

Abstract
Malignant hyperthermia (MH) and central core disease (CCD) are disorders of skeletal muscle Ca2+ homeostasis that are linked to mutations in the type 1 ryanodine receptor (RyR1). Certain RyR1 mutations result in an MH-selective phenotype (MH-only), whereas others result in a mixed phenotype (MH + CCD). We characterized effects on Ca2+ handling and excitation-contraction (EC) coupling of MH-only and MH + CCD mutations in RyR1 after expression in skeletal myotubes derived from RyR1-null (dyspedic) mice. Compared to wild-type RyR1-expressing myotubes, MH + CCD- and MH-only-expressing myotubes exhibited voltage-gated Ca2+ release (VGCR) that activated at more negative potentials and displayed a significantly higher incidence of spontaneous Ca2+ oscillations. However, maximal VGCR was reduced only for MH + CCD mutants (Y4795C, R2435L, and R2163H) in which spontaneous Ca2+ oscillations occurred with significantly longer duration (Y4795C and R2435L) or higher frequency (R2163H). Notably, myotubes expressing these MH + CCD mutations in RyR1 exhibited both increased [Ca2+]i and reduced sarcoplasmic reticulum (SR) Ca2+ content. We conclude that MH-only mutations modestly increase basal release-channel activity in a manner insufficient to alter net SR Ca2+ content (“compensated leak”), whereas the mixed MH + CCD phenotype arises from mutations that enhance basal activity to a level sufficient to promote SR Ca2+ depletion, elevate [Ca2+]i, and reduce maximal VGCR (“decompensated leak”).
INTRODUCTION
In skeletal muscle, excitation-contraction (EC) coupling involves a unique, presumably physical interaction between two different types of Ca2+ channels: voltage-gated L-type Ca2+ channels (L-channels, dihydropyridine receptors (DHPRs)) located in the sarcolemma and Ca2+ release channels (ryanodine receptors (RyR1s)) of the sarcoplasmic reticulum (SR) (Melzer et al., 1995,). In response to sarcolemmal depolarization, DHPRs undergo a conformational change that activates nearby RyR1s. The subsequent massive release of SR Ca2+ into the myoplasm activates the contractile machinery (a process termed “orthograde coupling”) (Nakai et al., 1996; Dulhunty et al., 2002). The DHPR-RyR1 mechanical interaction is bidirectional since RyR1 proteins dramatically enhance the ability of the DHPR to function efficiently as a Ca2+-conducting ion channel (termed “retrograde coupling”; Nakai et al., 1996; Grabner et al., 1999; Avila and Dirksen, 2000; Dirksen, 2002).
Four clinically distinct hereditary human muscle disorders are known to be associated with point mutations and deletions in the gene encoding RyR1: malignant hyperthermia (MH), central core disease (CCD), multiminicore disease (MmD), and nemaline rod myopathy (NM) (for recent reviews, see Jurkat-Rott et al., 2002; Taratuto, 2002; Lueck et al., 2004). MH is a pharmacogenetic syndrome in which hyperthermia, acidosis, hypermetabolism, and muscle rigidity are triggered in MH-susceptible (MHS) individuals after exposure to inhalation anesthetics (e.g., halothane) or depolarizing skeletal muscle relaxants (e.g., succinylcholine). CCD, also called Shy-Magee syndrome, is the first described congenital myopathy and is characterized by proximal muscle weakness and skeletal deformities of the lower limbs (Taratuto, 2002; Lueck et al., 2004). Diagnosis of CCD is determined through histological identification of single, large amorphous areas of reduced oxidative enzyme activity in central or peripheral regions of type 1 muscle fibers (i.e., central cores). MacLennan and colleagues suggested that elevated resting Ca2+ levels in CCD muscle may cause Ca2+-mediated destruction of mitochondria in the center of the muscle fiber and that cores represent a cellular defense mechanism designed to protect the rest of the cell from further Ca2+-induced damage (Loke and MacLennan, 1998). However, in some CCD individuals, cores are not centrally located, but rather are found in peripheral regions of the muscle fiber. Additionally, resting Ca2+ levels are apparently not elevated by CCD mutations that result in EC uncoupling (Avila et al., 2001, 2003b), indicating that an elevation in resting Ca2+ is not an absolute requirement for core formation. Specific variants of MmD (Ferreiro et al., 2002; Jungbluth et al., 2002) and NM (Monnier et al., 2000; Scacheri et al., 2000) have very recently been shown to be also associated with mutations in the RyR1 gene. Skeletal muscle biopsies of patients suffering from MmD display multifocal, poorly circumscribed and short core-like lesions, whereas skeletal muscle obtained from individuals with NM exhibit rod-like structures when visualized using Gomori's trichrome stain in both type I and II skeletal muscle fibers (Monnier et al., 2000; Scacheri et al., 2000; Lueck et al., 2004).
Significant clinical overlap exists between these related muscle disorders. For example, early-stage CCD may first present as a minicore myopathy (Ferreiro et al., 2002), nemaline rods have been found adjacent to central cores in biopsies of some CCD patients (Scacheri et al., 2000), and CCD patients are often found to be MHS. A particularly interesting example of the clinical/genetic overlap between MH and CCD is the finding that mutation of a single highly conserved arginine in RyR1 to a cysteine residue (R2163C—all numbering here and throughout corresponds to the rabbit RyR1 amino acid sequence; accession P11716) results in MHS in the absence of a clinical CCD phenotype in one family, whereas mutation of the same amino acid to a histidine residue (R2163H) results in MHS and CCD coincidence in another family (Manning et al., 1998). For simplicity, in this report we refer to RyR1 mutations that result only in increased MHS as MH-only, RyR1 mutations that result in CCD diagnosis in the absence of a documented increase in MHS as CCD-only, and RyR1 mutations that result in the coincidence of MHS and clinical CCD as MH + CCD.
At least 61 different autosomal dominant mutations/deletions in RyR1 are associated with MH and/or CCD (see Dirksen and Avila, 2004, for review). Among these, at least 20 mutations result in MHS in the absence of clinical CCD (i.e., MH-only) and only 10 mutations have been unequivocally shown to lead to a mixed MH and CCD phenotype (i.e., MH + CCD). MH susceptibility has not been confirmed/tested for many of the other CCD mutations. Parallel studies characterizing functional effects of RyR1 mutations associated with MH-only, CCD-only, and MH + CCD are required to probe possible distinct cellular/molecular mechanisms associated with each disorder. Thus, we set out to characterize the functional consequences of several MH-only (R615C, R2163C, ΔE2347, and T4825I) and MH + CCD (R2163H, R2435L, T4636A, and Y4795C) mutations in RyR1 on resting Ca2+, SR Ca2+ content, and the orthograde/retrograde signals of EC coupling after expression in dyspedic myotubes. Our results indicate that the R2163H, R2435L, and Y4795C MH + CCD mutations in RyR1 lead to a parallel increase in resting myoplasmic Ca2+ levels and decrease in SR Ca2+ content, whereas MH-only mutations do not significantly alter these parameters. In addition, maximal voltage-gated SR Ca2+ release during EC coupling is significantly decreased only in MH + CCD-expressing dyspedic myotubes. Interestingly, we also found that both MH-only- and MH + CCD-expressing myotubes exhibited spontaneous, nontriggered Ca2+ oscillations. However, myotubes expressing MH + CCD mutations in RyR1 that result in steady-state SR Ca2+ depletion exhibited spontaneous Ca2+ oscillations that occurred with significantly higher frequency (R2163H) or longer duration (R2435L and Y4795C) than those observed for wild-type (WT) RyR1-expressing myotubes. Our results suggest that the MH + CCD mutations in RyR1 are unique in that they enhance basal release-channel activity in a manner sufficient to promote steady-state SR Ca2+ store depletion. Taken together, these findings indicate that MH and CCD coincidence arises from a subgroup of MH mutations that exhibit increased basal release-channel activity that leads to uncompensated SR Ca2+ unloading and a subsequent reduction in voltage-gated Ca2+ release during EC coupling.
