig. 3 A). As seen, the E461A, E462A, and D463A (EED) mutants displayed Ba2+-dependent kinetics that were faster than the CaV1.2 (XhoI) wt and more clearly voltage-dependent (Fig. 3 B). The behavior of the EED cluster contrasts with the relatively normal VDI kinetics of the neighboring L464A mutant. The faster VDI kinetics of E461A, E462A, and D463A were further echoed in the lower residual currents of 0.14 ± 0.02 (10), 0.09 ± 0.02 (6), and 0.07 ± 0.01 (8), respectively, of their isochronal inactivation curve (Fig. 3 C) (Table 1). Altogether, these data suggest that the cluster of negatively charged residues in the N-terminal end of the AID helix could account for the slower VDI kinetics of CaV1.2. Although the VDI kinetics of the D463A mutant were significantly faster than the wild-type channel, the D463R mutant (not shown) behaved like the wild-type channel, indicating that the effect of the arginine mutation is specific to position 462.
FIGURE 3.

Alanine mutations in the EED locus accelerates the VDI kinetics. (A) Whole-cell current traces are shown for CaV1.2 mutants E461A, E462A, D463A, and L464A (from left to right). (B) Corresponding r300 values are shown mean ± SE from 0 to +20 mV for CaV1.2 (XhoI) wt, E461A, E462A, D463A, and L464A (from left to right). The r300 ratios varied from 0.73 ± 0.01 at 0 mV to 0.67 ± 0.02 at +20 mV (23) for CaV1.2 (XhoI); 0.53 ± 0.02 at 0 mV to 0.44 ± 0.03 at +20 mV (11) for E461A; 0.67 ± 0.02 at 0 mV to 0.49 ± 0.02 at +20 mV (7) for E462A; 0.67 ± 0.02 at 0 mV to 0.53 ± 0.03 at +20 mV (9) for D463A; and 0.80 ± 0.04 at 0 mV to 0.74 ± 0.02 at +20 mV (3) for L464A. At +20 mV, r300 were significantly different between CaV1.2 (XhoI) wt and E461A, E462A, and D463A at p < 10−5. (C) Fraction of noninactivating Ba2+ current increased from 0.07 ± 0.01 (8) for D463A and 0.09 ± 0.02 (6) for E462A, to 0.14 ± 0.02 (10) for E461A, and to 0.31 ± 0.05 (5) for L464A. Fit values are shown in Table 1.
Positive and neutral residues at position E462 in the AID region increase VDI gating
The structural requirements for increased VDI kinetics at position E462 were investigated with E462R, E462K, E462Q, E462N, E462A, E462G, and E462D mutants. The salient features are shown in Fig. 4 A. As previously reported, E462R inactivated significantly faster than the wild-type CaV1.2 and the CaV1.2 (XhoI) wt channel (p < 10−16) between 0 and + 20 mV (Berrou et al., 2001). Herein we further show that E462R displayed faster VDI gating than any other point mutation including the positively charged E462K (Fig. 4 B). The positively charged E462K nonetheless inactivated significantly faster than the parent CaV1.2 (XhoI) wt channel at all voltages (p < 10−4), although the increased VDI kinetics were heightened by depolarization to + 20 mV. The VDI gating of the neutral-substituted residues E462A, E462N, E462Q, and E462G varied widely. The hydrophobic E462A behaved mostly like the positively charged E462K, whereas E462G was closer to the parent channel. The VDI kinetics of hydrophilic E462N and E462Q were similar to the parent channel at 0 mV, but depolarization to + 20 mV specifically increased the kinetics of E462N to the level of E462K and E462A. Finally, the E462D mutant inactivated significantly slower than the parent channel (p < 10−4). The changes in the VDI kinetics occurred without any significant shift in the voltage dependence of activation (Table 1).
FIGURE 4.

Positive and neutral residues at position 462 accelerate the VDI kinetics in CaV1.2. (A) Whole-cell current traces are shown for CaV1.2 mutants: E462R, E462K, E462A, and E462D (from left to right). (B) R300 values are shown mean ± SE from 0 to +20 mV for CaV1.2 wt, CaV1.2 (XhoI) wt, E462R, E462K, E462A, E462N, E462Q, E462G, and E462D (from left to right). The r300 ratios varied from 0.74 ± 0.01 at 0 mV to 0.72 ± 0.01 at +20 mV (45) for CaV1.2 wt; 0.73 ± 0.01 at 0 mV to 0.67 ± 0.02 at +20 mV (23) for CaV1.2 (XhoI); 0.38 ± 0.02 at 0 mV to 0.29 ± 0.01 at +20 mV (20) for CaV1.2 E462R; 0.62 ± 0.02 at 0 mV to 0.54 ± 0.02 at +20 mV (16) for CaV1.2 E462K; 0.67 ± 0.02 at 0 mV to 0.49 ± 0.02 at +20 mV (7) for CaV1.2 E462A; 0.73 ± 0.02 at 0 mV to 0.58 ± 0.02 at +20 mV (6) for CaV1.2 E462N; 0.74 ± 0.02 at 0 mV to 0.70 ± 0.02 at +20 mV (13) for CaV1.2 E462Q; 0.79 ± 0.02 at 0 mV to 0.73 ± 0.02 at +20 mV (8) for CaV1.2 E462G; and 0.86 ± 0.01 at 0 mV to 0.79 ± 0.01 at +20 mV (19) for CaV1.2 E462D. At + 20 mV, r300 were significantly different between CaV1.2 (XhoI) wt and E462R at p < 10−16 ; E462K, E462A, and E462D at p < 10−4; E462N and E462Q at p < 0.05 but were not statistically significantly different between CaV1.2 wt, CaV1.2 (XhoI) wt and E462G (p > 0.5). (C) Voltage dependence of inactivation was not significantly different for CaV1.2 wt (21), CaV1.2 (XhoI) wt (19), E462K (14), E462A (6), E462N (4), and E462R (19) channels but were shifted slightly to the right for E462Q, E462G, and E462D. The fraction of noninactivating Ba2+ current decreased from 0.36 ± 0.03 (12) for E462D, to 0.27 ± 0.01 (7) for E462G, to 0.26 ± 0.06 (4) for E462N, to 0.21 ± 0.02 (11) for E462Q, to 0.22 ± 0.02 (14) for E462K, to 0.20 ± 0.02 (19) for CaV1.2 (XhoI), to 0.09 ± 0.02 (6) for E462A, and 0.09 ± 0.01 (19) for E462R at + 10 mV. Isochronal data points for CaV1.2 wt; CaV1.2 (XhoI) wt and E462K were superimposed. Fit values are shown in Table 1.
