

Abstract
Using methods of time-resolved two-dimensional infrared (2D-IR) spectroscopy, we approach the problem of the structural characterization of small polypeptide systems in a membrane environment. The 2D-IR spectra recorded for a model dipeptide in different environments demonstrated a significant change in the homogeneous and inhomogeneous broadenings of the amide I resonances when the molecule inserts either into a surfactant or a phospholipid membrane. Besides the change in the diagonal features in the 2D-IR response, we observe both intramolecular and intermolecular crosspeaks between the carbonyls of the dipeptide and the phospholipid. Considering the character of the diagonal peaks and the presence of the crosspeaks, we discuss the localization of the dipeptide moieties in the membrane. Using both the anisotropy and relative intensity of the observed intramolecular crosspeaks between the two amide I modes, we provide observables that help to determine the φ/ψ-dihedral angles for the backbone of the dipeptide. Time dependent studies revealed slower conformational fluctuations of the dipeptide backbone in a membrane as compared to that in an aqueous environment.
INTRODUCTION
Our view of protein structural properties relies on a quantitative determination of the static architecture, probed by nuclear magnetic resonance (NMR) and/or x-ray scattering, and on complimentary reconstructions of structural dynamics, achieved by molecular dynamics (MD) simulations. This approach shows a great resolving power mostly in the subclass of relatively large water-soluble proteins. The second subclass of proteins consists of membrane integral proteins. The cellular or organelle membrane integrated proteins assist the necessary transport and secretion functions in a living cell. The third subclass comprises a variety of relatively small polypeptides: toxins and amphipathic segments that are associated with a membrane in a dynamical manner. A clear view of the nature of lipid-protein interactions is of fundamental importance for the general understanding of living cell molecular biology and of specific importance for the particular rationalization of membrane-mediated protein folding, assembly, partitioning, aggregation, and other macrostructural events. In contrary to the case of the water-soluble proteins, our knowledge on membrane associated polypeptides is rather limited. NMR techniques work best in the liquid phase, where motional line narrowing effects ensure sharp resonances. For the structural characterization of membrane associated proteins, in contrast, MD simulations (Tieleman et al., 1998; Shepherd et al., 2001; Grossfield and Woolf, 2002; MacCallum et al., 2003) acquire an exclusive place among other techniques such as fluorescent labeling (Weiss, 1999) and newly developed solid-state NMR techniques (Hu et al., 1993; Patrick et al., 2002; de Planque et al., 2003; Opella et al., 2001). It is to our concern, however, that MD results must be supported by independent experiments.
Recently, two-dimensional infrared (2D-IR) spectroscopy has been established (Asbury et al., 2003; Asplund et al., 2000; Golonzka et al., 2001; Hamm et al., 1998; Krummel et al., 2003; Yeremenko et al., 2003), which has the potential to access experimental information on selected structural aspects of small polypeptides (Cheatuma and Tokmakoff, 2004; Hamm et al., 1999; Zanni et al., 2001; Woutersen and Hamm, 2001, 2002; Woutersen et al., 2002). Moreover, 2D-IR spectroscopy is inherently fast, and every time level (from femtoseconds to milliseconds) in the hierarchy of structural dynamics (Frauenfelder et al., 1991) may be accessed (Woutersen et al., 2001; Bredenbeck et al., 2003). For an explicit explanation of the principle of 2D-IR spectroscopy, we may consider a molecular system that contains a pair of coupled (C=O) oscillators. A narrow band excitation of one of the oscillators results in a diagonal response at the frequency of the excited oscillator and in an off-diagonal response (crosspeak) at the frequency of the coupled second oscillator. A quantitative analysis of the intensity of the crosspeak reveals information on both the dipole moments' orientations and the coupling between the two oscillators. When scanning the frequency of the narrow band excitation across the spectral resonances, a series of spectra is collected and assembled to reveal a 2D-IR spectrum. It has been shown that this information is sufficient to determine the structure of the peptide backbone (Woutersen and Hamm, 2001). Besides the sensitivity to molecular structural and dynamical properties, 2D-IR spectroscopy gives a unique opportunity to probe and characterize the relatively narrow line-shape function of infrared active moieties in heterogeneous environments, such as a membrane. In this article, we report on our first steps in applying 2D-IR spectroscopy to structurally and dynamically characterize a small dipeptide in a membrane environment.
Biological membranes are very complex systems. Thus, for simplicity, we use membrane models composed of one molecular constituent. Specifically, we used micelles of sodium dodecyl sulfate (SDS) and bilayer liposomes of phospholipid (i.e., 1-palmitoyl-2-linoleyl phosphatidylcholine, PLPC). Fig. 1 shows the chemical structures of the SDS anionic surfactant and the zwitter-ionic phospholipid molecules. The headgroup of the surfactant molecule is a sulfate anion. The headgroup of the phospholipid is a positively charged choline that is linked to the glycerol backbone through a negatively charged phosphate moiety. In result, the interface of the phospholipid liposomes is structurally more complex and thicker. The SDS surface, on the other hand, is stronger curved. Fig. 1 also represents schematically the structures of a SDS micelle and of a bilayer phospholipid liposome.
FIGURE 1.
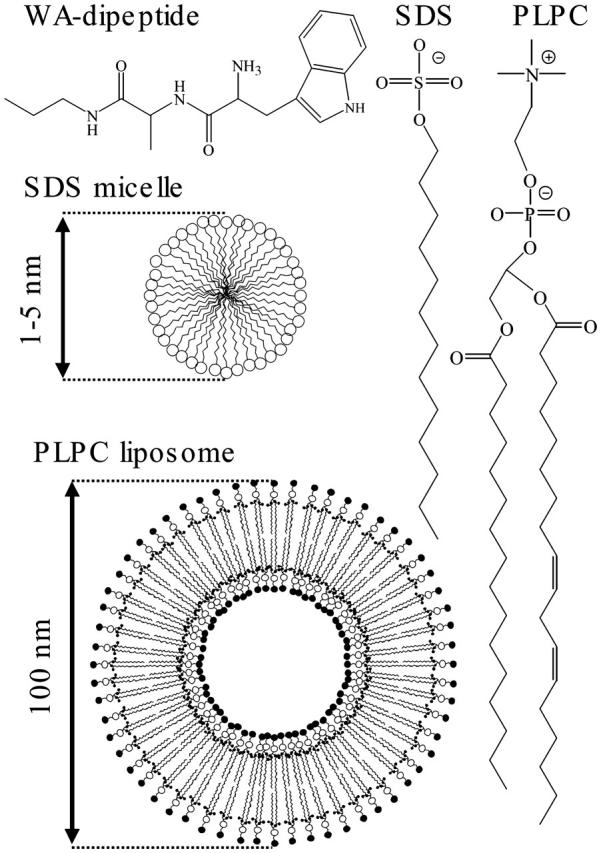
The structures of the WA-didpeptide, SDS and its micelle, and PLPC phospholipids and its liposome.
As a model peptide, we used Trp-Ala*-NH-CH2-CH2-CH3 (WA-dipeptide), the primary structure of which was chosen for the following reasons: An statistical search through the SWISS-PROT release (Jones et al., 1994) showed that tryptophan residua have a high preference to localize in terminal regions of transmembrane protein spans. Thus, both the localization and orientation of indole molecular derivates, (Grossfield and Woolf, 2002; MacCallum et al., 2003; Persson et al., 1998; Yau et al., 1998) as well as of tryptophan residua of large polypeptides on membrane surfaces, (Hu et al., 1993; Patrick et al., 2002; de Planque et al., 2003) are of great research interest. Considering this, we selected tryptophan as a first (N-terminal) residue. This gave us an additional diagnostics, the fluorescence of the indole moiety, which is sensitive to the local environment. The relatively small and hydrophobic character of the methyl moiety suggested alanine for the second residue in the dipeptide. We used 13C labeling for the carbonyl of the alanine to unambiguously assign the amide I modes in the molecule. To assure the directionality of the insertion into the membrane and to avoid unnecessary polar interactions, the dipeptide was synthesized with a -CH2-CH2-CH3 tail (hydrophobic anchor) instead of a carboxyl terminal. The alkyl tail was linked through an additional NH group to establish the second peptide unit in the molecule.
This article is organized as follows: To set the stage, we first interrogated the fluorescence properties of the indole moiety, which gave us an estimate of the binding efficiency of the dipeptide to the membrane and the approximate localization across the membrane interface. This was necessary to assess the sensitivity of 2D-IR spectroscopy to the change of the local environment of each residue upon insertion into the membrane. Second, analyzing intramolecular 2D crosspeaks between the peptide and the phospholipid, we developed a tentative interpretation of the localization of the dipeptide in the membrane. Third, determining the strength and anisotropy of intramolecular crosspeaks between both amide I modes, we provide observables that help to determine the φ/ψ-dihedral angles of the peptide backbone. Finally, we probed the dipeptide backbone conformational fluctuations in different environments by analyzing the 2D-IR line shapes and measuring the cross-relaxation rate between both amide modes.