METHODS
Preparation and cDNA microinjections of dyspedic myotubes
Dyspedic myotubes were prepared as described previously (Avila and Dirksen, 2000). Each of the human MH-only and MH + CCD mutations used in this study were introduced into a rabbit RyR1 cDNA (accession #X15750) using a standard two-step site-directed mutagenesis strategy (Avila and Dirksen, 2001). The human MH/CCD amino acid mutations (R614C, R2163H, R2163C, ΔE2347, R2435L, T4637A, Y4796C, and T4826I—accession #J05200) investigated here correspond to R615C, R2163H, R2163C, ΔE2347, R2435L, T4636A Y4795C, and T4825I, respectively, in the rabbit RyR1 amino acid sequence (accession #P11716). Four to 6 days after initial planting of myoblasts, nuclei of dyspedic myotubes were microinjected with cDNAs encoding CD8 (0.2 μg/μl) and the appropriate RyR1 expression plasmid (0.5 μg/μl). Expressing myotubes were identified 2–4 days after cDNA microinjection after incubation with CD8 antibody beads.
Intracellular Ca2+ measurements in intact myotubes
Intact (non-patch clamped) myotubes were loaded with the Ca2+-sensitive ratiometric dye Indo-1 AM (Molecular Probes, Eugene, OR) and resting intracellular Ca2+, as well as maximal responses to caffeine and cyclopiazonic acid (CPA) were measured as previously described (Avila et al., 2001, 2003a). Briefly, myotubes grown on glass coverslips were exposed for 45 min in rodent Ringer's solution (see Recording solutions) containing 6 μM Indo-1 AM. Myotubes were then rinsed several times with Indo-1 AM-free Ringer's solution and incubated an additional 20 min at 37°C to allow for deesterification of the dye. A small rectangular region of individual Indo-1 AM-loaded expressing myotubes was excited at 350 nm using a DeltaRam illumination system (Photon Technology International, Lawrenceville, NJ). Fluorescence emission at 405 nm and 485 nm was monitored using a 40× (1.35 NA) oil immersion objective, collected with a photomultiplier detection system, acquired at 100 Hz, and the results presented as the ratio (R) of F405/F485. Resting fluorescence ratio (Rbase) for each myotube was converted to [Ca2+]i using an in situ calibration approach described previously (Avila et al., 2001). Briefly, after recording the resting fluorescence ratio in Indo-1-AM-loaded myotubes, cells were whole-cell patch-clamped with patch pipette solutions containing (in mM): 145 CsCl, 5 MgCl2, 10 BAPTA, 0–2.75 CaCl2, 10 HEPES, 0.006 K5Indo-1, pH 7.4. Values of free Ca2+ in the patch pipette ranged from 0–150 nM. After equilibration with the pipette solution, a new ratio at the known free Ca2+ level was then recorded. Multiple experiments at five different free Ca2+ levels (0 nM, 30 nM, 90 nM, 120 nM, and 150 nM) were used to generate a standard in situ resting Ca2+ calibration curve. To measure SR Ca2+ content, maximal concentrations of caffeine (10 mM) or cyclopiazonic acid (CPA, 30 μM) were locally applied to myotubes using a rapid perfusion system (Warner Instruments, Hamden, CT) that permits local application/removal of agonists. Peak intracellular Ca2+ responses to each agent are expressed as ΔRatio (Ragonist − Rbase). Data were analyzed using FeliX (Photon Technology International, Lawrenceville, NJ) and SigmaPlot 2000 (SPSS, Chicago, IL) software packages. For the data presented in Figs. 1, B–D, and 2, C–E, statistical significance from WT RyR1 across multiple groups was assessed using a one-way analysis of variance (ANOVA) and a posthoc Dunnett test (P < 0.05). A chi-squared test was used to evaluate the statistical significance of effects of MH-only and MH + CCD mutations in RyR1 on the frequency of myotubes exhibiting spontaneous Ca2+ oscillations (Fig. 2 B).
FIGURE 1.

MH + CCD mutations in RyR1 induce parallel and opposing changes in resting Ca2+ and releasable SR Ca2+ content. (A) Representative caffeine-induced Ca2+ responses obtained from intact Indo-1 AM-loaded dyspedic myotubes expressing WT RyR1, R2163H (MH + CCD), or R2163C (MH-only). Intracellular Ca2+ levels were recorded in the absence (resting Ca2+) and presence of 10 mM caffeine (shaded bar). For each panel, a dashed line representing the average resting fluorescence ratio for WT RyR1-expressing myotubes is shown for comparison. (B) Average resting Ca2+ levels for dyspedic myotubes expressing either WT RyR1 (black bar), four different MH + CCD mutant RyR1s (shaded bars), and four different MH-only mutant RyR1s (white bars). Number of experiments is given above each bar. (C and D) Average peak responses to application of maximal concentrations of caffeine (C, 10 mM) and CPA (D, 30 μM) in dyspedic myotubes expressing wild-type RyR1 (black bar) and each of the MH + CCD (shaded bars) and MH-only (white bars) mutations in RyR1 illustrated in B. A one-way ANOVA analysis (p < 0.05) revealed that mean WT RyR1 data in B–D were statistically indistinguishable from the corresponding MH-only group data, whereas MH + CCD group data were statistically different from both WT RyR1 and MH-only group data. Asterisks indicate significant differences compared to WT RyR1 using ANOVA/Dunnett analysis (P < 0.05). In this and subsequent figures, RyR1 mutants for each group (MH + CCD and MH-only) are presented in increasing order for elevating resting Ca2+ as shown in B.
FIGURE 2.
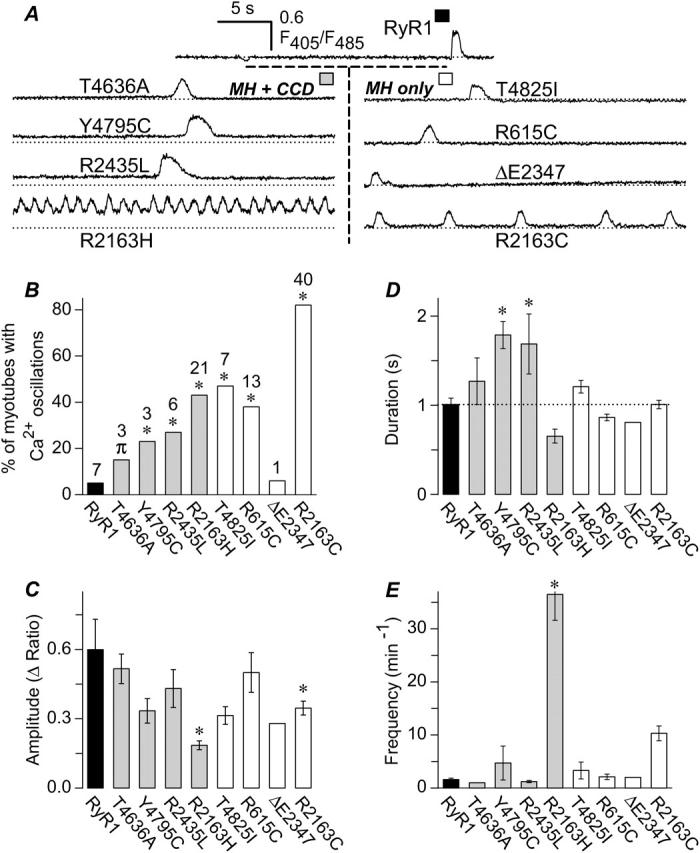
MH-only- and MH + CCD-expressing dyspedic myotubes exhibit increased incidence of spontaneous fluctuations in intracellular Ca2+ (Ca2+ oscillations). (A) Representative spontaneous Ca2+ oscillations in intact Indo-1 AM-loaded myotubes expressing WT RyR1 (top) and each of the eight different MH + CCD (left) and MH-only mutants (right). For comparison, a dashed line representing the average resting fluorescence ratio for WT RyR1-expressing myotubes is shown for each construct. (B) Global intracellular Ca2+ levels were continuously recorded for all of the cells analyzed in Fig. 1 B and spontaneous Ca2+ oscillations were analyzed for the percentage of myotubes exhibiting Ca2+ oscillations during the first 60 s of recording (numbers above each bar represent the number of myotubes from Fig. 1 B that exhibited spontaneous oscillations for each construct). Statistical significance in the percentage MH + CCD- and MH-only-expressing myotubes exhibiting spontaneous Ca2+ oscillations compared to that of WT RyR1-expressing myotubes was assessed using a nonparametric chi-squared test (*p < 0.05, πp < 0.1). (C–E) Spontaneous Ca2+ oscillation amplitude (C), duration (D), and frequency (E) analyzed from WT RyR1-, MH + CCD-, and MH-only-expressing myotubes. Asterisks indicate significant differences compared to RyR1 using ANOVA/Dunnett analysis (P < 0.05). MH/CCD mutations significantly increased the duration (Y4795C and R2435L) and frequency (R2163H) of spontaneous Ca2+ oscillations, whereas both MH-only and MH/CCD mutations in R2163 (R2163C and R2163H) resulted in a significant reduction in Ca2+ oscillation amplitude.