The voltage dependence of inactivation was measured after 5 s depolarizing prepulses (Fig. 4 C). The midpotential of inactivation was not significantly altered for most E462 mutations with values ranging from −19 ± 1 mV (7) for E462G channels to −24 ± 1 mV (19) for E462R. Inactivation was more complete for the E462R and the E462A channels than for the parent CaV1.2 (XhoI) wt channel, echoing the inactivation kinetics data obtained at +10mV. E462D was the least inactivated mutant with fractional residual currents of 0.36 ± 0.03 (12), whereas E462G, E462N, and E462Q clustered around a fractional current of ≈0.26 (Table 1). Altogether, these results depict the complex relations between the VDI kinetics and the charge, the size, and the hydrophilicity of the residue at position 462. The arginine-substituted E462R displayed faster VDI gating than the similarly positively charged E462K followed closely by the neutral and hydrophobic E462A mutant. E462G behaved like the wild-type channel between 0 and +20 mV. The VDI kinetics of the neutral and hydrophilic E462N and E462Q were similar at 0 mV but differed significantly at +20 mV (p < 0.01). In contrast, the conservative mutation E462D resulted in a channel with slower VDI kinetics than the wild-type channel.
E462R restores fast VDI gating to the CEEE chimera
The hinged-lid mechanism supposes that the I-II linker will dock onto its receptor site, thereby stopping the flow of ions. To investigate the structural determinants involved in the docking of the inactivating particle, E462 mutations were introduced in the slow CEEE chimera. The CEEE chimera encompasses domain I + part of the I-II linker of CaV1.2, including the whole AID region with its EED cluster in the N-terminal end, inserted into the CaV2.3 host channel. Typical recordings are shown in Fig. 5 A for the CEEE, CEEE + E462R, CEEE + Q473K, and CEEE+ E462R+ Q473K constructs with the corresponding r300 analysis in Fig. 5 B. As seen in CaV1.2, the E462R and E462K mutations significantly accelerated the inactivation kinetics of CEEE at 10−10 < p < 10−15 for voltages between 0 and + 20 mV, but E462R remained more potent than E462K at Vm = −10 and 0 mV (p < 10−3). The introduction of the E462R mutation significantly hyperpolarized the voltage dependence of inactivation from E0.5 = −19 ± 1 (13) mV for CEEE to E0.5 = −35 ± 1 (17) mV for CEEE + E462R, and decreased the fraction of the noninactivating current from 0.15 ± 0.01 (13) to 0.02 ± 0.01 (17) (Fig. 5 C). The midpotential of inactivation in CEEE + E462R channels remained, however, more positive than the midpotential of inactivation in the wild-type CaV2.3, suggesting that all four domains are required to fully account for the voltage dependence of inactivation. Furthermore, the stability of the inactivated state was not significantly affected by the introduction of the E462R mutation in the CEEE background. The time course of recovery from inactivation was well described by a sum of two exponential functions, in both cases with a dominant fast time constant τREC = 73 ± 5 ms (6) for CEEE + E462R that is comparable with the fast τREC = 63 ± 4 ms (4) published previously for CEEE (Bernatchez et al., 2001b). The Q473K mutation had little impact on the VDI kinetics and voltage dependence of CEEE, but the double E462R + Q473K mutation produced channels with VDI kinetics and voltage dependence that are intermediary between CEEE and CEEE + E462R. The complete set of values is shown in Table 2. Altogether, mutations at position E462 altered to the same extent the VDI gating of the CEEE chimera and the CaV1.2 channel. These data indicate that the properties of the docking site are not rate limiting for VDI gating. Alternatively, it could also suggest that the docking site is either contained within domain I or else is strictly conserved between CaV1.2 and CaV2.3.
FIGURE 5.

E462R increases the VDI kinetics and voltage dependence of the CEEE chimera. (A) Whole-cell current traces are shown for CEEE mutants: CEEE; CEEE + E462R; CEEE + Q473K; and CEEE + E462R + Q473K (CEEE + ER + QK) in 10 mM Ba2+ (from left to right). (B) r300 values are shown mean ± SE from −10 to +20 mV for CEEE, CEEE+ E462R; CEEE + E462K; CEEE + Q473K; and CEEE + E462R + Q473K (from left to right). The r300 ratios were strongly voltage-dependent for all constructs. The E462R (29) and E462K (14) point mutations significantly increased the inactivation kinetics of CEEE (26) at p < 10−15 and p < 10−10, respectively. In contrast the inactivation kinetics of CEEE (26) and CEEE + Q473K (12) were not significantly different (p > 0.1). (C) Voltage dependence of inactivation was estimated from isochronal inactivation data points measured after 5 s conditioning pulses. Point mutations at position E462 shifted significantly the voltage dependence of inactivation toward negative potentials from E0.5 = −19 ± 1 mV (13) for CEEE to E0.5 = −35 ± 1 mV (17) for CEEE + E462R (p < 10−4); E0.5 = −30 ± 1 mV (8) for CEEE + E462R + Q473K (p < 10−3); and E0.5 = −28 ± 1 mV (10) for E462K (p < 10−3). In contrast, the Q473K mutation did not significantly alter the voltage dependence of inactivation with E0.5 = −23 ± 1 mV (8). Complete set of fit values are shown in Table 2.
