

Abstract
A detailed molecular dynamics study of the haemagglutinin fusion peptide (N-terminal 20 residues of the HA2 subunits) in a model bilayer has yielded useful information about the molecular interactions leading to insertion into the lipids. Simulations were performed on the native sequence, as well as a number of mutant sequences, which are either fusogenic or nonfusogenic. For the native sequence and fusogenic mutants, the N-terminal 11 residues of the fusion peptides are helical and insert with a tilt angle of ∼30° with respect to the membrane normal, in very good agreement with experimental data. The tilted insertion of the native sequence peptide leads to membrane bilayer thinning and the calculated order parameters show larger disorder of the alkyl chains. These results indicate that the lipid packing is perturbed by the fusion peptide and could be used to explain membrane fusion. For the nonfusogenic sequences investigated, it was found that most of them equilibrate parallel to the interface plane and do not adopt a tilted conformation. The presence of a charged residue at the beginning of the sequence (G1E mutant) resulted in a more difficult case, and the outcomes do not fall straightforwardly into the general picture. Sequence searches have revealed similarities of the fusion peptide of influenza haemagglutinin with peptide sequences such as segments of porin, amyloid αβ peptide, and a peptide from the prion sequence. These results confirm that the sequence can adopt different folds in different environments. The plasticity and the conformational dependence on the local environment could be used to better understand the function of fusion peptides.
INTRODUCTION
Many processes in living cells require the recognition and merging of membranes of different organelles and the mixing of their aqueous contents. One of the much-studied fusion mechanisms involves virus-cell fusion, which constitutes an essential step in the infectious process of all enveloped animal viruses. These events are mediated by fusion glycoproteins, which require a triggering event like a change in pH or the binding to a receptor to induce conformational changes that are required for membrane fusion. One important consequence of these changes is the exposure of a highly conserved stretch of hydrophobic amino acids, the fusion peptide, and its interaction with the target membrane. For the influenza virus, the fusion glycoprotein is the haemagglutinin (HA) (Skehel and Wiley, 2000; Cross et al., 2001). HA is a homotrimer with each monomer consisting of a receptor-binding (HA1) domain and a membrane-anchoring (HA2) domain linked by a single disulphide bond. The virus binds to sialic acid-containing receptors on the cell plasma membrane and is internalized into endosomes. Endosomal acidification triggers large conformational rearrangements (Skehel et al., 1982; Bullough et al., 1994; Bizebard et al., 1995), converting the metastable pH form into a low pH form with a lower energy. The HA1 subunits, although retaining their structure, are dissociated (Skehel et al., 1982; Graves et al., 1983; Bullough et al., 1994; Chen et al., 1998), and the trimeric coiled-coil of HA2 is elongated with the fusion peptide exposed at one end. These changes also place the C-terminal membrane anchor at the same end of the rod-shaped molecule as the fusion peptide, suggesting that these two hydrophobic regions are responsible for bringing the endosomal and viral membranes into proximity to initiate membrane fusion. The crucial role of the fusion peptide (Epand, 2003) in virus-cell fusion has been shown by site-directed mutagenesis (Gething et al., 1986), leakage of the liposomal contents of artificial lipid membranes (Wharton et al., 1988), and reverse genetics (Cross et al., 2001). Although influenza haemagglutinin is a structurally well-characterized fusion protein (Wilson et al., 1981; Chen et al., 1998; Skehel and Wiley, 2000), not much is known about the structure of HA-lipid complexes that may be required for fusion. Mutants of HA with amino acid substitutions in the amino-terminal region of HA2 (Wharton et al., 1988; Steinhauer et al., 1995) have been used to investigate the role of the conserved spacing of glycine residues within the first 10 amino acids, and the possible substitutions in this first segment that can retain the ability to fuse. NMR investigations in membrane-like environments have provided some structural insights into synthetic fusion peptides and their membrane interactions (Dubovskii et al., 2000; Han et al., 2001; Li et al., 2003). These studies demonstrate a high percentage of α-helical secondary structure and interactions of the charged residues with the phosphate groups of the lipids. This structure is different from the corresponding region in the native structure, suggesting a structural plasticity of the sequence that may be crucial for the fusion process. Spin-label electron paramagnetic resonance (EPR) (Macosko et al., 1997; Han et al., 2001) and 15NMR studies (Bradshaw et al., 1998) have shown that HA fusion peptides insert obliquely into the membrane. Neutron diffraction studies of the fusion peptide from simian immunodeficiency virus gp41 have also demonstrated oblique insertion, which has been correlated with the ability of the peptide to increase membrane curvature (Bradshaw et al., 1998). Despite their shortness, these peptides appear to have the ability to perturb the phospholipid bilayer, which is sensitive to small changes in their sequence. Although the previously mentioned studies have shed light on important aspects of the fusion peptide-membrane interactions, no description at a molecular level of the mechanism of insertion into lipid bilayers of fusogenic