EXPERIMENTAL METHODS
The Trp-Ala*-NH-CH2-CH2-CH3 dipeptide has been synthesized by Biosyntan GmbH (Berlin, Germany) using standard solid-state peptide synthesis. The purity was better then 95%. We removed the trifluoroacetic acid (TFA, left after reverse phase high-performance liquid chromatography) by lyophilizing the acquired substance under low pH (pD) condition three times. We double-checked by 1H-NMR spectroscopy and energy dispersive x-ray spectroscopy (EDS) that the residual trifluoroacetic acid is negligible.
Sodium dodecyl sulfate was from Sigma-Aldrich (St. Louis, MO). We prepared 0.5 M SDS normal micelles in D2O under pD 1 and under pD 7. The adjustment of pD was achieved with 35 wt % DCl from Aldrich (Milwaukee, WI). The dipeptide was added after preparation of SDS micelles.
L-α-phosphatidylcholine (soy: 95%) extract was obtained from Avanti Polar Lipids (Alabaster, AL). 1-Palmitoyl-2-linoleyl phosphatidylcholine is the main molecular component (97.2%) of the extract. The preparation of a suspension of 100 nm phospholipid liposomes was done with a miniextruder from Avanti Polar Lipids using 100 nm pore polycarbonate membranes and following the methodology according to the description and references provided at http://www.avantilipids.com/Extruder.html. Extrusion was repeated at least 11 times until the suspension was clear. According to the preparation protocol from Avanti, the sample contained liposomes with a narrow size distribution of ∼80 nm. We prepared phospholipid liposomes in D2O under both pD 1 and pD 7. The dipeptide was added after extrusion. The addition of dipeptide did not cause any change in optical properties of the sample.
A sample volume of ∼2 μl was needed for the infrared experiments. The dipeptide concentration was ∼0.05 M in an attempt to satisfy several crucial conditions: the preservation of a reasonable ratio between the dipeptide and surfactant (phosphatidylcholine), establishment of a high enough optical absorption for the IR experiment, and maintaining an excellent optical quality of the sample. The preparation of the SDS surfactant system required several trials as it proved to be quite unstable (particularly under low pD condition). This is due to the relatively high surfactant concentration and electrostatic interaction with the dipeptide molecules. In contrast, the phospholipid liposome samples proved to be very stable.
Emission studies were accomplished with a Perkin-Elmer (Foster City, CA) luminescence spectrometer LS 50B. Most of emission measurements were done in suspensions where the concentration of surfactant (phosphatidylcholine in liposomes) was the same as in the infrared experiment, whereas the concentration of the dipeptide was ∼100 times lower. To insure the validity of the emission studies, we measured and confirmed the same emission properties in a 2-μl droplet of a high concentration sample (as used in the IR experiment). We recorded the steady-state IR spectra using a BioRad (Hercules, CA) 175C Fourier transform (FT) spectrometer. The time resolved 2D-IR and hole burning experiments were accomplished using an ultrafast IR spectrometer, the details of which are provided elsewhere (Hamm et al., 1998; Woutersen et al., 2001).
It is important to note that pD dependent FTIR spectra of the WA-dipeptide indicate that weak pairing, stacking, or even micellization of the molecule itself might occur in an aqueous suspension under neutral pD (see Appendix). Thus, in our comparative structural study, we discuss the results of the 2D-IR studies on WA-dipeptide in D2O under low pD conditions only. When in membrane under pD 7, indolyl stacking is unlikely to occur since it would require antiparallel orientation of the moieties (Subramannian and Sahayamary, 1989). This is because the membrane supports a parallel orientation with the indol moiety at the interface.
STRUCTURAL CHARACTERIZATION USING THE TRYPTOPHAN FLUORESCENCE
Absorption and fluorescence spectroscopies of the tryptophan residue, when in different environments, allow for a conventional and independent (to 2D-IR) experimental approach to determine the dipeptide binding efficiency to the membrane and obtain an approximate view on its localization in the interface. Fig. 2 shows excitation and emission spectra of the tryptophan residue when the dipeptide is in a D2O environment (Fig. 2 A), in SDS micelles (Fig. 2 B), and in phospholipid liposomes (Fig. 2 C) at pD 1 (thick solid line) and pD 7 (dashed lines), respectively. The maximum of the indole emission in the D2O sample is at 353 nm and 348.5 nm under pD 7 and pD 1, respectively. In the SDS micelles, the indole emission shows a hypsochromic shift of 14 nm at pD 7 and 12 nm at pD 1. In the case of the phospholipid liposome samples, the shift is 8 nm at pD 7 and 6 nm at pD 1, respectively. This indicates that the aromatic side group of the dipeptide is buried in the interface of the SDS micelles and the phospholipid liposomes (Lakowicz, 1983).
FIGURE 2.

The excitation and emission spectra of the indole moiety in the dipeptide in D2O (A), in SDS micelles (B), and in phospholipid liposomes (C). The solid and the dashed lines for each case show the responses at pD 1 and pD 7, respectively. The numbers indicate the emission maxima. In liposomes (C), the line given in triangles represents the emission after quenching with iodide and before subsequent quenching with acrylamide (see text). The connected circles (○ and • for the measurements under pH 7 and pH 1, respectively) in A–C show the wavelength of the emission maxima in dependence of the excitation wavelength according to the right vertical axis, and the arrows mark the excitation wavelength. The emission quenching in D2O, in SDS micelles, and in phospholipid liposomes is shown in D–F, respectively. Here, the results of quenching with acrylamide are depicted as ○ and with iodide as ▪. For the quenching experiments, the aqueous and SDS samples were prepared at pD 1 and the phospholipid sample at pD 7.
Red edge excitation shift effect
In SDS micelles (under both pD), we observed a bathochromic shift (up to 12 nm) of the emission maxima upon the systematic increment of the excitation wavelength (connected circles, Fig. 2 B). This is the so-called red edge excitation shift effect (REES) (Galley and Purkey, 1970; Chattopadhyay and Mukherjee, 1993), which is observed whenever the fluorescence lifetime of a chromophore is comparable or shorter then the dipolar relaxation of a solvent. Under such conditions, a narrow band excitation selects a subpopulation of chromophores, whose frequency is determined by the degree of solvation. Thus, a strong REES effect of the chromophore in SDS micelle suspension arises due to a wide distribution of sites across a membrane interface between the hydrophobic core and aqueous surrounding. Consistently, the tryptophan emission is independent on the excitation wavelength, when the dipeptide is in D2O solution (Fig. 2 A). This is due to the fast solvation dynamics in the bulk solution.
Interestingly, we did not observe any REES effect when the dipeptide is in phospholipid liposomes under neutral pD, although the sample under low pD shows the expected dependence on the excitation wavelength (Fig. 2 C). The lack of any REES effect may indicate a sharp distribution of indole location sites in the phospholipid liposome bilayer, in contrast to SDS micelles or in phospholipid liposomes under low pD.
Fluorescence quenching
Selected results of quenching studies are shown in Fig. 2, D–F. We used the Lehrer law to estimate quenching constants and accessibility factors (i.e., the fraction of emission centers that are accessible to the quencher; Lehrer, 1971; Eftink, 1991). The evaluation of the tryptophan fluorescence quenching by iodide and acrylamide yielded quenching constants of 40 M−1 and 8 M−1, respectively, when the dipeptide is in D2O (Fig. 2 D). The accessibility factor was close to unity in both cases. These values are comparable or somewhat larger than that reported for well-exposed tryptophan (Eftink and Ghiron, 1976).
We were not able to perform iodide quenching in SDS micelles due to a rapid phase separation. We used acrylamide instead, for which we found a decay of the tryptophan fluorescence with the quenching constant of 17 M−1 (Fig. 2 E) and the accessibility factor close to one. Since acrylamide is known to not go deep into the hydrophobic core of the membrane (Raja et al., 1999), one may conclude that the indole moiety of the dipeptide localizes on the surface of the SDS micelles.