TABLE 2.
Biophysical properties of CEEE mutants
| With α2bδ in 10 Ba2+ | Inactivation (5 s) E0.5 (mV)
|
Activation E0.5 (mV)
|
Peak IBa (μA)
|
|||
|---|---|---|---|---|---|---|
| −β | +β3 | −β | +β3 | −β | +β3 | |
| CEEE | −17 ± 1 (3) z = 2.6 | −19 ± 1 (13) z = 2.6 | −4 ± 1 (6) z = 4.1± 0.3 | −7 ± 0.5 (23) z = 4.4 ± 0.2 | −0.39 ± 0.02 (6) | −1.9 ± 0.2 (23) |
| CEEE + E462R | −32.2 ± 0.4 mV (16) z = 2.8 | −35 ± 1 (17) z = 3.1 | −7 ± 1 (12) z = 4.0 ± 0.5 | −9 ± 1 (18) z = 4.0 ± 0.3 | −1.4 ± 0.2 (7) | −2.1 ± 0.4 (13) |
| CEEE + E462K | −19 ± 1 (4) z = 2.2 | −28 ± 1 (10) z = 2.5 | −4 ± 1 (6) z = 4.0 ± 0.2 | −10 ± 1 (14) z = 4.3 ± 0.3 | −0.6 ± 0.5 (6) | −1.5 ± 0.2 (14) |
| CEEE + Q473K | n.d. | −23 ± 1 (8) z = 3.1 | n.d. | −12 ± 1 (5) z = 4.7 ± 0.5 | n.d. | −1.9 ± 0.5 (5) |
| CEEE + E462R + Q473K | −25 ± 1 (5) z = 2.6 | −30 ± 1 (8) z = 2.9 | −3 ± 1 (6) z = 4.0 ± 0.3 | −6 ± 1 (7) z = 3.7 ± 0.2 | −1.1 ± 0.1 (6) | −0.8 ± 0.1 (7) |
Biophysical parameters of the CEEE chimera and its derivative mutant channels expressed in Xenopus oocytes in the presence of CaVα2bδ and CaVβ3 subunits. Whole-cell currents were measured in 10 mM Ba2+ throughout. The voltage dependence of inactivation was determined from the peak currents measured at 0 mV after 5 s pulses from −100 to +50 mV. Relative currents were fitted to Boltzmann Eq. 1. Activation data were estimated from the mean I/V relationships and fitted to Boltzmann Eq. 2. Peak IBa was determined from I/V relationships for the corresponding experiments. The data are shown with the mean ± SE and the number n of samples appears in parentheses.
n.d., not determined.
CaVβ2a abolished whereas CaVβ2a C3S + C4S restored the fast VDI gating of E462R
Coexpression of the auxiliary subunit CaVβ2a with HVA α1 subunits such as CaV1.2 (Chien and Hosey, 1998; Takahashi et al., 2003), CaV2.1 (Restituito et al., 2000), CaV2.2 (Stephens et al., 2000; Takahashi et al., 2003), and CaV2.3 (Parent et al., 1997; Qin et al., 1998) is known to nearly eliminate VDI. The CaVβ2a-mediated decrease in VDI kinetics results from the formation of a thioester bond with 3C and 4C in the N-terminus of CaVβ2a (Chien et al., 1996). The palmitoylation of CaVβ2a occurs in mammalian cells (Chien et al., 1996) as well as in Xenopus oocytes (Qin et al., 1998). The C3S + C4S mutations in CaVβ2a eliminated the membrane anchoring of CaVβ2a (Chien et al., 1996) while recovering the fast inactivation kinetics of CaV2.1 (Restituito et al., 2000), CaV2.2 (Stephens et al., 2000), and CaV2.3 (Qin et al., 1998).
To test the hypothesis that E462R promotes the mobility of the inactivation gate, the inactivation properties of CaV1.2 E462R and CEEE + E462R were tested with either CaVβ2a or CaVβ2a C3S + C4S mutant (CaVβ2a CS) as the auxiliary subunit (Fig. 6). As compared with CaVβ3, coexpression with CaVβ2a significantly decreased the VDI kinetics of CaV1.2, CaV1.2 E462R, CEEE, and CEEE + E462R (Fig. 6, C and D) and nearly abolished their voltage dependence (Table 3). Despite the retardation effect of CaVβ2a, the VDI gating of the E462R mutant in CaV1.2 and the CEEE + E462R channel remained faster than their respective parent channels under the same conditions with 0.05 < p < 0.001 (Fig. 6, C and D). Although the VDI kinetics of CaV1.2 E462R were faster in the presence of CaVβ2a CS than in the presence of the wild-type CaVβ2a, they remained, however, significantly slower than VDI kinetics measured with CaVβ3 especially at 0 and +10 mV (p < 0.001) (Fig. 6 C).
FIGURE 6.