Simultaneous measurements of macroscopic L-currents and Ca2+ transients
The whole-cell patch-clamp technique (Hamill et al., 1981) in conjunction with a Ca2+-sensitive dye (fluo-3) was used to simultaneously measure voltage-gated L-currents and intracellular Ca2+ transients in RyR1-, MH-only-, and MH + CCD-expressing myotubes. A 1-s prepulse to −20 mV delivered immediately before each test pulse was used to inactivate sodium and T-type Ca2+ channels without producing significant L-channel inactivation. L-currents were subsequently elicited by brief (30-ms) test depolarizations applied from a holding potential of −80 mV. Capacitative currents were minimized to ∼10% using the capacitance cancellation feature of the patch-clamp amplifier. Remaining linear components were leak-subtracted using −P/3 protocol delivered from the holding potential before each test pulse. Peak L-currents were normalized to total cell capacitance (pA/pF), plotted as a function of test potential, and fitted according to the following Boltzmann equation:
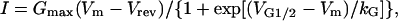 |
(1) |
where Gmax is maximal L-channel conductance, Vm is the test potential, Vrev is the extrapolated reversal potential, VG1/2 is the potential for half-maximal activation of Gmax, and kG is a slope factor. Relative changes in intracellular Ca2+ during each test pulse were recorded in fluo-3 dialyzed myotubes as described previously (Avila et al., 2003a). All patch-clamp experiments were carried out after allowing an ∼5-min period of dialysis after establishment of the whole-cell configuration. Fluo-3-dialyzed myotubes were excited at 480 nm and fluorescence emission measured at 535 nm was analog filtered (τ = 0.5 ms) before digitization at 10 kHz. A computer-controlled shutter was used to eliminate dye illumination during intervals between test pulses. Relative changes in Ca2+ were expressed as ΔF/F ([Fpeak − Fbase]/Fbase) at the end of each test pulse, plotted as a function of Vm, and fitted according to:
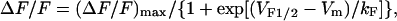 |
(2) |
where (ΔF/F)max is the maximal fluorescence change, VF1/2 is the potential for half-maximal activation of (ΔF/F)max, and kF is a slope factor. Pooled current-voltage (I-V) and fluorescence-voltage (ΔF/F-V) data in Table 1 were expressed as mean ± SE. Statistical significance from WT RyR1-expressing myotubes was determined using a Student's two-tailed t-test. For all analyses, differences were considered statistically significant at P < 0.05.
TABLE 1.
Parameters of fitted I - V and ΔF/F - V curves
| I - V data
|
ΔF/F - V data
|
|||||||
|---|---|---|---|---|---|---|---|---|
| n | Gmax | VG1/2 | kG | Vrev | (ΔF/F)max | VF1/2 | kF | |
| ns/nF | mV | mV | mV | mV | mV | |||
| RyR-1 | 31 | 193 ± 14 | 15.6 ± 0.7 | 5.6 ± 0.1 | 75 ± 1.0 | 2.9 ± 0.3 | 7.5 ± 1.0 | 5.6 ± 0.3 |
| MH+CCD | ||||||||
| T4636A | 10 | 198 ± 18 | 16.6 ± 1.5 | 6.2 ± 0.2 | 76 ± 1.7 | 2.2 ± 0.4 | 3.4 ± 2.4 | 4.6 ± 0.6 |
| Y4795C | 9 | 229 ± 41 | 15.4 ± 1.1 | *6.6 ± 0.2 | 75 ± 1.3 | *1.6 ± 0.2 | *−7.0 ± 1.9 | 7.1 ± 0.3 |
| R2435L | 8 | 213 ± 29 | 17.7 ± 1.8 | 5.9 ± 0.2 | 72 ± 2.5 | 2.0 ± 0.4 | *0.7 ± 1.5 | 4.9 ± 0.3 |
| R2163H | 8 | 144 ± 18 | *8.6 ± 1.5 | *7.5 ± 0.3 | 75 ± 2.1 | *1.6 ± 0.7 | *−13.5 ± 1.1 | 6.8 ± 1.7 |
| MH−only | ||||||||
| T4825l | 13 | 180 ± 19 | *19.4 ± 1.7 | *6.4 ± 0.2 | 78 ± 2.3 | 2.4 ± 0.3 | 5.1 ± 2.0 | 4.5 ± 0.4 |
| R615C | 9 | 188 ± 15 | 13.9 ± 1.3 | *6.6 ± 0.4 | 72 ± 1.7 | 3.3 ± 1.0 | *−0.1 ± 1.6 | 5.2 ± 0.8 |
| ΔE2347 | 9 | 231 ± 20 | 13.4 ± 1.1 | *6.4 ± 0.1 | 74 ± 2.4 | 3.1 ± 0.7 | *−3.8 ± 1.7 | 5.0 ± 0.4 |
| R2163C | 14 | 167 ± 14 | 12.8 ± 1.1 | *7.2 ± 0.2 | 74 ± 1.1 | 3.1 ± 0.3 | *−7.0 ± 1.5 | 5.5 ± 0.2 |
Compared to RyR-1 p < 0.05.
Recording solutions
Myoplasmic Ca2+ levels in intact myotubes were determined in the presence of a normal rodent Ringer's solution consisting of (in mM): 145 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, and 10 HEPES. For patch-clamp experiments, the bath solution contained (in mM) 145 TEA-Cl, 10 CaCl2, and 10 HEPES and the internal solution contained (in mM) 145 Cs-aspartate, 10 CsCl, 0.1 Cs2EGTA, 1.2 MgCl2, 5 Mg-ATP, 0.2 K5-fluo-3, and 10 HEPES. The pH of all solutions was adjusted to 7.4.
RESULTS
Only MH + CCD mutations in RyR1 increase resting Ca2+ and promote SR Ca2+ depletion
We have previously shown that dyspedic myotubes expressing certain MH + CCD mutations in RyR1 exhibit reduced voltage-gated SR Ca2+ release as a result of a partial depletion of SR Ca2+ content (Avila and Dirksen, 2001; Dirksen and Avila, 2002; Avila et al., 2003b). Interestingly, Tong et al. (1999) found that the Ca2+ content of the endoplasmic reticulum was reduced after transient expression of both MH-only and MH + CCD mutations in MH/CCD regions 1 and 2 in HEK-293 cells. To determine if a similar effect on SR Ca2+ store content occurs after expression within a skeletal muscle context, we compared the functional consequences (on resting Ca2+ and SR Ca2+ content) of Indo-1 AM-loaded dyspedic myotubes expressing several different MH-only (R615C, R2163C, ΔE2347, and T4825I) and MH + CCD (R2163H, R2435L, T4636A, and Y4795C) mutant RyR1 proteins (Fig. 1). In these experiments, releasable SR Ca2+ content was measured following exposure to maximal concentrations of either 10 mM caffeine (which rapidly releases SR Ca2+ by directly activating RyRs) or 30 μM CPA (which increases cytoplasmic Ca2+ by inhibiting SR Ca2+-ATPase activity and thereby preventing Ca2+ reuptake lost through passive Ca2+ leak pathways). Together, caffeine and CPA experiments provide two mechanistically distinct means of assessing SR Ca2+ content in WT-, MH-only-, and MH + CCD-expressing myotubes. In this regard, mutations to R2163 of RyR1 are of particular comparative interest as substitution by a histidine residue at this position results in an MH + CCD phenotype, whereas a cysteine mutation of the same residue results in an MH-selective phenotype (Manning et al., 1998).