The quenching studies in phospholipid liposomes brought a clear picture of the partitioning of the indole moiety in the membrane. Under neutral pD, the addition of iodide quenches the emission down to 90% of the original count, but not further, regardless of how much more iodide we added. This first phase of quenching (with iodide) occurs with the constant comparable to that in D2O solution and an accessibility factor of 0.14. The resulting emission band showed a higher energy maximum compared to that of the unquenched sample (the line given in triangles in Fig. 2 C). To investigate the distribution of unquenched aromatic groups located deeper in the phospholipid liposome membrane, we continued (right after the iodide series in the same sample) with quenching using acrylamide. We could reduce the emission down to 20% of the original with a constant of 9 M−1 and an access factor of 0.93 (Fig. 2 F). The residual emission (which we were not able to quench with acrylamide) indicated that there is a subpopulation of dipeptide molecules localized deeply in the hydrophobic compartment of the phospholipid membrane.
The interpretation of the REES and quenching results in the phospholipid liposome sample is the following: At neutral pD, 90% of the aromatic groups protrude through the surface of the phospholipid liposome at least till the indole is on the level of the negatively charged phosphate linker. This phospholipid liposome bound subpopulation of indoles splits further, and 80% (from 90%) localize not lower then the phospholipid carbonyl, and the remaining 20% are buried deep in the hydrophobic core.
The situation in phospholipid liposomes at low pD is different. In this case, we were able to quench the full load of dipeptide with iodide with a very low constant. This suggests that either the ionized form of the dipeptide does not penetrate deep under the surface of the phospholipid liposome bilayer or iodide may protrude further under the phospholipid liposome interface.
The results of the quenching experiments are consistent with the results of the REES effect studies. At pD 7, both the outer population (of 10%) and the deeper buried one do not contribute into the REES effect due to the fast solvation and to the narrow site distribution, respectively. At pD 1, the REES shows a broad site distribution across the interface, since phospholipid liposomes under low pD (due to structural properties) cannot sustain a stable and specifically located population of the dipeptide.
In summary, fluorescence studies may provide a very approximate picture of the localization and partitioning of the indole moiety in the membrane. The emission and quenching properties from the side group of the tryptophan residue indicate a very effective binding of the WA-dipeptide to a membrane surface. In the case of a phospholipid membrane, ∼90% of the aromatic side groups localize at the level (or below) of the phosphate group in the lipid, and the remaining 10% of the indole moieties are in an aqueous environment above the membrane surface. Conventional optical spectroscopy cannot characterize any further the structural properties of the dipeptides across the interface of the liquid membrane. Neither can it probe the specific localization of the structural components of the dipeptide beside the indole group nor their relative orientation in respect to each other and to the structural components of membrane interface. Therefore, we utilize methods of 2D-IR spectroscopy on the dipeptide in different environments in a comparative manner in the next section.
STRUCTURAL CHARACTERIZATION USING 2D-IR SPECTROSCOPY
Fig. 3 shows overview 2D-IR spectra of the dipeptide in D2O (pD 1), SDS micelles (pD 7), and phospholipid liposomes (pD 7), respectively. The upper parts in the figure represent the corresponding steady-state absorption spectra. The multidimensional responses were recorded with a 1-ps delay time of pump and probe pulse. Both linear and 2D spectra clearly show the resonances of the carbonyls of alanine at 1600 cm−1 and tryptophan at 1675 cm−1. The additional high frequency band at 1732 cm−1 in the liposome sample is due to the carbonyl group of the phospholipid.
FIGURE 3.

The absorption and overview 2D-IR spectra (with parallel and perpendicular polarization of pump and probe pulse) of the dipeptide in D2O, SDS liposomes, and a phospholipid membrane.
Fig. 4 magnifies the weak crosspeaks for the phospholipid liposome sample. Fig. 4 shows horizontal cuts through the 2D-IR spectra at selected pump frequencies resonant with the phospholipid carbonyl group at 1740 cm−1 (Fig. 4 A), the tryptophan at 1687 cm−1 (Fig. 4 B), and the alanine at 1610 cm−1 (Fig. 4 C). Fig. 4 D shows the weighted differences between the 2D-IR spectra recorded with parallel and perpendicular polarizations of pump and probe pulse. This procedure enhances crosspeaks that cannot be seen on the scale of Fig. 3, relative to the otherwise dominating diagonal peaks (Woutersen and Hamm, 2000). A complete set of crosspeaks is obtained: intermolecular crosspeaks between the amide I bands of the dipeptide and the carbonyl group of the phospholipid, as well as intramolecular crosspeaks between both amide I bands of the dipeptide. The intermolecular crosspeaks serve as markers that help to analyze the localization of the dipeptide in the membrane, whereas the intramolecular crosspeaks allow us to estimate the backbone structure of the dipeptide itself.
FIGURE 4.
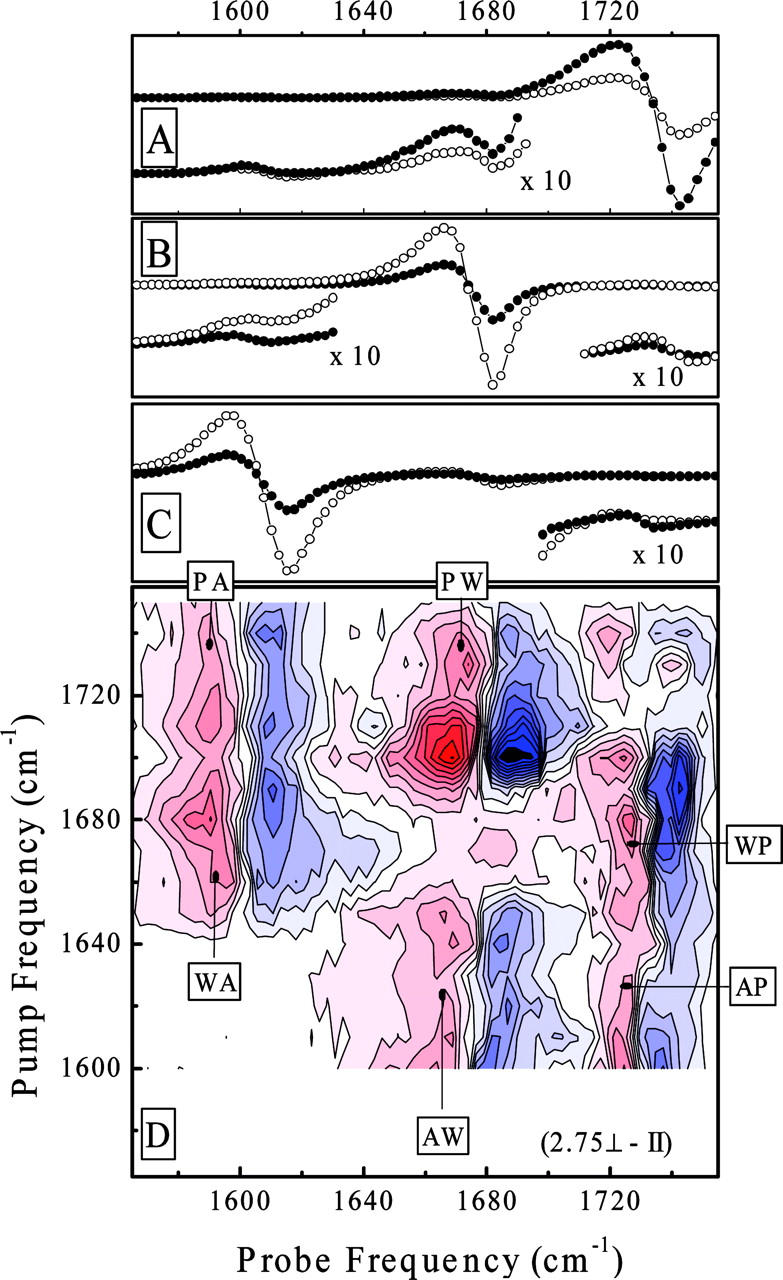
Vertical cut through the 2D-IR spectra of the didpeptide in the phospholipid membranes at pump-pulse frequencies of 1740 cm−1 (A), 1687 cm−1 (B), and 1610 cm−1 (C) for parallel (•) and perpendicular polarizations (○) of pump and probe pulses. The corner inserts show a 10× magnified spectral response in the crosspeak spectral range. D represents the weighted difference between perpendicular and parallel polarization 2D-IR spectra. The markers indicate the crosspeaks between alanine (A), tryptophan (W), and phospholipids (P).