Faster VDI of E462R are abolished by CaV β2a and restored in part by CaVβ2a C3S + C4S. (A) Whole-cell current traces are shown for CaV1.2 (XhoI) wt and CaV1.2 E462R after coexpression with the palmitoylated-deficient CaVβ2a C3S + C4S (CaVβ2a CS) mutant in 10 mM Ba2+. (B) Whole-cell current traces are shown for the CEEE chimera and CEEE + E462R (CEEE + ER) after coexpression with CaVβ2a CS. (C) Bar graph displaying the r300 values mean ± SE from 0 to +20 mV for CaV1.2 (XhoI) wt and CaV1.2 E462R contrasting the VDI kinetics in the presence of CaVβ3 (data shown in Fig 2) (solid black and white bars) of CaVβ2a (black and white bars with vertical stripes), or of CaVβ2a CS (crossed black and white bars). In the presence of CaVβ2a CS, r300 were significantly different at 10−7 < p <10−3 between E462R and CaV1.2 (XhoI) wt with values decreasing from 0.87 ± 0.01 at 0 mV to 0.68 ± 0.06 at +20 mV (4) for CaV1.2 (XhoI) wt and 0.57 ± 0.01 at 0 mV to 0.36 ± 0.05 at +20 mV (7) for CaV1.2 E462R. The statistical significance, however, decreased with depolarization. The r300 for CaV1.2 E462R with CaVβ2a CS were not significantly different from r300 measured with CaVβ3 except at 0 mV (p < 0.05) (D) Bar graph displaying the r300 values mean ± SE from 0 to +20 mV for CEEE and CEEE + E462R contrasting the VDI kinetics in the presence of CaVβ3 (solid black and white bars), of CaVβ2a (black and white bars with vertical stripes), or of CaVβ2a CS (crossed black and white bars). In the presence of CaVβ2a CS, r300 were significantly different at 10−5 < p < 10−3 between CEEE + E462R and CEEE with values ranging from 0.86 ± 0.03 at 0 mV to 0.71 ± 0.05 at +20 mV (7) for CEEE and 0.33 ± 0.04 at 0 mV to 0.29 ± 0.07 at +20 mV (14) for CEEE+ E462R. The statistical significance decreased with depolarization. The r300 for CEEE + E462R with CaVβ2a CS remained significantly different from r300 measured with CaVβ3 at p < 10−3. The complete set of data is shown in Table 3.
TABLE 3.
Biophysical properties of E462R mutants with CaVβ2a wt and CS
| Inactivation (5 s) E0.5 (mV)
|
Activation E0.5 (mV)
|
Peak IBa (μA)
|
||||
|---|---|---|---|---|---|---|
| With α2bδ in 10 Ba2+ | +β2a | +β2aCS | +β2a | +β2aCS | +β2a | +β2aCS |
| CEEE | −18 ± 1 (5) z = 2.3 | n.d. | −4.6 ± 0.7 (2) z = 4.6 ± 0.3 | −5.1 ± 0.6 (6) z = 3.8 ± 0.1 | −0.51 ± 0.03 (2) | −0.66 ± 0.06 (6) |
| CEEE + E462R | −27.3 ± 0.5 mV (12) z = 2.5 | −33.6 ± 0.7 mV (6) z = 2.6 | −9.0 ± 0.4 (9) z = 5.0 ± 0.3 | −5.7 ± .5 (14) z = 4.0 ± 0.2 | −4.6 ± 0.9 (9) | −1.6 ± 0.2 (14) |
| CaV1.2 (XhoI) wt | n.d. | n.d. | −8.8 ± 0.6 (8) z = 3.9 ± 0.3 | −5.9 ± 0.7 (4) z = 3.7 ± 0.2 | −0.8 ± 0.1 (8) | −0.58 ± 0.05 (4) |
| CaV1.2 E462R | n.d. | n.d. | −11 ± 1 (8) z = 4.2 ± 0.3 | −7.7 ± 0.9 (7) z = 4.9 ± 0.6 | −0.9 ± 0.2 (8) | −0.63 ± 0.08 (7) |
Biophysical parameters of CaV1.2 and CEEE mutants expressed in Xenopus oocytes in the presence of CaVα2bδ and CaVβ2a or CaVβ2a C3S + C4S subunits. Whole-cell currents were measured in 10 mM Ba2+ throughout. The voltage dependence of inactivation was determined from the peak currents measured at 0 mV after 5 s pulses from −100 to +50 mV. Relative currents were fitted to Boltzmann Eq. 1. Activation data were estimated from the mean I/V relationships and fitted to Boltzmann Eq. 2. Peak IBa was determined from I/V relationships for the corresponding experiments. The data are shown with the mean ± SE and the number n of samples appears in parentheses.
n.d., not determined.
Similar observations were made for the CEEE + E462R chimera (Fig. 6 B). The VDI properties of CEEE were similar, whether measured with CaVβ2a and the palmitoylated-deficient CaVβ2a subunit (Fig. 6 D). Coinjection with CaVβ2a CS significantly accelerated the VDI kinetics as well as significantly shifted by −10 mV the voltage dependence of inactivation of CEEE + E462R when comparing the same construct with the wt CaVβ2a (Table 3). Nonetheless, the coinjection with CaVβ2a CS did not restore the VDI kinetics of CEEE + E462R to the level of CaVβ3-injected oocytes. A similar observation has been reported for the CaV2.3 channel, where the time course of inactivation of CaV2.3 + CaVβ2a CS channels remained slower than with CaV2.3 + CaVβ3 channels (Qin et al., 1998) indicating that palmitoylation alone does not account completely for the difference between CaVβ2a and CaVβ3. The difference between CaVβ2a CS- and CaVβ3- inactivated channels was carried over in the voltage dependence of inactivation with E0.5 values decreasing from −27.3 ± 0.5 mV (12) with CaVβ2a, to E0.5 = −32.2 ± 0.4 mV (16) without CaVβ, to E0.5 = −33.6 ± 0.7 mV (6) with CaVβ2a CS, and to E0.5 = −35 ± 1 mV (17) with CaVβ3. Altogether, these observations confirm that CaVβ2a and E462R modulate VDI through common structural determinants and further suggest that the presence of a positively charged Arg residue at the fifth position of the AID region could promote the mobility of the I-II linker in CaV1.2.