As reflected in the representative traces shown in Fig. 1 A, compared to wild-type RyR1-expressing myotubes (left), resting Ca2+ was elevated and maximal caffeine-induced Ca2+ release was reduced in R2163H-expressing (MH + CCD) myotubes (middle). However, myotubes expressing an MH-only mutation of the same residue (R2163C) were without effect on either resting Ca2+ or maximal caffeine-induced Ca2+ release (right). On average, the R2163H mutation significantly increased resting Ca2+ from ∼50 nM (RyR1, n = 143) to ∼90 nM (R2163H, n = 49), whereas resting Ca2+ was not significantly altered in R2163C-expressing myotubes (R2163C, n = 49) (Fig. 1 B). In addition, average maximal caffeine- (10 mM, Fig. 1 C) and CPA-induced (30 μM, Fig. 1 D) responses were significantly reduced in R2163H-expressing, but not R2163C-expressing, myotubes. These results are striking in that mutation of a single RyR1 residue associated with CCD (R2163H) increased resting Ca2+ and reduced caffeine and CPA responses, whereas these parameters were essentially unaffected by an MH-only mutation of the same residue (R2163C). Accordingly, two of the other MH + CCD mutations tested (R2435L and Y4795C) also significantly increased resting Ca2+ levels and reduced maximal caffeine and CPA responses. In contrast, none of the MH-only mutants tested (T4825I, R615C, ΔE2347, and R2163C) significantly modified either resting Ca2+ or maximal caffeine/CPA responses (Fig. 1, B–D).
MH-only- and MH + CCD-expressing myotubes exhibit increased incidence of spontaneous Ca2+ oscillations
During the course of these experiments, we noted that MH-only- and MH + CCD-expressing myotubes exhibited a significant degree of spontaneous, slow fluctuations in intracellular Ca2+ (i.e., Ca2+ oscillations). Thus, we compared the properties (incidence, peak amplitude, duration, and frequency) of spontaneous Ca2+ oscillations for dyspedic myotubes expressing WT RyR1, MH + CCD mutant RyR1 proteins, and MH-only mutant RyR1 proteins (Fig. 2). Representative spontaneous Ca2+ oscillations recorded from dyspedic myotubes expressing WT RyR1 and each of the eight different disease mutations in RyR1 are shown in Fig. 2 A. Compared to RyR1, the proportion of myotubes exhibiting spontaneous Ca2+ oscillations within the first 60 s of recording was higher for most of the MH-only (T4825I, R615C, and R2163C) and MH + CCD (T4636A, Y4795C, R2435L, and R2163H) mutants (Fig. 2 B). Specifically, spontaneous Ca2+ oscillations were observed in only 7 out of 143 WT RyR1-expressing myotubes (∼5%). A higher incidence of spontaneous Ca2+ oscillations (ranging from 15 to 82%) was observed for all of the different disease mutations in RyR1, except ΔE2347 (6%; 1 out of 17). Fig. 2 also summarizes the effects of MH-only and MH + CCD mutations in RyR1 on the amplitude (Fig. 2 C), duration (Fig. 2 D), and frequency (Fig. 2 E) of spontaneous Ca2+ oscillations.
Interestingly, mutations to residue R2163 were unique in that significant decreases in the amplitude of spontaneous Ca2+ oscillations were observed for both R2163H- (MH + CCD) and R2163C-expressing (MH-only) myotubes (Fig. 2 C). Importantly, MH/CCD mutations were the only mutations that caused a significant alteration in either the duration (Y4795C and R2435L) or frequency (R2163H) of spontaneous Ca2+ oscillations duration compared to that of WT RyR1 (Fig. 2, D–E). Taken together, the results from Fig. 2 suggest that the MH + CCD mutations in RyR1 promote spontaneous Ca2+ oscillations that last significantly longer (e.g., Y4795C and R2435L; Fig. 2 D) or occur at a much higher frequency (e.g., R2163H; Fig. 2 E) than those attributable to WT RyR1. Thus, the increased incidence, duration, and frequency of nontriggered activity of the MH + CCD mutant release channels likely contributes to the steady-state depletion of SR Ca2+ content observed for these mutants (Fig. 1).
Effects of MH-only and MH + CCD mutations on bidirectional DHPR-RyR1 mechanical coupling
We have previously shown that CCD mutations in MH/CCD regions 1 and 2 promote parallel reductions in SR Ca2+ content, maximal voltage-gated SR Ca2+ release, and the voltage required for half-maximal Ca2+ release (Avila and Dirksen, 2001; Dirksen and Avila, 2002; Avila et al., 2003b). Since the data shown in Fig. 1 suggest that store depletion occurs only for MH + CCD mutations in RyR1 and not MH-only mutations, we compared the magnitude and sensitivity of voltage-gated SR Ca2+ release in MH + CCD- and MH-only-expressing myotubes. To accomplish this goal, we simultaneously monitored macroscopic L-type Ca2+ currents (L-currents) and voltage-gated Ca2+ transients in whole-cell voltage-clamp experiments. Fig. 3, A and B, demonstrates that L-current magnitude, kinetics, and voltage dependence are similar for WT RyR1- (black circles), R2163H- (shaded circles), and R2163C-expressing (open circles) myotubes. No significant differences in maximal L-channel conductance were found between these or any of the nine different RyR1 constructs (Fig. 3 C; see also Table 1). Moreover, the few differences observed in the voltage dependence of L-channel activation (VG1/2, and kG) were modest in magnitude and did not reflect any ordered or systematic dependence (Table 1). Thus, all of the different MH-only and MH + CCD mutant RyR1 proteins augmented DHPR L-channel activity to a similar degree (i.e., support retrograde coupling), results which demonstrate that the mutant release channels are synthesized, targeted to SR-sarcolemmal release sites, and functionally interact with junctional voltage sensors.
FIGURE 3.

The MH + CCD and MH-only mutations in RyR1 fully restore retrograde coupling with sarcolemmal DHPRs. (A) Representative whole-cell L-type Ca2+ currents recorded from RyR1- (left), R2163H- (middle), and R2163C-expressing (right) dyspedic myotubes. Current traces were elicited in response to brief (30-ms) membrane depolarizations to the indicated membrane potentials. Dashed lines represent the zero current level. (B) Average (±SEM) peak current-voltage (I-V) relationships for dyspedic myotubes expressing wild-type RyR1 (black circles; n = 31), R2163H (shaded circles, top; n = 8), and R2163C (open circles, bottom; n = 14). The average values (±SEM) for the parameters obtained by fitting each myotube within a group separately to Eq. 1 are given in Table 1 (I-V data). The solid lines through the data were obtained using Eq. 1 and the corresponding parameters given in Table 1 (I-V data). (C) Comparison of maximal L-channel conductance (Gmax) obtained from RyR1- (black bar), MH + CCD- (shaded bars), and MH-only-expressing (white bars) myotubes. Number of experiments is given above each bar.