Localization in the membrane interface
The intermolecular crosspeaks between the carbonyls of the dipeptide and the phospholipid (Fig. 4) suggest that the corresponding groups are in close proximity. Since the forces giving rise to intermolecular crosspeaks are relatively short ranged, their presence enables us to narrow the localization of the molecule in the membrane with relatively high precision. For example, within the transition dipole model (Krimm and Yasuaki, 1972), the crosspeak intensity decays quickly with the distance between the molecular groups (1/r3) and with the detuning of the resonances 1/Δω2 (Hamm et al., 1999). In addition, the coupling strength depends strongly on the relative orientation given by the angle dependence of the dipole-dipole coupling term. Given the large energy splitting of the alanine-phospholipid crosspeak, we conclude that these moieties must be in very close proximity. The separation between the carbonyls of alanine and the phospholipid can be estimated to be <≈3 Å. Comparatively, the intensity of the crosspeaks between the near-in-frequencies resonances for tryptophan and phospholipid, which are about equally strong, indicates a somewhat larger distance and/or a less favorable orientation. This suggests that the carbonyl group of the dipeptide first residue is above that of the phospholipid. Fig. 5 shows a qualitative sketch of the possible placement of the dipeptide in the phospholipid liposome membrane, as obtained from the 2D-IR spectrum. The interpretation is in perfect agreement with the fluorescence quenching results from the previous paragraph.
FIGURE 5.

Schematic representation of the localization of the WA-dipeptide in the phospholipid membrane.
Backbone structure
We have recently shown that we can determine the structural distribution of small peptides in aqueous solution in great detail by combining 2D-IR spectroscopy, 1D-NMR spectroscopy, and MD simulations (Woutersen et al., 2002). We suggested for trialanine (Ala-Ala-Ala) in D2O that the central peptide bond is predominantly in the so-called PII structure, and only a small fraction (20%) exists in different conformations. Alternatively, the structural distribution can be probed using a combined method of polarized Raman and circular dichroism studies (Schweitzer-Stenner et al., 2001; Eker et al., 2003a,b), pointing to a larger contribution from more extended β-sheetlike structures. In this article, we report on our first attempt to use methods of 2D-IR spectroscopy to investigate the structural and dynamic properties of a peptide upon partitioning into the interface of a liquid membrane.
Fig. 6 shows pump-probe transients recorded with the center frequency of the pump pulse resonant with the amide I band of the tryptophan residue at 1675 cm−1 for perpendicular and parallel polarizations of pump and probe pulses. These spectra represent horizontal cuts through 2D-IR spectra and highlight the essential information. For a better comparison, the spectra are scaled in the crosspeak region by a factor 10. The anisotropy of the crosspeaks is close to zero in all samples, and the intensity of the crosspeaks decreases when the dipeptide is in a membrane. As the frequencies of the two modes in WA-dipeptide are well separated, the crosspeak anisotropy r = 1/5(3cos2θ − 1) that may be used to determine the angle θ between the transition dipole moments of the carbonyl groups can readily be read off (Woutersen and Hamm, 2001). Furthermore, the absolute value of the coupling constants (|β|) can be estimated from the relative intensities of the diagonal and off-diagonal peaks (Hamm et al., 1999). In all cases, the higher frequency amide I band carries the stronger oscillator strength in the absorption spectra (Fig. 3, top). Assuming that the intrinsic intensities of the site amide I vibrators is the same, the exciton model suggests that the product β × cosθ must be positive in this case (see Appendix in Bredenbeck and Hamm, 2003). This reduces the ambiguity in the determination of the angle θ and the sign of the coupling constant β to two possibilities, (|β|, θ) or (−|β|, 180° − θ) with 0° < θ < 90°. The results are summarized in Table 1. Interestingly, the coupling strength β changes significantly when transferring the WA-peptide from water into micelles or membranes, whereas the angle θ between the transition dipoles are practically identical. Torii and Tasumi (1998) have shown that the dependence of the coupling strength β on the dihedral angles (φ, ψ) is very steep in the regions of interest, which explains why rather small structural changes my cause relatively large changes in β.
FIGURE 6.
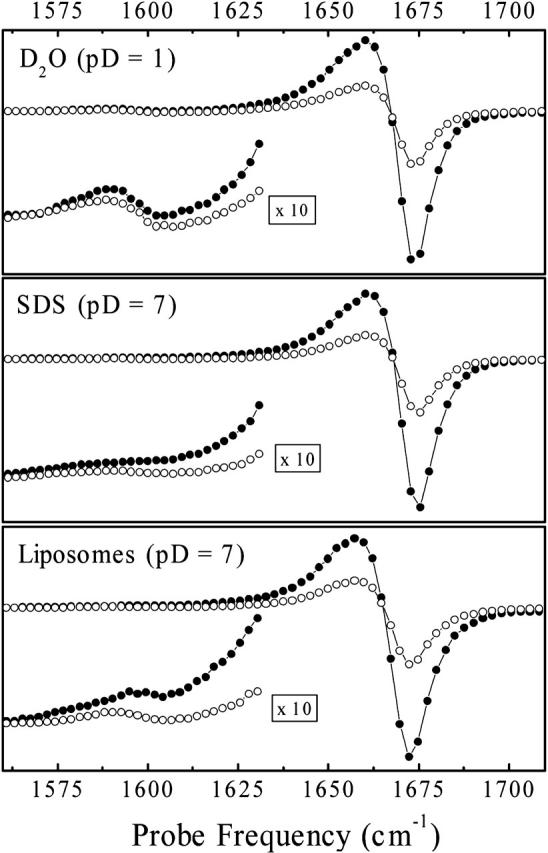
Spectral transients recorded with a pump-pulse frequency of 1675 cm−1 for parallel (•) and perpendicular polarizations (○) of pump and probe pulses. The left lower corner inserts show the 10× magnified spectral response in the crosspeak spectral range.
TABLE 1.
Structural parameters derived from 2D-IR spectroscopy (Fig. 6) and 1H-NMR spectroscopy
| Sample | Crosspeak anisotropy | θ (°) | Coupling strength (cm−1) | 3JHNCαH (Hz) |
|---|---|---|---|---|
| D2O (pD 1) | 0.06 ± 0.05 | 49° (or 131°) | 8 ± 2 (or −8) | 6.35 |
| Micelles (pD 7) | 0.03 ± 0.05 | 52° (or 128°) | 5.5 ± 1.5 (or −5.5) | 6.8 |
| Liposomes (pD 7) | 0.04 ± 0.05 | 51° (or 129°) | 6.5 ± 2 (or −6.5) | – |
If one assumes that the molecules exists in one predominant structure, one may match these observables with the coupling map constructed by Torii and Tasumi (1998) and propose possible dihedral (φ, ψ) angles of the central peptide bond (an alternative coupling map has recently been published by Cho and co-workers (Choi et al., 2003)). Two regions in the Ramachandran plot are consistent with the 2D-IR results: In the area of a strongly folded β-segment, we obtain (φ, ψ) ≈ (−55°, 64°) for the aqueous system and (φ, ψ) ≈ (−70°, 40°) in the membrane. A second solution is principally possible with (φ, ψ) ≈ (−120°, −80°) for the aqueous system (pD 1) and (φ, ψ) ≈ (−140°, −50°) in both types of membranes. However, it is localized in a sterically rather unlikely region of the Ramachandran plot.
These results can be cross-checked and/or refined with the help of the 3JHNCαH coupling obtained from 1H-NMR spectroscopy. 3J coupling between the amide proton and the Cα-proton is a commonly used simple measure of the φ-dihedral angle of the peptide bond (Karplus, 1959). The measurement is straight forward in D2O and in SDS micelles but cannot be done in the phospholipid membranes due to slower orientational relaxation of the liposomes. We obtained in the aqueous environment 3J = 6.35 Hz at pD 1 and in SDS micelles 3J = 6.8 Hz, revealing two possibilities: φ ≈ −77° in D2O and φ ≈ −80° in SDS micelles and φ ≈ −163° in D2O and φ ≈ −160° in SDS micelles, respectively. Hence, the NMR results seem to contradict the 2D-IR results with the mismatch being specifically large for the aqueous sample. We have applied the same method to trialanine and found converging structural parameters from both 2D-IR and 1H-NMR spectroscopy (Woutersen et al., 2002). The failure in this case may be due to two reasons. First, it may arise from the fact that the peptide ensemble exists in several conformational substates. Both 2D-IR and NMR techniques would average over these substates in a completely different, highly nonlinear manner and thus yield seemingly contradicting results. A distribution of structures is not unexpected; in fact, it was a surprising result of our trialanine work that this molecule seems to exists predominantly (80%) in one conformation. Second, an additional systematic error due to the difference in the oscillator strengths of both amide I vibrators may obscure the numeric evaluation (the mixing due to exciton coupling is not sufficient to explain the difference in intensities of the two amide I vibrators observed in the FTIR spectra in Fig. 3). The different intensities of the two transitions are normalized out in the determination of both the coupling strength β and the angle θ between the transition dipoles. However, the coupling map of Torii and Tasumi (1998) intrinsically assumes identical intensities of both transitions, which is why our determination of the (φ, ψ) dihedral angles is not totally self-consistent. A more specific structural refinement would require MD simulations of the dipeptide in the particular environment, averaging the trajectory in the way specific to either 2D-IR or NMR spectroscopy, and refining the MD results until agreement with the measured values for the anisotropy r and the coupling strength β (see Table 1) is obtained. This procedure has recently been demonstrated for trialanine (Mu and Stock, 2002).