DISCUSSION
The role of the EED cluster in the VDI gating of CaV1.2
Before this work, the role of the I-II linker in the VDI gating of CaV1.2 was embodied in the single-point mutation E462R that was shown by the group of Catterall (Herlitze et al., 1997) and later by us (Berrou et al., 2001) to promote faster Ba2+-dependent inactivation. The crystal structures of the AID-CaVβ complex from L-type CaV1.1 and CaV1.2 channels (Van Petegem et al., 2004; Chen et al., 2004; Opatowsky et al., 2004) have confirmed that the side chain of E462 projects in the direction opposite to CaVβ and could hence participate to VDI through a hinged-lid type mechanism. In addition to E462, the side chains of Q458, Q459, E461, K465, L468, D469, and T472 in the AID region were also found to line the hydrophilic face of the AID helix where they could also potentially contribute to a hinged-lid type mechanism. Herein we have shown that charge mutations in the C-terminal end of the AID helix, namely K465N, L468R, D469A, and T472D, failed to increase significantly the VDI gating kinetics. In contrast, charge mutations in the EED cluster (E461, E462, D463) increased VDI kinetics, suggesting that negatively charged residues in the N-terminal end of the AID helix could account for the slower VDI kinetics of CaV1.2. The increase in the rate of inactivation of E462R was observed without any significant change in the rate of recovery from inactivation (Bernatchez et al., 2001b). Despite the increased VDI gating of E461A, E462R, and D463A, their voltage dependence of activation and their voltage dependence of inactivation were not significantly altered. It is worth noting that we had previously reported (Berrou et al., 2001) a steep increase in the slope and a −10 mV shift in the voltage dependence of inactivation for the E462R mutant that were never reproduced in the course of the last four years.
The concentration of negative charges in the EED cluster makes it an interesting candidate for an inactivating particle that could sense changes in the local electrical field. One can envision that the EED cluster interacts with another region of the channel under resting conditions. The stability of the AID helix-channel interaction could be enhanced by negatively charged residues and could consequently modulate the rate at which the I-II linker detaches itself from the interaction site and blocks the channel. The observation that the kinetics of recovery from inactivation were not affected suggests indeed that the EED mutations were not altering the affinity between the inactivating particle and the pore anchoring region (Isacoff et al., 1991; Zhou et al., 2001).
The role of the EED cluster appears to be unique to the L-type CaV1.2. In CaV2.1 channels, positive or neutral residues at positions 387 and 388, equivalent to E462 and D463 in CaV1.2, were found to increase VDI gating only when measured in the absence of CaVβ (Sandoz et al., 2004), in contrast to the observations reported here. In CaV2.3 channels, the decrease in VDI gating observed with negatively charged residues at R378 (Berrou et al., 2001, 2002) was restricted to that residue since the VDI gating of adjacent residues E377A and E379A was normal (L. Berrou, Y. Dodier, A. Raybaud, A. Tousignant, O. Dafi, J. Pelletier, and L. Parent, unpublished data). In contrast, our data suggest that even neutral substitutions in the EED locus could promote a faster VDI gating of CaV1.2 channels.
Within the EED cluster, D463 is the only residue facing the CaVβ subunit (Van Petegem et al., 2004; Chen et al., 2004; Opatowsky et al., 2004). It thus remains possible that the increased VDI kinetics we observed with D463A result from a change in the interaction between the AID region and CaVβ3. Charged mutations are indeed likely to modify the formation of hydrogen bonds that exist between D463 and CaVβ3 (Chen et al., 2004) or at least the nature of electrostatic interactions between the two proteins. It remains to be seen whether the Asp to Ala mutation at this position could alter the protein conformation to such an extent that it could release the side chain of D463 from the CaVβ fold. Evidently, the accelerating effect observed with a CaVβ-interacting AID residue appears to be specific to D463. When measured under the same experimental conditions, the VDI gating of the interacting residues L464, G466, Y467, and I47I were not altered by mutations with an alanine residue (Fig. 3 and O. Dafi, Y. Dodier, and L. Parent, unpublished data). Furthermore, there is no information available hinting that mutations at the fifth position of the AID region could significantly alter CaVβ subunit binding onto CaV1.2. At least under denaturing conditions, the Arg to Glu mutation (R378E) at the same position in CaV2.3 did not decrease the [35S]-CaVβ3 subunit overlay binding to GST-AIDE fusion proteins in contrast to mutations of the conserved Trp residue that disrupted both the CaVβ subunit binding and CaVβ subunit modulation of CaV2.3 (Berrou et al., 2002).
Mutations of E462 restore fast VDI gating to the CEEE chimera
E462 mutations significantly increased VDI gating to the CEEE chimera following the same order of potency seen with CaV1.2, namely E462R > E462K ≈ E462A. This observation confirms that a positively charged Arg at the fifth position in the AID motif is critical to confer fast VDI gating in both CaV1.2 and CaV2.3 and strongly indicates that the I-II linker is the single most important determinant in this process even when possibly acting in concert with other cytoplasmic linkers (Sandoz et al., 2004). Furthermore, since the increase in VDI kinetics was similar in both channel backgrounds our data suggest that the interaction between the AID helix and the channel pore is either not the rate limiting step for VDI gating or else that this interaction is taking place within domain I.
The E462R mutation increased the voltage dependence of inactivation by imparting a −15 mV shift in steady-state inactivation of CEEE, whereas the same mutation failed to affect the inactivation curves of CaV1.2. The observation that the voltage dependence of inactivation of E462R was affected by the host channel could stem from the intrinsic voltage-dependent properties in CaV1 versus CaV2 channels. Mutations in IVS5, IIS6, IIIS6, and IVS6 were reported to decrease VDI kinetics of CaV1.2 without any significant change in its voltage dependence of inactivation (Bodi et al., 2002; Shi and Soldatov, 2002). In one case, the acceleration of VDI kinetics brought by the F823A mutation in the IIS6 region of the rat CaV1.2 was accompanied by the hyperpolarization of both the voltage dependence of activation and the voltage dependence of inactivation (Stotz and Zamponi, 2001). Moreover, Herlitze and co-workers pointed out that the conversion of QXXEE to QXXER in CaV1.2 (α1C) produced “effects (that) are not as large as the effects of the converse mutation in CaV2.1 (α1A)” (Herlitze et al., 1997). Hence, in contrast to CaV1.2, the charge mutations at the fifth position of the AID region in CaV2.3 and CaV2.1 channels caused a significant decrease in VDI kinetics accompanied by a robust +20 mV shift in the voltage dependence of inactivation (Herlitze et al., 1997).