Although retrograde coupling was not significantly altered by MH-only and MH + CCD mutations in RyR1 (Fig. 3), significant effects on orthograde coupling were observed (Fig. 4). Fig. 4 A shows representative voltage-gated Ca2+ transients elicited by 30-ms voltage steps to the indicated potentials (left) in dyspedic myotubes expressing either RyR1 (black circles), R2163H (shaded circles), or R2163C (open circles). These data demonstrate that mutation of R2163 to either a histidine or cysteine residue results in a similar hyperpolarizing shift in the threshold for voltage-gated SR Ca2+ release (i.e., in Fig. 4 A detectable release occurs at −20 mV for both R2163H and R2163C, but not WT RyR1). However, maximal voltage-gated SR Ca2+ release was significantly reduced only in R2163H-expressing myotubes. Thus, although both mutations caused a similar increase in the sensitivity to activation by voltage, maximal voltage-gated Ca2+ release was significantly reduced only for the MH + CCD mutation in R2163. On average, maximal voltage-gated SR Ca2+ release (ΔF/F)max was reduced 45% in R2163H-expressing myotubes (Fig. 4, B and C; see also Table 1). These results demonstrate that although both mutations in residue R2163 enhance the inherent voltage sensitivity of the Ca2+ release mechanism, the MH + CCD mutation is unique in also causing a reduction in the maximum magnitude of voltage-gated release. Accordingly, maximal voltage-gated release was reduced to a similar degree inY4795C-expressing myotubes (and to a lesser degree in R2435L-expressing myotubes), but was essentially unaltered in myotubes expressing MH-only mutations in RyR1 (Fig. 4 C and Table 1). Furthermore, the voltage sensitivity of Ca2+ release mechanism (VF1/2) was significantly reduced by most (six out of eight) of the MH + CCD and MH-only mutations in RyR1 (Fig. 4 D and Table 1). Taken together, the results of Fig. 4 indicate that increased release-channel sensitivity to activation by voltage correlates with increased MH susceptibility, whereas a reduction in the magnitude of voltage-gated SR Ca2+ release represents a unique distinguishing feature of the CCD phenotype.
FIGURE 4.

Voltage-gated SR Ca2+ release is preferentially reduced in MH + CCD-expressing myotubes. (A) Representative intracellular Ca2+ transients elicited by 30-ms test pulses to the indicated potentials. Representative Ca2+ transients are displayed for dyspedic myotubes expressing RyR1 (left), R2163H (middle), and R2163C (right). Dashed lines represent basal fluorescence before depolarization. (B) Average voltage-dependence of intracellular Ca2+ transients (measured at the end of each test pulse) for dyspedic myotubes expressing wild-type RyR1 (black circles; n = 31), R2163H (shaded circles, top; n = 8), and R2163C (open circles, bottom; n = 14). The average values (±SEM) for the parameters obtained by fitting each myotube within a group separately to Eq. 2 are given in Table 1 (ΔF/F-V data). The solid lines through the data were obtained using Eq. 2 and the corresponding parameters given in Table 1 (ΔF/F-V data). For clarity, average values (±SEM) for maximal voltage-gated SR Ca2+ release ([ΔF/F]max) and the voltage required for half-maximal activation of release (VF1/2) for WT RyR1 (black bar/symbol), MH + CCD mutant RyR1s (shaded bars/symbols), and MH-only mutant RyR1s (white bars/symbols) are plotted in C and D, respectively. Dashed lines in C and D correspond to mean data for WT RyR1. *p < 0.05, πp < 0.1 compared to RyR1.
DISCUSSION
This study presents the first direct comparison of the functional effects on skeletal muscle Ca2+ homeostasis and orthograde/retrograde DHPR/RyR1 coupling of mutations in RyR1 that result in either an MH-selective phenotype or the coincidence of both MH and CCD. Our results demonstrate that MH-only and MH + CCD mutant RyR1 proteins fully support retrograde coupling with sarcolemmal DHPRs (as indicated by normal RyR1-mediated restoration of maximal L-channel conductance or Gmax, Fig. 3 and Table 1). However, compared to WT RyR1-expressing myotubes, MH-only- and MH + CCD-expressing myotubes exhibit both an increased incidence of nonevoked or spontaneous Ca2+ oscillations (Fig. 2) and enhanced sensitivity of the voltage-gated Ca2+ release mechanism (i.e., a hyperpolarized shift in VF1/2, Fig. 4 and Table 1). Importantly, the MH + CCD mutations in RyR1 differed from MH-only mutations in several important aspects in that only MH + CCD-expressing myotubes also exhibited 1), elevated resting Ca2+ levels; 2), a reduction in maximal caffeine/CPA-induced Ca2+ release; 3), altered duration/frequency of spontaneous Ca2+ oscillations; and 4), a reduction in the magnitude of maximal voltage-gated SR Ca2+ release. A similar rank order for MH + CCD mutations in RyR1 was observed for elevations in resting Ca2+ (R2163H > Y4795C, R2435L > T4636A; Fig. 1 B), SR Ca2+ depletion (R2163H > Y4795C, R2435L > T4636A; Fig. 1, C and D), and reduced voltage-gated Ca2+ release (R2163H, Y4795C > R2435L > T4636A; Fig. 4 C). These results suggest that SR Ca2+ unloading underlies the observed reduction in Ca2+ release during EC coupling in MH + CCD-expressing myotubes. In agreement with this notion, resting Ca2+ levels, releasable SR Ca2+ content, and maximal voltage-gated SR Ca2+ were all unaltered in MH-only-expressing myotubes. Taken together, our results indicate that SR Ca2+ depletion and increased basal Ca2+ levels are preferentially associated with RyR1 mutations that result in MH and CCD coincidence.
Distinct alterations in Ca2+ handling caused by MH-only and MH + CCD mutations in RyR1
A number of results presented here and elsewhere (Tong et al., 1999; Avila and Dirksen, 2001; Avila et al., 2003b) indicate that certain CCD mutations in RyR1 (including R164C, Y523S, R2163H, R2435H, R2435L, and Y4795C) promote SR/ER Ca2+ depletion. First, compared to wild-type RyR1-expressing cells, the magnitude of Ca2+ release in response to application of maximal concentrations of caffeine is reduced after both heterologous (Tong et al., 1999) and homologous expression of these CCD mutant RyR1 proteins (Avila and Dirksen, 2001; Avila et al., 2003b; Fig. 1, A and C). Second, since mutations in RyR1 could alter caffeine-induced Ca2+ release via a mechanism distinct from a change in SR Ca2+ load, we also assessed SR Ca2+ load using CPA (Avila and Dirksen, 2001; Avila et al., 2003b; Fig. 1 D), an agent that increases myoplasmic Ca2+ in a manner independent of RyR1 activation (Inesi and Sagara, 1994). The reduced CPA responses of MH + CCD-expressing myotubes (but not MH-only-expressing myotubes) provides further evidence that MH + CCD mutations in RyR1 preferentially promote significant SR Ca2+ unloading (Fig. 1 D). Third, we recently demonstrated that SR Ca2+ depletion of Y523S-expressing myotubes is abolished after incorporation of an additional mutation in the RyR1 pore region (I4897T) that renders the channel poorly permeable to Ca2+ (Avila et al., 2003b). Finally, control experiments in normal myotubes indicate that caffeine and CPA operate on identical Ca2+ storage compartments since CPA does not alter myoplasmic Ca2+ levels after the application of a maximal concentration of caffeine (data not shown). Taken together, these results provide compelling evidence that MH + CCD mutations in RyR1 are unique in promoting a significant reduction in steady-state SR Ca2+ load.
The putative impact of MH-only mutations in RyR1 on resting myoplasmic Ca2+ remains controversial (Mickelson and Louis, 1996). Conflicting reports that MH-only mutations either increase (e.g., Lopez et al., 1986, Wehner et al., 2002) or exert no effect on (Iaizzo et al., 1988; Censier et al., 1998; Tong et al., 1999; this study) resting Ca2+ levels may arise from differences in the methodologies used to quantify intracellular Ca2+ (e.g., Ca2+-sensitive intracellular microelectrodes versus Ca2+-sensitive fluorescence dyes). Another potential source of variation in these studies is the biological background used to investigate the mutant SR Ca2+ release channels (e.g., native cells versus heterologous or homologous expression systems). However, it is well established that intracellular Ca2+ levels are significantly elevated by certain MH + CCD mutations in RyR1 (Tong et al., 1999; Avila and Dirksen, 2001; Avila et al., 2003b; Fig. 1 B). In this context, our experiments in which resting Ca2+ levels were compared using a single assay conducted under identical conditions in the context of a uniform muscle expression system provide strong support for the notion that MH + CCD mutations in RyR1 produce a more profound alteration in resting Ca2+ than that observed for MH-only mutations.