In any case, the anisotropy and coupling strength obtained from the 2D-IR experiment as well as the 3J coupling obtained from the 1H-NMR experiment of the WA-peptide in aqueous solution are different from that of AAA (Woutersen et al., 2002; Schweitzer-Stenner et al., 2001) and other tripeptides with a central alanine residue (e.g., SAA and KAA; Eker et al., 2003a). Hence, independent of the uncertainty in determining the (φ, ψ) angles, the structures of both molecules must be different as well. One might suspect that the bulky W-residue, as well as the missing charge at the carboxyl terminal, affects the conformational propensity of the central alanine.
DYNAMICAL CHARACTERIZATION USING 2D-IR SPECTROSCOPY
2D-IR spectroscopy not only allows extracting structural properties but also reveals important information about the dynamics of the systems under study. In this experiment, we may utilize two observables: i), cross-relaxation between both amide I resonances of the dipeptide and ii), the 2D line shape of the diagonal peaks. The first reports on fluctuations of the dipeptide backbone and the second on homogeneous and inhomogeneous broadening of the amide I resonances in their local membrane environment.
Backbone fluctuations
Measuring cuts through 2D-IR spectra at different delay times between pump and probe pulses, we examined the cross-relaxation between the amide I modes of the dipeptide molecule in the various samples (Figs. 7 and 8). Cross-relaxation has been investigated in detail for trialanine in D2O and has attributed to fluctuations of the peptide backbone (Woutersen et al., 2001). More specifically, the cross-relaxation rate measures the spectral density of the autocorrelation function of the coupling constant β(t) at a frequency given by the splitting of both resonances. Since the coupling constant β is a direct measure of the backbone structure, the cross-relaxation can be directly related to backbone fluctuations. Since furthermore the energy splitting between both amide I modes is relatively large (75 cm−1), the method is particularly sensitive to the inertial backbone fluctuations, which occur on an ultrafast 100-fs timescale.
FIGURE 7.
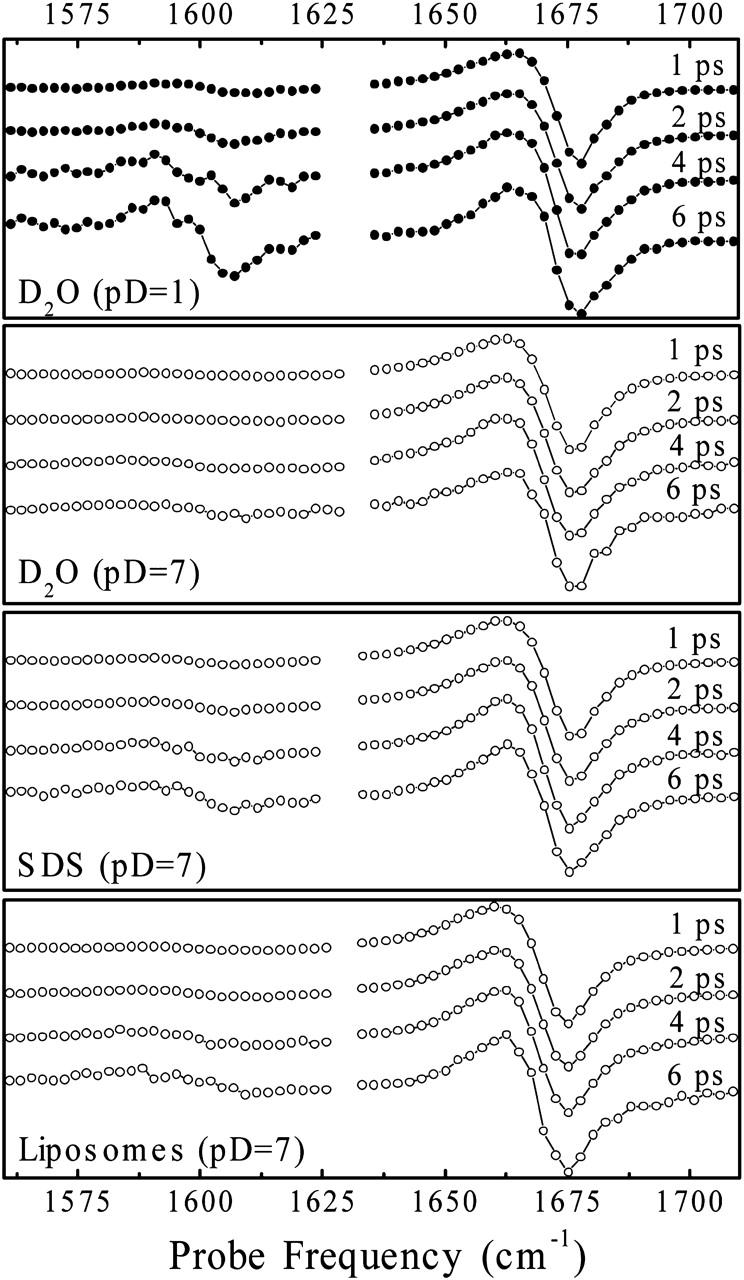
Transient spectra detected as a function of mixing time (1, 2, 4, and 6 ps, from top to bottom) in the indicated samples for a pump pulse frequency of 1675 cm−1. The transients are rescaled to correct for vibrational relaxation out of the initially excited amide I mode.
FIGURE 8.
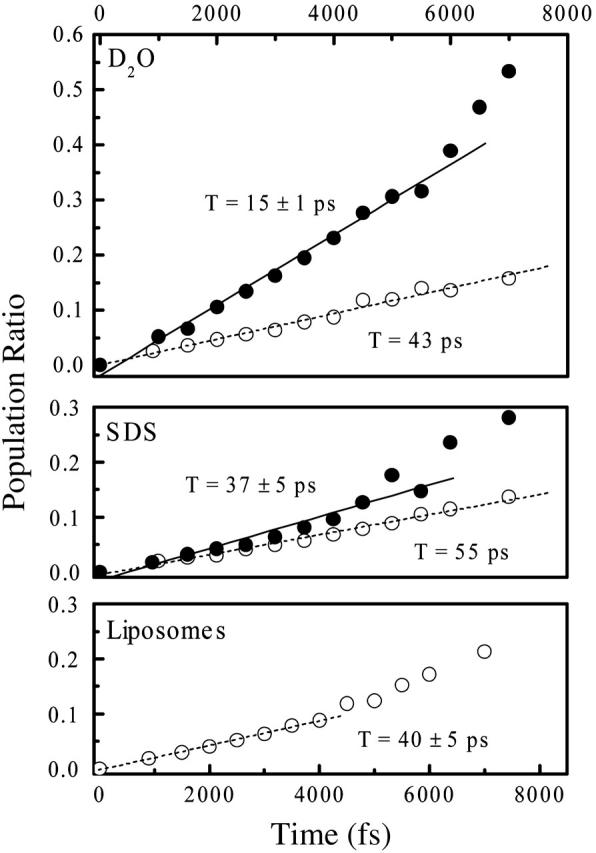
Population ratios as a function of mixing time, as deduced from Fig. 7.
Transient spectra as a function of the mixing time, as well as population ratios deduced from these spectra, are compared in Fig. 7 and Fig. 8, respectively. The cross-relaxation rate in the dipeptide in an aqueous environment at low pD (0.07 ps−1) is comparable to that reported for trialanine (Woutersen et al., 2001). In the Appendix, we show that the dipeptide molecules are free and nonaggregated only under low pD conditions, which is why we use this value as a reference. We find a threefold decrease when the didpeptide is inserted into either the SDS micelles of the phospholipid membrane. Hence, in particular, the fast inertial motion of the peptide backbone is significantly reduced in the membrane.
MD-simulations suggest that the interior of a membrane is significantly more rigid compared to the structural dynamics of bulk water (de Groot and Grubmüller, 2001; see also a computer animation at http://www.mpibpc.mpg.de/abteilungen/071/bgroot/gallery.html). Water motion is known to be super fast with components in the sub-100-fs regime (Fecko et al., 2003). Hence, in a water environment, it is the continuous collisions of surrounding water molecules with the peptide that render the peptide backbone highly flexible as well. In fact, MD simulations of trialanine dissolved in water could almost quantitatively explain the observed cross-relaxation rate assuming the mechanism sketched above (Woutersen et al., 2001). When brought into the more rigid membrane environment, however, the peptide backbone motion will also be considerably slowed down. In particular, the fast inertial motion is mostly frozen in the highly viscous interior of the membrane, explaining the large reduction in the cross-relaxation rate.