Mutations of E462 confers greater mobility to the inactivation gate
To confirm the hypothesis that E462 constitutes an intrinsic component of the inactivation gate, the inactivation properties of CaV1.2 E462R and CEEE + E462R were tested with CaVβ2a and its nonpalmitoylated form CaVβ2a CS. CaVβ2a locks the CaVα1 subunit in a rigid conformation that slows down VDI gating, whereas the CaVβ2a CS mutant could effectively counter that effect (Chien et al., 1996). Coexpression with CaVβ2a was shown herein to abolish the faster VDI kinetics of E462R in both CaV1.2 and the CEEE chimera, suggesting that E462R was not sufficient to counteract the slowing effect of CaVβ2a. The fast inactivation kinetics of the CaV1.2 E462R and the CEEE + E462R mutants were, however, restored to some extent when using the nonpalmitoylated form of CaVβ2a CS, indicating that CaVβ2a and E462R modulate VDI through a common pathway in which the mobility of the I-II linker is likely to play a significant role.
Our observations further suggest that positively charged residues are required at the fifth position of the AID helix of the HVA CaV1-2α1 subunits for CaVβ2a CS to promote VDI gating. Coinjection with CaVβ2a CS did not significantly increase the VDI gating of the wild-type CaV1.2 channel expressed in Xenopus oocytes (our data) or in mammalian cells (Chien et al., 1996). In addition, coinjection with CaVβ2a CS did not significantly increase the VDI gating of the CEEE chimera even though it is in fact 90% identical to CaV2.3. The charge mutation (Glu to Arg) was, however, sufficient to reestablish the accelerating effect of CaVβ2a CS on VDI gating, suggesting that the accelerating effect of CaVβ2a CS requires positively charged residues at position 462. Indeed, the Arg residue is strictly conserved within the CaV2 channel family for which the accelerating effect of CaVβ2a CS has been thoroughly documented (Qin et al., 1998; Restituito et al., 2000; Stephens et al., 2000).
Structural requirements of VDI at position E462
The mutational analysis carried out at position 462 confirmed that positively charged and/or neutral residues speed up inactivation kinetics in CaV1.2. The increase in the VDI kinetics was significantly larger with the Arg residue than with the Lys residue even though both residues carry a net positive charge at physiological pH. The VDI gating of E462K (positive) and E462A (neutral) was similar, whereas the E462D substitution that switched two negatively charged residues significantly decreased VDI. The E462N channel was similar to E462Q at 0 mV but behaved more like E462A at + 20 mV. Altogether, our results indicate that positively charged residues yield faster VDI kinetics than negatively substituted mutants. The volume of the residue further modulates the VDI response such that small positively charged residues yielded faster VDI kinetics than larger ones, whereas the reverse was observed for negatively substituted mutants. Neutral hydrophobic residues displayed faster VDI gating than hydrophilic neutral ones. In fact, the VDI kinetics of the small but neutral and hydrophobic substituted E462A mutant were similar to the positively charged E462K at most voltages. Among residues of similar hydrophilicity, smaller residues were also more likely to speed up VDI kinetics as seen with the distinct kinetics obtained with E462N and E462Q at + 20 mV. Finally, the glycine-substituted mutant behaved like a negatively charged mutant as documented before for CaV2.3 (Berrou et al., 2001). Hence, our observations are compatible with a molecular model where negatively charged residues in the N-terminal region of the AID helix region decrease the mobility of the I-II loop.
Acknowledgments
We thank Dr. Ed Perez-Reyes for the CaVβ3, CaVβ2a, and the CaV1.2 clones as well as for stimulating discussions; Gérald Bernatchez for preliminary experiments; Nicole Isaac and Stéphanie Bourbonnais for DNA work; Julie Verner for assistance with oocyte culture; and Claude Gauthier for artwork.
This work was completed with a grant of the Canadian Heart and Stroke Foundation and grant MOP13390 from the Canadian Institutes of Health Research to L.P.