Potential limitations of estimations in resting Ca2+
As discussed above, a difference in methodologies used to estimate resting Ca2+ (i.e., Ca2+-sensitive microelectrodes versus Ca2+-sensitive fluorescence dyes) likely contributes to the current controversy as to the effects of MH-only mutations in RyR1 on intracellular Ca2+ levels. Unfortunately, a significant number of limitations/assumptions are associated with both experimental approaches (Takahashi et al., 1999). We monitored resting Ca2+ levels using a fluorescence approach that does not exhibit the temporal limitations and potential for impalement-induced sarcolemmal damage that can occur when using ion-sensitive intracellular microelectrodes. Nevertheless, a number of other limitations/assumptions are associated with our indo-1 measurements of resting Ca2+. For example, since the Ca2+ affinity (i.e., Kd) and the fluorescence spectra of Indo-1 are influenced by relatively small changes in pH (Baker et al., 1994), differences in intracellular pH (pHi) of WT-, MH-only-, and MH + CCD-expressing myotubes could confound the accuracy of our intracellular Ca2+ measurements. Additionally, changes in the cellular redox state resulting from expression of MH-only and/or MH + CCD mutant RyR1 proteins could also alter the properties of indo-1 and/or autofluorescence of a redox-sensitive cellular constituent (e.g., NADH, which is also excited at 350 nm; Shorte and Bolsover, 1999; Takahashi et al., 1999). Finally, by virtue of its Ca2+ chelator activity, high-affinity Ca2+ indicators like indo-1 may buffer small changes in myoplasmic Ca2+ when loaded into myotubes at high concentrations.
In addition to potential direct effects on the properties of indo-1, changes in the levels of important myoplasmic constituents (e.g., pH, ATP, glutathione) caused by MH-only and/or MH + CCD mutations in RyR1 could indirectly influence myoplasmic Ca2+ by altering regulation of RyR1 activity (for a recent review see Fill and Copello, 2002). Thus, it is difficult to distinguish between direct effects of disease mutations on RyR1 function and alterations in RyR1 regulation that are secondary to changes in myoplasmic pHi, redox, and/or ATP levels. Consequently, observed differences in resting indo-1 fluorescence could reflect alterations in one or more of these myoplasmic factors rather than a genuine change in resting free Ca2+ per se. Moreover, dialysis with a uniform internal solution during our in situ calibration procedure would tend to eliminate differences resulting from changes in these factors. Clearly, future work will be needed (e.g., studies that combine the strengths of both Ca2+-sensitive microelectrode and fluorescence approaches) before a definitive conclusion can be made with regard to the influence of MH-only mutations in RyR1 on resting Ca2+ levels in skeletal muscle.
Pathophysiological implications
How might mutations in RyR1 result in release-channel hypersensitivity and malignant hyperthermia susceptibility (MH-only), whereas other RyR1 mutations promote the coincidence of MHS with muscle weakness and the development of central cores (MH + CCD)? Our results suggest that this dichotomy might arise from fundamentally distinct changes in SR Ca2+ release-channel function induced by MH-only and MH + CCD mutations in RyR1. Fig. 5 summarizes one possible means by which distinct functional defects in Ca2+ handling and EC coupling observed in this study might result in phenotype-specific manifestations of MH and CCD.
FIGURE 5.

Schematic depicting fundamentally distinct cellular consequences of mutations in RyR1 that result in MH-only (compensated leak), MH + CCD (decompensated leak), and CCD-only (EC uncoupled) mutations in RyR1. In this scheme, MH in the absence of CCD arises from RyR1 mutations that result in overactive/supersensitive release channels that fail to cause a net change in steady-state SR Ca2+ content (compensated leak), but predispose muscle to increased MHS upon exposure to triggering agents. The primary distinguishing feature of CCD mutations is a reduction in maximal voltage-gated release that results either from a net reduction in steady-state SR Ca2+ content or EC uncoupling (functional uncoupling of depolarization from the release of Ca2+ from a full SR store). MH and CCD coincidence occurs for mutations in RyR1 that enhance RyR1 activity/sensitivity to an extent sufficient to result in a partial depletion of SR Ca2+ stores decompensated leak).
Our findings, along with those of others (Tong et al., 1997, 1999; Dietze et al., 2000; Avila and Dirksen, 2001; Yang et al., 2003), suggest that both MH-only and MH + CCD mutations in RyR1 cause release channels to exhibit increased/overactive basal channel activity and enhanced sensitivity to activation by both endogenous (i.e., voltage sensors) and exogenous (e.g., caffeine, halothane, 4-chloro-m-cresol) RyR1 triggering agents. However, results presented here suggest that increased release-channel activity/sensitivity resulting from MH-only mutations in RyR1 are subtle enough that cellular compensatory mechanisms (e.g., enhanced SR Ca2+ reuptake via increased sarco/endoplasmic reticulum Ca2+ pump expression/activity) are sufficient to ensure maintenance of normal SR Ca2+ content and voltage-gated SR Ca2+ release during EC coupling (termed Compensated leak in Fig. 5). On the other hand, MH + CCD mutations in RyR1 exhibit a greater degree of increased basal activity (i.e., increased duration or frequency of spontaneous Ca2+ oscillations) that are severe enough that compensatory mechanisms are unable to prevent partial steady-state SR Ca2+ store depletion and a subsequent reduction in the magnitude of Ca2+ release during EC coupling (termed Decompensated leak in Fig. 5). Under this scenario, MH + CCD mutations in RyR1 represent an extreme example of release-channel hyperactivity that leads to net SR Ca2+ unloading. We must emphasize, however, that the scheme depicted in Fig. 5 is an extrapolation of our experimental results and represents only one view of how our data could be interpreted within the clinical context of MH and CCD.
We have previously suggested that mutations in the pore-lining region of RyR1 reduce Ca2+ release during EC coupling in a manner that does not arise from increased basal release-channel activity or SR Ca2+ unloading, but rather by reducing the gating and/or Ca2+ permeation of activated release channels (termed “EC uncoupled” in Fig. 5) (Avila et al., 2001, 2003b). Consequently, the EC uncoupling mechanism would not be expected to result in increased MH susceptibility (i.e., CCD-only; Fig. 5). Consistent with this prediction, no anesthetic complications were noted after exposure to MH-triggering anesthetics for 19 CCD-related individuals possessing the I4898T mutation in the pore-lining region of the channel (Lynch et al., 1999).
SR Ca2+ depletion does not result from increased sensitivity to activation by voltage
Voltage-clamp experiments from our laboratory have revealed that certain MH + CCD mutations in RyR1 (R164C, Y523S, R2163H, R2435H, R2435L, and Y4795C) significantly enhance release-channel sensitivity to activation by voltage (Avila and Dirksen, 2001; Avila et al., 2003b; this report). Interestingly, six of the eight disease mutants in RyR1 investigated in this study (MH-only: R614C, R2163C, and ΔE2347; MH + CCD: R2163H, R2435L, and Y4796C) exhibited varying degrees (7–21 mV hyperpolarizing shift in VF1/2) of increased sensitivity to activation by the voltage sensor (Fig. 4 and Table 1). Consistent with these data, an increased sensitivity of voltage-gated SR Ca2+ release attributable to the porcine R615C MH-selective mutation in RyR1 is also well-documented (Gallant and Lentz, 1992; Gallant and Jordan, 1996; Dietze et al., 2000). Additionally, Yang et al. (2003) recently found that KCl-stimulated Ca2+ release occurred at lower concentrations of KCl in dyspedic myotubes expressing seven different MH mutations in RyR1 (R164C, G342R, R615C, R2163C, V2168M, R2458H, and T4825I), four of which were also studied here (R164C, R615C, R2163C, and T4825I). Thus, it is clear that the voltage sensitivity of the Ca2+ release mechanism is increased by both MH-only and MH + CCD mutations in RyR1. A similar finding was recently reported for a novel MH-causing mutation in the III-IV linker of the skeletal muscle DHPR (R1086H) (Weiss et al., 2004).