We show in the Appendix that the dipeptide itself might aggregate, stack, or even micellize in water under neutral pD conditions due to the indole moiety. In agreement with this interpretation, the cross-relaxation rate is also considerably reduced in water at neutral pD to a value similar to that in membranes.
Flexibility of the environment
The tilt of a 2D diagonal peak is a direct measure of the ratio between homogeneous and (quasi-)inhomogeneous broadening (Kwac and Cho, 2003), where the term “(quasi-) inhomogeneous broadening” refers to the distribution of structures on the timescale of the measurement. (Quasi-)inhomogeneous broadening is not necessarily static. Both homogeneous and inhomogeneous broadening originate from the interaction of a vibrational transition with its immediate surrounding.
In D2O, the 2D-IR diagonal peaks of the carbonyl resonances of alanine and tryptophan are similar in shape. Specifically, both bands contain some inhomogeneous contribution on the 1-ps timescale (i.e., the pump-probe delay time) but are dominated by homogeneous broadenings with a width of ∼15–20 cm−1 (estimated from the hole width). When the peptide is brought into SDS micelles, or into phospholipid liposomes, the 2D-IR peak of alanine changes dramatically in shape. It is strongly skewed along the diagonal due to a large inhomogeneous distribution, whereas the homogenous width is very small (its precise measurement is limited by the spectral width of the pump pulses). The response of tryptophan, on the other hand, is similar to that in water. It is interesting to note that the linear absorption spectra do not change very much in the different samples. Only nonlinear spectroscopy may separate the broadening contributions that are hidden in the linear responses.
At first sight, these results seem to be counterintuitive. Seemingly, the tryptophan carbonyl group, which is located closer to the outside of the membrane, experiences less inhomogeneous broadening than the alanine carbonyl group, which is in the interior of the more rigid core of the membrane. Since the core is hydrophobic, one might expect a narrower distribution of vibrational frequencies for the alanine carbonyl group, whereas the tryptophan carbonyl group might still experience outside water molecules through hydrogen bonding. Hydrogen bonding typically is a source of strong inhomogeneous broadening. In fact, neutron scattering experiments (Jacobs and White, 1989) and MD simulations (Grossfield and Woolf, 2002; MacCallum et al., 2003) demonstrate that water molecules do distribute into polar compartments of the membrane interface but not further than the level of the phospholipid carbonyl.
We shall first discuss the situation in the phospholipid liposomes, for which we have a clear picture of the localization of the peptide in the membrane (Fig. 5). The crosspeak between the alanine amide I mode and the phospholipid carbonyl group shows that the alanine is not really located in the hydrophobic core of the membrane but experiences a strong interaction with the polar carbonyl group of the phospholipid. Nevertheless, the environment might already be rigid. This would provide exactly the conditions needed to obtain a band that is strongly inhomogeneously broadened, as observed by the strong tilt of the corresponding 2D diagonal peak. The tryptophan carbonyl group, on the other hand, is located in an environment whose polarity might be similar to that of the alanine group. However, the tryptophan carbonyl group is in a region that is still affected by highly mobile water molecules that might introduce ultrafast components to the frequency fluctuations of the tryptophan amide I band. Hence, although the instantaneous distribution of frequencies 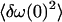 might be similar in both cases, the decay of the correlation function
might be similar in both cases, the decay of the correlation function 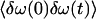 might contain very fast components only in the case of the tryptophan amide I band. Any fast component in the frequency fluctuation correlation function would convert a distribution of frequencies into homogeneous broadening. The interpretation is in agreement with the localization of the peptide in the membrane sketched in Fig. 5.
might contain very fast components only in the case of the tryptophan amide I band. Any fast component in the frequency fluctuation correlation function would convert a distribution of frequencies into homogeneous broadening. The interpretation is in agreement with the localization of the peptide in the membrane sketched in Fig. 5.
In the SDS micelle, we know much less about the localization of the dipeptide in the membrane. For the tryptophan amide I band, the same explanation as in the case of phospholipid liposomes should apply. However, since the polar headgroup of SDS is smaller than that of PLPC, and since the backbone structure of the dipeptide is stretched in roughly the same way in both cases (see Table 1), one might expect that the alanine carbonyl group is in fact located more in the hydrophobic environment of the membrane core than it is in the phospholipid membrane. Consistent with this interpretation, the inhomogeneous distribution is indeed somewhat smaller in the SDS micelle (i.e., the length of the 2D peak along the diagonal direction is somewhat less). The homogeneous width is still small, reflecting little fluctuations on the fast 100-fs timescale.
However, one should keep in mind that vibrational dephasing is not necessarily dominated by Coloumbic forces, but van der Waals interactions may contribute to an equal extent (Rossend and Hynes, 1998). In that sense, one does not even expect a dramatic difference between hydrophobic and polar environments. Hence, the difference in the 2D line shape of alanine and tryptophan amide I bands in both membranes merely reflects the rigidity or flexibility of the corresponding environments and consequently the partitioning between homogenous and inhomogeneous broadening. The sum of homogenous and inhomogeneous broadening is roughly constant in both cases, explaining the similar bandwidth of the absorption spectra of all samples.
CONCLUSION
We have explored the structural and dynamical properties of a simple dipeptide in a membrane environment using methods of 2D-IR spectroscopy. To assess securely the sensitivity of the 2D-IR response to the change of the local environment, we used the fluorescence properties of the indole moiety in a first step to confirm the effective binding of the dipeptide to the membrane and define the approximate localization across the interface. The key results of the time-resolved 2D-IR experiments results are as follows:
First, when the dipeptide inserts into phospholipid liposomes, we observed a complete set of intra- and intermolecular crosspeaks between the amide I modes of the dipeptide as well as between the dipeptide modes and the phospholipid carbonyl. In particular the intermolecular crosspeaks turn out to be extremely valuable since their presence helps us to estimate the localization of the molecule in the membrane. As a result, we suggested a schematic view of the arrangement of the WA-dipeptide in the interface of a liquid phospholipid membrane (Fig. 5).
Second, analyzing the anisotropy and the relative intensity of intramolecular crosspeaks between both amide I modes of the dipeptide, we determined possible (φ, ψ) dihedral angular pairs accessible to the dipeptide backbone. A comparison with 3JHNCαH couplings obtained from 1H-NMR spectroscopy indicated a nontrivial structural distribution of the peptide backbone both in aqueous solution and in SDS micelles. Thus, a more specific structural refinement would be possible only in connection with MD simulations, which are beyond the scope of this article.
Third, investigating the cross-relaxation rate between the two amide modes, we could confirm the intuitive expectation that conformational fluctuations of the peptide backbone are considerably reduced when the dipeptide molecule is inserted into the membrane interface. We assign this effect to the slower molecular motion in the hydrophobic compartment of the membrane and to the depletion of water in the polar interface of the membrane. Furthermore, we observe a significant change in the 2D line shape of the diagonal peaks of the alanine resonance upon binding of the dipeptide to the membrane. Also, this result can be understood by the rigidity of the interior of the membrane.
Our results demonstrate the resolving power of 2D-IR spectroscopy in probing structural and dynamical properties of molecules in heterogeneous (liquid membrane) environment. Two-dimensional infrared spectroscopy might play an exclusive role in investigating fluctuating structural properties of a variety of relatively small polypeptides, toxins, and amphipathic segments, which are associated with membranes in a dynamic manner.
Most remarkable is the observation of intermolecular crosspeaks between noncovalently bound molecules, i.e., the carbonyls of the dipeptide and the phospholipid (Fig. 4). A similar observation has been made recently by Zanni and co-workers (Mukherjee et al., 2004). The intermolecular crosspeak reveals the close proximity of certain molecular groups. Hence, the spirit of measuring vibrational intermolecular couplings with the help of 2D-IR spectroscopy is similar to that of florescence resonance energy transfer (FRET) between fluorescence labels. The latter is an extremely sensitive method that is extensively applied in biophysical studies of protein structure and dynamics as well as protein-protein interactions (Weiss, 1999). Intermolecular couplings of vibrational transitions are different in two aspects: i), they are much shorter ranged (<5 Å) and hence allow for a more precise localization, and ii), they don't require artificial labels but use molecular groups that are present anyway. Hence, 2D-IR spectroscopy might develop into a powerful, complimentary spectroscopic tool for studying molecular aggregation and recognition.