References
- Berjukow, S., and S. Hering. 2001. Voltage-dependent acceleration of Ca(v)1.2 channel current decay by (+)- and (−)-isradipine. Br. J. Pharmacol. 133:959–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernatchez, G., L. Berrou, Z. Benakezouh, J. Ducay, and L. Parent. 2001a. Role of Repeat I in the fast inactivation kinetics of the CaV2.3 channel. Biochim. Biophys. Acta. 1514:217–229. [DOI] [PubMed] [Google Scholar]
- Bernatchez, G., R. Sauvé, and L. Parent. 2001b. State-dependent inhibition of inactivation-deficient CaV1.2 and CaV2.3 channels by mibefradil. J. Membr. Biol. 184:143–159. [DOI] [PubMed] [Google Scholar]
- Bernatchez, G., D. Talwar, and L. Parent. 1998. Mutations in the EF-hand motif of the cardiac α1C calcium channel impair the inactivation of barium currents. Biophys. J. 75:1727–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrou, L., G. Bernatchez, and L. Parent. 2001. Molecular determinants of inactivation within the I-II linker of α1E (CaV2.3) Ca2+ channels. Biophys. J. 80:215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrou, L., H. Klein, G. Bernatchez, and L. Parent. 2002. A specific tryptophan in the I-II linker is a key determinant of β-subunit binding and modulation in CaV2.3 calcium channels. Biophys. J. 83:1429–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodi, I., S. E. Koch, H. Yamaguchi, G. P. Szigeti, A. Schwartz, and G. Varadi. 2002. The role of region IVS5 of the human cardiac calcium channel in establishing inactivated channel conformation: use-dependent block by benzothiazepines. J. Biol. Chem. 277:20651–20659. [DOI] [PubMed] [Google Scholar]
- Castellano, A., X. Wei, L. Birnbaumer, and E. Perez-Reyes. 1993. Cloning and expression of a third calcium channel β subunit. J. Biol. Chem. 268:3450–3455. [PubMed] [Google Scholar]
- Cens, T., S. Restituito, S. Galas, and P. Charnet. 1999. Voltage and calcium use the same molecular determinants to inactivate calcium channels. J. Biol. Chem. 274:5483–5490. [DOI] [PubMed] [Google Scholar]
- Chen, Y. H., M. H. Li, Y. Zhang, L. L. He, Y. Yamada, A. Fitzmaurice, Y. Shen, H. Zhang, L. Tong, and J. Yang. 2004. Structural basis of the alpha1-beta subunit interaction of voltage-gated Ca channels. Nature. 429:675–680. [DOI] [PubMed] [Google Scholar]
- Chien, A. J., K. M. Carr, R. E. Shirokov, E. Rios, and M. M. Hosey. 1996. Identification of palmitoylation sites within the L-type calcium channel β2a subunit and effects on channel function. J. Biol. Chem. 271:26465–26468. [DOI] [PubMed] [Google Scholar]
- Chien, A. J., and M. M. Hosey. 1998. Post-translational modifications of β subunits of voltage-dependent calcium channels. J. Bioenerg. Biomembr. 30:377–386. [DOI] [PubMed] [Google Scholar]
- deLeon, M., Y. Wang, L. Jones, E. Perez-Reyes, X. Wei, T. W. Soong, T. P. Snutch, and D. T. Yue. 1995. Essential Ca2+ -binding motif for Ca2+-sensitive inactivation of L-type Ca2+ channels. Science. 270:1502–1506. [DOI] [PubMed] [Google Scholar]
- Erickson, M. G., H. Liang, M. X. Mori, and D. T. Yue. 2003. FRET two-hybrid mapping reveals function and location of L-type Ca2+ channel CaM preassociation. Neuron. 39:97–107. [DOI] [PubMed] [Google Scholar]
- Ertel, E. A., K. P. Campbell, M. M. Harpold, F. Hofmann, Y. Mori, E. Perez-Reyes, A. Schwartz, T. P. Snutch, T. Tanabe, L. Birnbaumer, R. W. Tsien, and W. A. Catterall. 2000. Nomenclature of voltage-gated calcium channels. Neuron. 25:533–535. [DOI] [PubMed] [Google Scholar]
- Ferreira, G., E. Rios, and N. Reyes. 2003. Two components of voltage-dependent inactivation in Cav1.2 channels revealed by its gating currents. Biophys. J. 84:3662–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering, S., S. Aczel, M. Grabner, F. Doring, S. Berjukow, J. Mitterdorfer, M. J. Sinnegger, J. Striessnig, V. E. Degtiar, Z. Wang, and H. Glossmann. 1996. Transfer of high sensitivity for benzothiazepines from L-type to class A (BI) calcium channels. J. Biol. Chem. 271:24471–24475. [DOI] [PubMed] [Google Scholar]
- Hering, S., S. Berjukow, A. Aczel, and E. N. Timin. 1998. Ca2+ channel block and inactivation: common molecular determinants. Trends Pharmacol. Sci. 19:439–443. [DOI] [PubMed] [Google Scholar]
- Herlitze, S., G. H. Hockerman, T. Scheuer, and W. A. Catterall. 1997. Molecular determinants of inactivation and G protein modulation in the intracellular loop connecting domains I and II of the calcium channel α1A subunit. Proc. Natl. Acad. Sci. USA. 94:1512–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isacoff, E. Y., Y. N. Jan, and L. Y. Jan. 1991. Putative receptor for the cytoplasmic inactivation gate in the Shaker K channel. Nature. 353:86–90. [DOI] [PubMed] [Google Scholar]
- Kim, J., S. Ghosh, D. A. Nunziato, and G. S. Pitt. 2004. Identification of the components controlling inactivation of voltage-gated Ca(2+) channels. Neuron. 41:745–754. [DOI] [PubMed] [Google Scholar]
- Lacerda, A. E., E. Perez-Reyes, X. Wei, A. Castellano, and A. M. Brown. 1994. T-type and N-type calcium channels of Xenopus oocytes: evidence for specific interactions with β-subunits. Biophys. J. 66:1833–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. H., A. N. Daud, L. L. Cribbs, A. E. Lacerda, A. Pereverzev, U. Klockner, T. Schneider, and E. Perez-Reyes. 1999a. Cloning and expression of a novel member of the low voltage-activated T-type calcium channel family. J. Neurosci. 19:1912–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, A., S. T. Wong, D. Gallagher, B. Li, D. R. Storm, T. Scheuer, and W. A. Catterall. 1999b. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 399:155–159. [DOI] [PubMed] [Google Scholar]