Yang et al. (2003) also found that compared to WT RyR1, dyspedic myotubes expressing MH mutants exhibited reduced SR Ca2+ release in response to maximal concentrations of direct channel activators (i.e., caffeine and 4-chloro-m-cresol) and a paradoxical increase in Ca2+ release in response to 60 mM KCl depolarization. These results are in contrast to our observations that neither maximal caffeine-induced Ca2+ release (Fig. 1 C) nor maximal voltage-gated Ca2+ release (Fig. 4 C and Table 1) was significantly altered after expression in dyspedic myotubes of four different MH-only mutant RyR1 proteins (R615C, R2163C, ΔE2347, and T4825I). One possible explanation for this discrepancy is that the study of Yang et al. (2003) inferred effects on maximal orthograde coupling based on 10-s applications of a high concentration (60 mM) of extracellular KCl. In addition, activation of SR Ca2+ release after transient application of 60 mM KCl was apparently not sufficient to provide a saturating response (see Fig. 2 in Yang et al., 2003). The use of saturating KCl concentrations in these experiments is important since as voltage-gated Ca2+ release is shifted to more negative potentials for MH-expressing myotubes, an anomalous increase in voltage-gated Ca2+ release would be expected when using submaximal KCl concentrations. Another possible contributing factor is that our measurements are restricted to rapid voltage-gated release events obtained at the end of very brief (30-ms) and uniform voltage steps. By contrast, Yang et al. (2003) estimated voltage-gated release as the integral of the Ca2+ signal in response to much longer (10-s) KCl depolarizations. Additionally, our caffeine-induced Ca2+ measurements were obtained at the peak of the response, whereas Yang et al. (2003) estimated peak caffeine and 4-chloro-m-cresol responses using a time-integrated method. Differences in release-channel inactivation, slow myoplasmic Ca2+ clearance mechanisms, and/or Ca2+ influx pathways could differentially influence results obtained using these two methodologies.
Our results (Fig. 4 D) suggest that SR Ca2+ unloading is not directly caused by enhanced release-channel activation by voltage since certain MH-only mutations in RyR1 produced hyperpolarizing shifts in VF1/2 that were similar to or larger in magnitude than that observed for some MH + CCD mutations that promote SR Ca2+ depletion (e.g., compare VF1/2 data for R2163C and R2435L data in Fig. 5 D). These findings suggest that the increased frequency (R2163H) and prolonged duration (Y4795C and R2435L) of spontaneous Ca2+ oscillations and SR Ca2+ depletion (R2163H, Y4785C, and R2435L) observed for MH + CCD mutant release channels are not directly attributable to increased release-channel sensitivity to activation by voltage.
Taken together, our results demonstrate that a significant reduction in the magnitude of voltage-gated SR Ca2+ release represents a central distinguishing feature of CCD; either via decompensated leak and store depletion (Avila and Dirksen, 2001; this study) or by EC uncoupling (Avila et al., 2001, 2003b). Moreover, our results also indicate that RyR1 mutations that result in increased MHS (and not CCD) produce release-channel defects that promote subtle increases in release-channel overactivity/sensitivity that occur in the absence of steady-state store depletion (compensated leak), whereas MH and CCD coincidence results from mutations in RyR1 that enhance RyR1 activity/sensitivity to an extent sufficient to result in partial SR Ca2+ store depletion (decompensated leak). A decade ago, Quane et al. (1993) suggested that “a mutation in RyR1 that causes both hypersensitive gating and diminished excitation-contraction coupling could cause both MH and CCD in an individual.” Our results provide compelling experimental support for this assertion.
Acknowledgments
We thank Drs. Kurt G. Beam and Paul D. Allen for providing access to the dyspedic mice used in this study, Sanjeewa Goonasekera for suggestions to improve the manuscript, and Linda Groom for excellent technical assistance.
This work was supported in part by a grant from the National Institute of Health (AR44657 to R.T.D.) and a Neuromuscular Research Grant from the Muscular Dystrophy Association (to R.T.D.).
References
- Avila, G., and R. T. Dirksen. 2000. Functional impact of the ryanodine receptor on the skeletal muscle L-type Ca2+ channel. J. Gen. Physiol. 115:467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila, G., and R. T. Dirksen. 2001. Functional effects of central core disease mutations in the cytoplasmic region of the skeletal muscle ryanodine receptor. J. Gen. Physiol. 118:277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila, G., E. H. Lee, C. F. Perez, P. D. Allen, and R. T. Dirksen. 2003a. FKBP12 binding to RyR1 modulates excitation-contraction coupling in mouse skeletal myotubes. J. Biol. Chem. 278:22600–22608. [DOI] [PubMed] [Google Scholar]
- Avila, G., J. J. O'Brien, and R. T. Dirksen. 2001. Excitation-contraction uncoupling by a human central core disease mutation in the ryanodine receptor. Proc. Natl. Acad. Sci. USA. 98:4215–4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila, G., K. M. S. O'Connell, and R. T. Dirksen. 2003b. The pore region of the skeletal muscle ryanodine receptor is a primary locus for excitation-contraction uncoupling in central core disease. J. Gen. Physiol. 121:277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, A. J., R. Brandes, J. H. Schreur, S. A. Camacho, and N. W. Weiner. 1994. Protein and acidosis alter calcium-binding and fluorescence spectra of the calcium indicator indo-1. Biophys. J. 67:1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Censier, K., A. Urwyler, F. Zorzato, and S. Treves. 1998. Intracellular calcium homeostasis in human primary muscle cells from malignant hyperthermia-susceptible and normal individuals. Effect of overexpression of recombinant wild-type and Arg163Cys mutated ryanodine receptors. J. Clin. Invest. 101:1233–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietze, B., J. Henke, H. M. Eichinger, F. Lehmann-Horn, and W. Melzer. 2000. Malignant hyperthermia mutation Arg615Cys in the porcine ryanodine receptor alters voltage dependence of Ca2+ release. J. Physiol. 526:507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirksen, R. T. 2002. Bi-directional coupling between dihydropyridine receptors and ryanodine receptors. Front. Biosci. 7:d659–d670. [DOI] [PubMed] [Google Scholar]
- Dirksen, R. T., and G. Avila. 2002. Altered ryanodine receptor function in central core disease: leaky or uncoupled Ca2+ release channels? Trends Cardiovasc. Med. 12:189–197. [DOI] [PubMed] [Google Scholar]
- Dirksen, R. T., and G. Avila. 2004. Pathophysiology of muscle disorders linked to mutations in the skeletal muscle ryanodine receptor. In Ryanodine Receptors: Structure, Function and Dysfunction in Clinical Disease. X. H. Wehrens and A. R. Marks, editors. Kluwer Academic Publishers, Dordrecht, The Netherlands. In press.