Acknowledgments
The work has been supported by the Swiss Science Foundation under contract 2100-067573.02/1.
APPENDIX
The indole moiety of tryptophan tends to pair or stack, which could lead to aggregation or even micellization in aqueous environments. Hydrogen bond mediated pairing has been reported for crystals of tryptophan containing tripeptides (Subramannian and Sahayamary, 1989), and a variety of stacking mechanisms have been described in Gervasio et al. (2002) and Thomas et al. (2002a,b). To stress this effect, we show in Fig. 9 A the pD dependence of IR absorption spectrum of the Ala-Trp-Ala tripeptide in the amide I spectral range. The spectra clearly show the insensitivity of the terminal carboxyl spectra to pD between 7 and 1.6, which normally shifts from 1590 cm−1 in the deprotonated from to 1720 cm−1 in the protonated form. As the pKa of the terminal carboxyl group is typically between 3.5 and 4, one would expect that shift to be complete between these two pD values. Fig. 9 B represents the concentration dependence of the IR response of Ala-Trp-Ala at pD 7. Upon dilution, pairing becomes less favorable, and the deprotonated carboxyl band increases as expected. The unexpected behavior is due to the fact that carboxyl terminal is bound through a hydrogen bond to the indole nitrogen in paired molecules (Subramannian and Sahayamary, 1989). We observed an analogous insensitivity to pD in Trp-Gly-Gly. A similar in character but not as pronounced behavior was monitored for Ala-Phe-Ala and Ala-Tyr-Ala. Since the nature of the aromatic group is different in these systems (there is no indole hydrogen), the “screening” of carboxyl in pairs or in aggregates of these molecules might be steric in nature (Gervasio et al., 2002; Thomas et al., 2002a,b).
FIGURE 9.
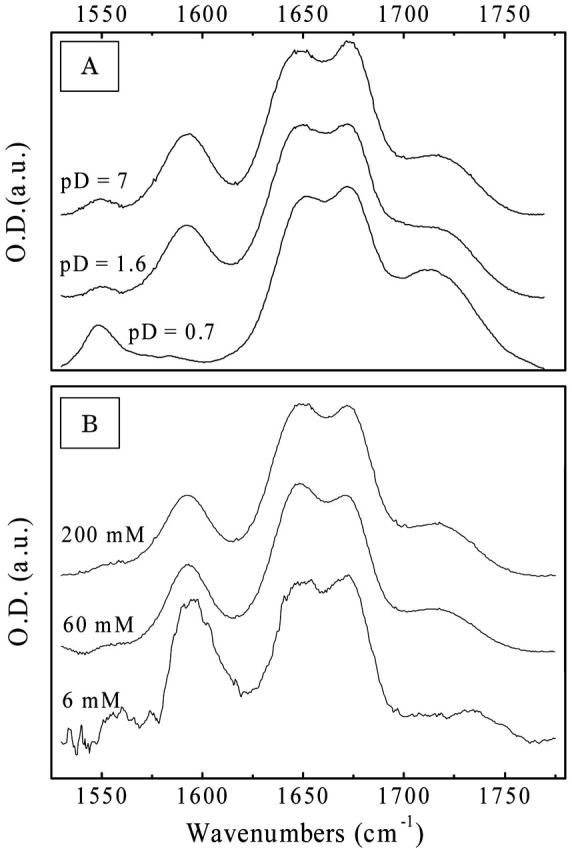
(A) pD-dependent absorption spectra of Ala-Trp-Ala at pD 7, 1.6, and 0.7 at a 100-mM concentration and (B) concentration dependent absorption spectra at 200 mM, 60 mM, and 6 mM at neutral pD.
We furthermore find that backbone fluctuations are reduced in D2O at neutral pD to a value similar to that in micelles and liposomes (Fig. 8), suggesting that even micellization of the dipeptide molecules might occur. Therefore, we limit our discussion on the structural properties of the WA-dipeptide in an aqueous environment to low pD 1 conditions, when dielectric screening eliminates aggregation.
References
- Asbury, J. B., T. Steinel, C. Stromberg, K. J. Gaffney, I. R. Piletic, A. Goun, and M. D. Fayer. 2003. Hydrogen bond dynamics probed with ultrafast infrared heterodyne detected multidimensional vibrational stimulated echoes. Phys. Rev. Lett. 91:237402–237405. [DOI] [PubMed] [Google Scholar]
- Asplund, M. C., M. T. Zanni, and R. M. Hochstrasser. 2000. Two dimensional infrared spectroscopy of peptides by phase controlled femtosecond vibrational photon echoes. Proc. Natl. Acad. Sci. USA. 97:8219–8224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredenbeck, J., and P. Hamm. 2003. Peptide structure determination by two-dimensional infrared spectroscopy in the presence of homogeneous and inhomogeneous broadening. J. Chem. Phys. 119:1569–1578. [Google Scholar]
- Bredenbeck, J., J. Helbing, R. Behrendt, C. Renner, L. Moroder, J. Wachtveitl, and P. Hamm. 2003. Transient 2D-IR spectroscopy: snapshots of the nonequilibrium ensemble during the picosecond conformational transition of a small peptide. J. Phys. Chem. B. 107:8654–8660. [Google Scholar]
- Chattopadhyay, A., and S. Mukherjee. 1993. Fluorophore environment in membrane-bound probes: a red edge excitation shift study. Biochemistry. 32:3804–3811. [DOI] [PubMed] [Google Scholar]
- Cheatuma, C. M., and A. Tokmakoff. 2004. Signatures of β-sheet secondary structures in linear and two-dimensional infrared spectroscopy. J. Chem. Phys. 120:8201–8215. [DOI] [PubMed] [Google Scholar]
- Choi, J. H., S. Y. Ham, and M. Cho. 2003. Local amide I mode frequencies and coupling constants in polypeptides. J. Phys. Chem. B. 107:9132–9138. [Google Scholar]
- de Groot, B. L., and H. Grubmüller. 2001. Water permeation across biological membranes: mechanism and dynamics of aquaporin-1 and GlpF. Science. 294:2353–2357. [DOI] [PubMed] [Google Scholar]
- de Planque, M. R. R., B. B. Bonev, J. A. A. Demmeris, D. V. Greathouse, R. E. Koeppe II, F. Separovic, A. Watts, and J. A. Killian. 2003. Interfacial anchor properties of tryptophan residues in transmembrane peptides can dominate over hydrophobic matching effects in peptide-lipid interactions. Biochemistry. 42:5341–5348. [DOI] [PubMed] [Google Scholar]
- Eftink, M. R. 1991. Fluorescence quenching: theory and application. In Topics in Fluorescence Spectroscopy. J. R. Lakowicz, editor. Plenum Press, New York. 53–126.