- Liang, H., C. D. DeMaria, M. G. Erickson, M. X. Mori, B. A. Alseikhan, and D. T. Yue. 2003. Unified mechanisms of Ca regulation across the Ca channel family. Neuron. 39:951–960. [DOI] [PubMed] [Google Scholar]
- Liu, Y., M. E. Jurman, and G. Yellen. 1996. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron. 16:859–867. [DOI] [PubMed] [Google Scholar]
- Monteil, A., J. Chemin, E. Bourinet, G. Mennessier, P. Lory, and J. Nargeot. 2000. Molecular and functional properties of the human α1G subunit that forms T-type calcium channels. J. Biol. Chem. 275:6090–6100. [DOI] [PubMed] [Google Scholar]
- Opatowsky, Y., C. C. Chen, K. P. Campbell, and J. A. Hirsch. 2004. Structural analysis of the voltage-dependent calcium channel beta subunit functional core and its complex with the alpha1 interaction domain. Neuron. 42:387–399. [DOI] [PubMed] [Google Scholar]
- Page, K. M., G. J. Stephens, N. S. Berrow, and A. C. Dolphin. 1997. The intracellular loop between domains I and II of the B-type calcium channel confers aspects of G-protein sensitivity to the E-type calcium channel. J. Neurosci. 17:1330–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent, L., M. Gopalakrishnan, A. E. Lacerda, X. Wei, and E. Perez-Reyes. 1995. Voltage-dependent inactivation in a cardiac-skeletal chimeric calcium channel. FEBS Lett. 360:144–150. [DOI] [PubMed] [Google Scholar]
- Parent, L., T. Schneider, C. P. Moore, and D. Talwar. 1997. Subunit regulation of the human brain α1E calcium channel. J. Membr. Biol. 160:127–140. [DOI] [PubMed] [Google Scholar]
- Patil, P. G., D. L. Brody, and D. T. Yue. 1998. Preferential closed-state inactivation of neuronal calcium channels. Neuron. 20:1027–1038. [DOI] [PubMed] [Google Scholar]
- Peterson, B. Z., C. D. DeMaria, J. P. Adelman, and D. T. Yue. 1999. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 22:549–558. [DOI] [PubMed] [Google Scholar]
- Piedras-Renteria, E. S., and R. W. Tsien. 1998. Antisense oligonucleotides against α1E reduce R-type calcium currents in calcium currents in cerebellar granule cells. Proc. Natl. Acad. Sci. USA. 95:7760–7765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, N., R. Olcese, M. Bransby, T. Lin, and L. Birnbaumer. 1999. Ca2+-induced inhibition of the cardiac Ca2+ channel depends on calmodulin. Proc. Natl. Acad. Sci. USA. 96:2435–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, N., D. Platano, R. Olcese, J. L. Costantin, E. Stefani, and L. Birnbaumer. 1998. Unique regulatory properties of the type 2a Ca2+ channel β subunit caused by palmitoylation. Proc. Natl. Acad. Sci. USA. 95:4690–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restituito, S., T. Cens, C. Barrere, S. Geib, S. Galas, W. M. De, and P. Charnet. 2000. The β2a subunit is a molecular groom for the Ca2+ channel inactivation gate. J. Neurosci. 20:9046–9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoz, G., I. Lopez-Gonzalez, S. Stamboulian, N. Weiss, C. Arnoult, and M. De Waard. 2004. Repositioning of charged I-II loop amino acid residues within the electric field by beta subunit as a novel working hypothesis for the control of fast P/Q calcium channel inactivation. Eur. J. Neurosci. 19:1759–1772. [DOI] [PubMed] [Google Scholar]
- Shi, C., and N. M. Soldatov. 2002. Molecular determinant of voltage dependent slow inactivation of Ca2+ channel. J. Biol. Chem. 277:6813–6821. [DOI] [PubMed] [Google Scholar]
- Stephens, G. J., K. M. Page, Y. Bogdanov, and A. C. Dolphin. 2000. The α1B Ca2+ channel amino terminus contributes determinants for subunit-mediated voltage-dependent inactivation properties. J. Physiol. (Lond.). 525:377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stotz, S. C., J. Hamid, R. L. Spaetgens, S. E. Jarvis, and G. W. Zamponi. 2000. Fast inactivation of voltage-dependent calcium channels: a hinged-lid mechanism. J. Biol. Chem. 275:24575–24582. [DOI] [PubMed] [Google Scholar]
- Stotz, S. C., S. E. Jarvis, and G. W. Zamponi. 2004. Functional roles of cytoplasmic loops and pore lining transmembrane helices in the voltage-dependent inactivation of HVA calcium channels. J. Physiol. 554:263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stotz, S. C., and G. W. Zamponi. 2001. Identification of inactivation determinants in the domain IIS6 region of high voltage activated calcium channels. J. Biol. Chem. 276:33001–33010. [DOI] [PubMed] [Google Scholar]
- Takahashi, S. X., S. Mittman, and H. M. Colecraft. 2003. Distinctive modulatory effects of five human auxiliary β2 subunit splice variants on L-type calcium channel gating. Biophys. J. 84:3007–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tareilus, E., M. Roux, N. Qin, R. Olcese, J. Zhou, E. Stefani, and L. Birnbaumer. 1997. A Xenopus oocyte β subunit: evidence for a role in the assembly/expression of voltage-gated calcium channels that is separate from its role as a regulatory subunit. Proc. Natl. Acad. Sci. USA. 94:1703–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Petegem, F., K. A. Clark, F. C. Chatelain, and D. L. Minor Jr. 2004. Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain. Nature. 429:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, M. E., D. H. Feldman, A. F. McCue, R. Brenner, G. Velicelebi, S. B. Ellis, and M. M. Harpold. 1992. Structure and functional expression of α1, α2, and β subunits of a novel human neuronal calcium channel subtype. Neuron. 8:71–84. [DOI] [PubMed] [Google Scholar]
- Zhou, M., J. H. Morais-Cabral, S. Mann, and R. MacKinnon. 2001. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 411:657–661. [DOI] [PubMed] [Google Scholar]
- Zuhlke, R. D., G. S. Pitt, K. Deisseroth, R. W. Tsien, and H. Reuter. 1999. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 399:159–162. [DOI] [PubMed] [Google Scholar]