- Dulhunty, A. F., C. S. Haarmann, D. Green, D. R. Laver, P. G. Board, and M. G. Casarotto. 2002. Interactions between dihydropyridine receptors and ryanodine receptors in striated muscle. Prog. Biophys. Mol. Biol. 79:45–75. [DOI] [PubMed] [Google Scholar]
- Ferreiro, A., N. Monnier, N. B. Romero, J. P. Leroy, C. Bonnemann, C. A. Haenggeli, V. Straub, W. D. Voss, Y. Nivoche, H. Jungbluth, A. Lemainque, T. Voit, J. Lunardi, M. Fardeau, and P. Guicheney. 2002. A recessive form of central core disease, transiently presenting as multi-minicore disease, is associated with a homozygous mutation in the ryanodine receptor type 1 gene. Ann. Neurol. 51:750–759. [DOI] [PubMed] [Google Scholar]
- Fill, M., and J. A. Copello. 2002. Ryanodine receptor calcium release channels. Physiol. Rev. 82:893–922. [DOI] [PubMed] [Google Scholar]
- Gallant, E. M., and R. C. Jordan. 1996. Porcine malignant hyperthermia: genotype and contractile threshold of immature muscles. Muscle Nerve. 19:68–73. [DOI] [PubMed] [Google Scholar]
- Gallant, E. M., and L. R. Lentz. 1992. Excitation-contraction coupling in pigs heterozygous for malignant hyperthermia. Am. J. Physiol. 262:C422–C426. [DOI] [PubMed] [Google Scholar]
- Grabner, M., R. T. Dirksen, N. Suda, and K. G. Beam. 1999. The II–III loop of the skeletal muscle dihydropyridine receptor is responsible for the bi-directional coupling with the ryanodine receptor. J. Biol. Chem. 274:21913–21919. [DOI] [PubMed] [Google Scholar]
- Hamill, O. P., A. Marty, E. Neher, B. Sakmann, and F. J. Sigworth. 1981. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391:85–100. [DOI] [PubMed] [Google Scholar]
- Iaizzo, P. A., W. Klein, and F. Lehmann-Horn. 1988. Fura-2 detected myoplasmic calcium and its correlation with contracture force in skeletal muscle from normal and malignant hyperthermia susceptible pigs. Pflugers Arch. 411:648–653. [DOI] [PubMed] [Google Scholar]
- Inesi, G., and Y. Sagara. 1994. Specific inhibitors of intracellular Ca2+ transport ATPases. J. Membr. Biol. 141:1–6. [DOI] [PubMed] [Google Scholar]
- Jungbluth, H., C. R. Muller, B. Halliger-Keller, M. Brockington, S. C. Brown, L. Feng, A. Chattopadhyay, E. Mercuri, A. Y. Manzur, A. Ferreiro, N. G. Laing, M. R. Davis, H. P. Roper, V. Dubowitz, G. Bydder, C. A. Sewry, and F. Muntoni. 2002. Autosomal recessive inheritance of RYR1 mutations in a congenital myopathy with cores. Neurology. 59:284–287. [DOI] [PubMed] [Google Scholar]
- Jurkat-Rott, K., H. Lerche, and F. Lehmann-Horn. 2002. Skeletal muscle channelopathies. J. Neurol. 249:1493–1502. [DOI] [PubMed] [Google Scholar]
- Loke, J., and D. H. MacLennan. 1998. Malignant hyperthermia and central core disease: disorders of Ca2+ release channels. Am. J. Med. 104:470–486. [DOI] [PubMed] [Google Scholar]
- Lopez, J. R., L. A. Alamo, D. E. Jones, L. Papp, P. D. Allen, J. Gerjely, and F. A. Sreter. 1986. [Ca2+]i in muscles of malignant hyperthermia susceptible pigs determined in vivo with Ca2+ selective microelectrodes. Muscle Nerve. 9:85–86. [PubMed] [Google Scholar]
- Lueck, J. D., S. Goonasekera, and R. T. Dirksen. 2004. Ryanodinopathies: muscle disorders linked to mutations in ryanodine receptors. Basic Appl. Myol. In press.
- Lynch, P. J., J. Tong, M. Lehane, A. Mallet, L. Giblin, J. J. Heffron, P. Vaughan, G. Zafra, D. H. MacLennan, and T. V. McCarthy. 1999. A mutation in the transmembrane/luminal domain of the ryanodine receptor is associated with abnormal Ca2+ release channel function and severe central core disease. Proc. Natl. Acad. Sci. USA. 96:4164–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickelson, J. R., and C. F. Louis. 1996. Malignant hyperthermia: excitation-contraction coupling, Ca2+ release channel, and cell Ca2+ regulation defects. Physiol. Rev. 76:537–592. [DOI] [PubMed] [Google Scholar]
- Manning, B. M., K. A. Quane, H. Ording, A. Urwyler, V. Tegazzin, M. Lehane, J. O'Halloran, E. Hartung, L. M. Giblin, P. J. Lynch, P. Vaughan, K. Censier, D. Bendixen, G. Comi, L. Heytens, K. Monsieurs, T. Fagerlund, W. Wolz, J. J. Heffron, C. R. Muller, and T. V. McCarthy. 1998. Identification of novel mutations in the ryanodine-receptor gene (RYR1) in malignant hyperthermia: genotype-phenotype correlation. Am. J. Hum. Genet. 62:599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzer, W., A. Herrmann-Frank, and H. C. Luttgau. 1995. The role of Ca2+ ions in excitation-contraction coupling of skeletal muscle fibres. Biochim. Biophys. Acta. 1241:59–116. [DOI] [PubMed] [Google Scholar]
- Monnier, N., N. B. Romero, J. Lerale, Y. Nivoche, D. Qi, D. H. MacLennan, M. Fardeau, and J. Lunardi. 2000. An autosomal dominant congenital myopathy with cores and rods is associated with a neomutation in the RYR1 gene encoding the skeletal muscle ryanodine receptor. Hum. Mol. Genet. 9:2599–2608. [DOI] [PubMed] [Google Scholar]
- Nakai, J., R. T. Dirksen, H. T. Nguyen, I. N. Pessah, K. G. Beam, and P. D. Allen. 1996. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature. 380:72–75. [DOI] [PubMed] [Google Scholar]
- Quane, K. A., J. M. Healy, K. E. Keating, B. M. Manning, F. J. Couch, L. M. Palmucci, C. Doriguzzi, T. H. Fagerlund, K. Berg, H. Ording, D. Bendixen, W. Mortier, U. Linz, C. R. Muller, and T. McCarthy. 1993. Mutations in the ryanodine receptor gene in central core disease and malignant hyperthermia. Nat. Genet. 5:51–55. [DOI] [PubMed] [Google Scholar]
- Scacheri, P. C., E. P. Hoffman, J. D. Fratkin, C. Semino-Mora, A. Senchak, M. R. Davis, N. G. Laing, V. Vedanarayanan, and S. H. Subramony. 2000. A novel ryanodine receptor gene mutation causing both cores and rods in congenital myopathy. Neurology. 55:1689–1696. [DOI] [PubMed] [Google Scholar]
- Shorte, S. L., and S. Bolsover. 1999. Imaging reality: understanding maps of physiological cell signals measured by fluorescence microsopy and digital imaging. In Fluorescent and Luminicent Probes for Biological Activity. W. T. Mason, editor. Academic Press, London. 94–106.
- Takahashi, A., P. Camacho, J. D. Lechleiter, and B. Herman. 1999. Measurement of intracellular calcium. Physiol. Rev. 79:1089–1125. [DOI] [PubMed] [Google Scholar]
- Taratuto, A. L. 2002. Congenital myopathies and related disorders. Curr. Opin. Neurol. 15:553–561. [DOI] [PubMed] [Google Scholar]
- Tong, J., T. V. McCarthy, and D. H. MacLennan. 1999. Measurement of resting cytosolic Ca2+ concentrations and Ca2+ store size in HEK-293 cells transfected with malignant hyperthermia or central core disease mutant Ca2+ release channels. J. Biol. Chem. 274:693–702. [DOI] [PubMed] [Google Scholar]
- Tong, J., H. Oyamada, N. Demaurex, S. Grinstein, T. V. McCarthy, and D. H. MacLennan. 1997. Caffeine and halothane sensitivity of intracellular Ca2+ release is altered by 15 calcium release channel (ryanodine receptor) mutations associated with malignant hyperthermia and/or central core disease. J. Biol. Chem. 272:26332–26339. [DOI] [PubMed] [Google Scholar]
- Wehner, M., H. Rueffert, F. Koenig, J. Neuhaus, and D. Olthoff. 2002. Increased sensitivity to 4-chloro-m-cresol and caffeine in primary myotubes from malignant hyperthermia susceptible individuals carrying the ryanodine receptor 1 Thr2206Met (C6617T) mutation. Clin. Genet. 62:135–146. [DOI] [PubMed] [Google Scholar]
- Weiss, R., K. M. S. O'Connell, B. E. Flucher, P. D. Allen, M. Grabner, and R. T. Dirksen. 2004. Functional analysis of the R1086H malignant hyperthermia mutation in the DHPR reveals an unexpected influence of the III–IV loop on skeletal muscle EC coupling. Am. J. Physiol. 287:C1094–C1102. [DOI] [PubMed] [Google Scholar]
- Yang, T., T. A. Ta, I. N. Pessah, and P. D. Allen. 2003. Functional defects in six ryanodine receptor isoform-1 (RyR1) mutations associated with malignant hyperthermia and their impact on skeletal excitation-contraction coupling. J. Biol. Chem. 278:25722–25730. [DOI] [PubMed] [Google Scholar]