- Eftink, M. R., and C. A. Ghiron. 1976. Exposure of tryptophan residues in proteins: quantitative determination by fluorescence quenching studies. Biochemistry. 15:672–680. [DOI] [PubMed] [Google Scholar]
- Eker, F., X. Cao, L. Nafie, Q. Huang, and R. Schweitzer-Stenner. 2003a. The structure of alanine based tripeptides in water and dimethyl sulfoxide probed by vibrational spectroscopy. J. Phys. Chem. B. 107:358–365. [Google Scholar]
- Eker, F., K. Griebenow, and R. Schweitzer-Stenner. 2003b. Stable conformations of tripeptide in aqueous solution studied by UV circular dichroism spectroscopy. J. Am. Chem. Soc. 125:8178–8185. [DOI] [PubMed] [Google Scholar]
- Fecko, C. J., J. D. Eaves, J. J. Loparo, A. Tokmakoff, and P. L. Geissler. 2003. Ultrafast hydrogen-bond dynamics in the infrared spectroscopy of water. Science. 301:1698–1702. [DOI] [PubMed] [Google Scholar]
- Frauenfelder, H., S. G. Sligar, and P. G. Wolynes. 1991. The energy landscape and motions of proteins. Science. 254:1598–1603. [DOI] [PubMed] [Google Scholar]
- Galley, W. C., and R. M. Purkey. 1970. Role of heterogeneity of the salvation site in electronic spectra in solution. Proc. Natl. Acad. Sci. USA. 67:1116–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervasio, F. L., R. Chelli, P. Procacci, and V. Schettino. 2002. The nature of intermolecular interactions between aromatic amino acid residues. Proteins. 48:117–125. [DOI] [PubMed] [Google Scholar]
- Golonzka, O., M. Khalil, N. Demirdoven, and A. Tokmakoff. 2001. Vibrational anharmonicities revealed by coherent two-dimensional infrared spectroscopy. Phys. Rev. Lett. 86:2154–2157. [DOI] [PubMed] [Google Scholar]
- Grossfield, A., and T. B. Woolf. 2002. Interaction of tryptophan analogs with POPC lipid bilayers investigated by molecular dynamics calculations. Langmuir. 18:198–210. [Google Scholar]
- Hamm, P., M. Lim, W. F. DeGrado, and R. M. Hochstrasser. 1999. The two-dimensional IR nonlinear spectroscopy of a cyclic penta-peptide in relation to its three dimensional structure. Proc. Natl. Acad. Sci. USA. 96:2036–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm, P., M. Lim, and R. M. Hochstrasser. 1998. Structure of the amide I band of peptides measured by femtosecond nonlinear-infrared spectroscopy. J. Phys. Chem. B. 102:6123–6138. [Google Scholar]
- Hu, W., K. C. Lee, and T. A. Cross. 1993. Tryptophans in membrane proteins: indole ring orientations and functional implications in the gramicidin channel. Biochemistry. 32:7035–7047. [DOI] [PubMed] [Google Scholar]
- Jacobs, R. E., and S. H. White. 1989. The nature of the hydrophobic binding of small peptide at the bilayer interface: implications for the insertion of transbilayer helices. Biochemistry. 28:3421–3437. [DOI] [PubMed] [Google Scholar]
- Jones, D. T., W. R. Taylor, and J. M. Thornton. 1994. A model recognition approach to the prediction of all-helical membrane protein structure and topology. Biochemistry. 33:3038–3049. [DOI] [PubMed] [Google Scholar]
- Karplus, M. 1959. Contact electron-spin coupling of nuclear magnetic moments. J. Chem. Phys. 30:11–15. [Google Scholar]
- Krimm, S., and A. Yasuaki. 1972. Intermolecular interaction effects in the amide I vibrations of β polypeptides. Proc. Natl. Acad. Sci. USA. 69:2788–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummel, A. T., P. Mukherjee, and M. T. Zanni. 2003. Inter and intrastrand vibrational coupling in DNA studied with heterodyned 2D-IR spectroscopy. J. Phys. Chem. B. 107:9165–9169. [Google Scholar]
- Kwac, K., and M. Cho. 2003. Molecular dynamics simulation studies of N-methylacetamide in water. II. Two-dimensional infrared pump-probe spectra. J. Chem. Phys. 119:2256–2263. [Google Scholar]
- Lakowicz, J. R. 1983. Introduction to Fluorescence Spectroscopy. Plenum Press, New York.
- Lehrer, S. S. 1971. Solute perturbation of protein fluorescence. The quenching of the tryptophyl fluorescence of model compounds and of lysozyme by iodide ion. Biochemistry. 10:3254–3263. [DOI] [PubMed] [Google Scholar]
- MacCallum, J. L., P. Mukhopadhyay, H. Luo, and D. P. Tieleman. 2003. Large scale molecular dynamics simulations of lipid-drug interactions. Proceedings of the 17th Annual International Symposium on High Performance Computing Systems and Applications and the OSCAR Symposium. David Senechal, editor. NRC Research Press, Ottawa, Canada.
- Mu, Y. G., and G. Stock. 2002. Conformational dynamics of trialanine in water: a molecular dynamics study. J. Phys. Chem. B. 106:5294–5301. [Google Scholar]
- Mukherjee, P., A. T. Krummel, E. C. Fulmer, I. Kass, I. T. Arkin, and M. T. Zanni. 2004. Site-specific vibrational dynamics of the CD3ζ membrane peptide using heterodyned two-dimensional infrared photon echo spectroscopy. J. Chem. Phys. 120:10215–10224. [DOI] [PubMed] [Google Scholar]
- Opella, S. J., C. Ma, and F. M. Marassi. 2001. Nuclear magnetic resonance of membrane-associated peptides and proteins. Methods Enzymol. 339:285–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick, C. A., E. van der Wel, J. A. Strandberg, J. Killian, and R. E. Koeppe II. 2002. Geometry and intrinsic tilt of a tryptophan-anchored transmembrane α-helix determined by 2H NMR. Biophys. J. 83:1479–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson, S., J. A. Killian, and G. Lindblom. 1998. Molecular ordering of interfacially localized tryptophan analogs in ester- and ether-lipid bilayers studies by 2H-NMR. Biophys. J. 75:1365–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja, S. M., S. S. Rawat, A. Chattopadhyay, and A. K. Lala. 1999. Localization and environment of tryptophans in soluble and membrane-bound state of a pore-forming toxin from Staphylococcus aureus. Biophys. J. 76:1469–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossend, R., and J. T. Hynes. 1998. Vibrational phase and energy relaxation of CN− in water. J. Chem. Phys. 108:142–153. [Google Scholar]
- Schweitzer-Stenner, R., F. Eker, Q. Huang, and K. Griebenow. 2001. Dihedral angles of trialanine in D2O determined by combining FTIR and polarized visible Raman spectroscopy. J. Am. Chem. Soc. 123:9628–9633. [DOI] [PubMed] [Google Scholar]
- Shepherd, C. M., K. A. Schaus, H. J. Vogel, and A. H. Juffer. 2001. Molecular dynamics study of peptide-bilayer adsorption. Biophys. J. 80:579–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramannian, E., and J. J. Sahayamary. 1989. Structure and conformation of linear peptides. Int. J. Pept. Protein Res. 34:134–138. [DOI] [PubMed] [Google Scholar]
- Thomas, A., R. Meurisse, and R. Brasseur. 2002a. Aromatic side-chain interactions in proteins. II. Near- and far-sequence Phe-X pairs. Proteins. 48:635–644. [DOI] [PubMed] [Google Scholar]
- Thomas, A., R. Meurisse, B. Charloteaux, and R. Brasseur. 2002b. Aromatic side-chain interactions in proteins. I. Main structural features. Proteins. 48:628–634. [DOI] [PubMed] [Google Scholar]
- Tieleman, D. P., L. R. Forrest, M. S. P. Sansom, and H. J. C. Berendsen. 1998. Lipid properties and the orientation of aromatic residues in OmpF, influenza M2, and alamethicin systems: molecular dynamics simulations. Biochemistry. 37:17554–17561. [DOI] [PubMed] [Google Scholar]
- Torii, H., and M. Tasumi. 1998. Ab initio molecular orbital study of the amide I vibrational interactions between the peptide groups in di- and tripeptides and considerations on the conformation of the extended helix. J. Raman. Spectrosc. 29:81–86. [Google Scholar]
- Weiss, S. 1999. Fluorescence spectroscopy of single biomolecules. Science. 283:1676–1683. [DOI] [PubMed] [Google Scholar]
- Woutersen, S., and P. Hamm. 2000. Structure determination of trialanine in water using polarization sensitive two-dimensional vibrational spectroscopy. J. Phys. Chem. B. 104:11316–11320. [Google Scholar]
- Woutersen, S., and P. Hamm. 2001. Isotope-edited two-dimensional vibrational spectroscopy of trialanine in aqueous solution. J. Chem. Phys. 114:2727–2737. [Google Scholar]
- Woutersen, S., and P. Hamm. 2002. Nonlinear two-dimensional vibrational spectroscopy of peptides. J. Phys. Condens. Matter. 14:R1035–R1062. [Google Scholar]
- Woutersen, S., Y. G. Mu, G. Stock, and P. Hamm. 2001. Subpicosecond conformational dynamics of small peptides probed by two-dimensional vibrational spectroscopy. Proc. Natl. Acad. Sci. USA. 98:11254–11258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woutersen, S., R. Pfister, P. Hamm, Y. G. Mu, D. S. Kosov, and G. Stock. 2002. Peptide conformational heterogeneity revealed from the nonlinear vibrational spectroscopy and molecular dynamics simulations. J. Chem. Phys. 117:6833–6840. [Google Scholar]
- Yau, W.-M., W. C. Wimley, K. Gawrisch, and S. H. White. 1998. The preference of tryptophan for membrane interfaces. Biochemistry. 37:14713–14718. [DOI] [PubMed] [Google Scholar]
- Yeremenko, S., M. S. Pshenichnikov, and D. A. Wiersma. 2003. Hydrogen-bond dynamics in water explored by heterodyne-detected photon echo. Chem. Phys. Lett. 369:107–113. [Google Scholar]
- Zanni, M. T., S. Gnanakaran, J. Stenger, and R. M. Hochstrasser. 2001. Heterodyned two-dimensional infrared spectroscopy of solvent-dependent conformations of acetylproline-NH2. J. Phys. Chem. B. 105:6520–6535. [Google Scholar]