peptides is yet available. The purpose of this study is twofold: 1), to analyze the sequence plasticity (meaning the capability of assuming different conformations upon changes of the local environment such as pH or solvent) of influenza haemagglutinin peptides by comparison with similar segments that are not fusion peptides; 2), to study the behavior of fusogenic and nonfusogenic mutant peptides by molecular dynamics (MD) simulations in explicit membrane environments. An atomic description of this structural plasticity can be performed by MD simulations with a realistic description of the lipidic environment mimicking the cellular membrane. A limited number of computational studies have been undertaken to date to simulate the interaction of influenza haemagglutinin fusion peptides with the membrane. Molecular modeling and Monte Carlo techniques have been applied to a series of analogs of the fusion peptide (Efremov et al., 1999), simulated in a membrane-mimicking environment based on atomic solvation parameters from gas-cyclohexane and gas-water transfer to describe the hydrophobic core of the bilayer and hydrated headgroups of the lipids. Benchor and Ben-Tal calculate the stability of different orientations of influenza haemagglutinin peptide with a mean-field approach (Benchor and Ben-Tal, 2001). To date, the only simulation of fusion peptides that has been done in explicit solvent is that of gp41 from human immunodeficiency virus (Kamath and Wong, 2002) and its mutants (Wong, 2003). These studies describe the mode of insertion and the specific interactions with water and the phosphate headgroups of the lipids. In this contribution, we present 5ns simulations of native fusion peptides from influenza haemagglutinin in an explicit palmitoyl-oleoyl-phosphatidylcholine (POPC) lipid bilayer together with some of their fusogenic and nonfusogenic mutant peptides and we attempt to investigate their different behavior in a lipid environment. We chose POPC because this is the lipid in which many of the activity measurements are performed. We are able to reproduce key features detected by different experimental techniques: the secondary-structure content, the tilting with respect to the membrane-water interface, and the depth of insertion in the membrane core.
METHODS
Sequence searching
Sequence searches for the FP (fusion peptide) “GLFGAIAGFIENGWEGMIDG” (HAFP) were performed on the Bioccelerator server (http://eta.embl-heidelberg.de:8000/) using the Smith-Waterman algorithm (Smith and Waterman, 1981) with the Blosum62 matrix (Henikoff and Henikoff, 1992) on the Swissprot (Boeckmann et al., 2003) database. Additional searches were performed using the program Quest (Taylor, 1998) with relaxed parameters (P-value range 0.001–1 and matrix range from Blosum62 to Blosum30) on a nonredundant database (Benson et al., 2002). Variants of FP scored at E-values between 1 × 10−20 and 2 × 10−9, whereas all other hits scored at E-values above 5 × 10−2. From the list of non-FPs, those matches with known structure and proven membrane association were extracted: Porin (2OMF), phosphofructokinase (6PFK), amyloid β peptide (1IYT, 1HZ3), glycerol facilitator (1LDF), aquaporin 1 (1J4N), and ABC transporter (1L7V). Multiple alignment and dendrogram construction of the extracted segments was performed using the program Praline (Heringa, 1999, 2002) with the Blosum35 matrix and gap penalties 8/2 (opening/extension). Contact accepted mutation (CAO) scores, which indicate the probability of substituting side-chain contacts and combine sequence and structure conservation information (Lin et al., 2003), were calculated using the CAO200 matrix.
Molecular dynamics simulations
To analyze the behavior of fusogenic and nonfusogenic sequences, we selected the following fusion peptides (Table 1): HA5 and G4E from the H3 influenza haemagglutinin, H9 from the H9 influenza haemagglutinin, and H2B from influenza B. For the H3 nonfusogenic mutant sequences we studied G1L, G4V, ΔG1, and G1E. These latter four peptide sequences have been shown to result in a loss of fusion activity in experiments using synthetic peptides and full-length expressed haemagglutinin (Steinhauer et al., 1995). However, in the case of G1L generated by reverse genetics it was possible to rescue the virus but this replicated to a significantly lower level than wild-type virus, therefore indicating that this mutant is greatly impaired but not totally fusion inactive in the virus (Cross et al., 2001). The initial structures for the simulations are the Protein Data Bank (PDB) files 1IBN (pH 5.0) and 1IBO (pH 7.4), hereafter denoted as HA5 and HA7 (Han et al., 2001). All the mutants have been modeled on the initial structure of HA5. The peptides were inserted in a fully equilibrated 128 POPC lipid bilayer (Tieleman et al., 1999). The Glu11 residue was placed in the bilayer at the level of the lipid phosphate groups (Han et al., 2001). The coordinates of the 128 POPC bilayer were used as a solvating box for the peptide; ∼10 lipid molecules were removed in the upper leaflet to accommodate the peptide for each of the systems. Given that the peptide is relatively small, more sophisticated procedures (Faraldo-Gomez et al., 2002) were not necessary in this case. Previous work on the fusion peptide gp41 (Kamath and Wong, 2002; Wong, 2003) used a similar approach. The peptides were simulated with charged N- and C termini for the native sequence (referred to as HA5_c); for all the other simulations neutral N- and C termini were used. Neutral termini allowed us to insert the peptide deeply into the membrane to see if the peptide orientation would equilibrate to the experimentally observed parameters. Thus, sampling was not biased toward the initial positioning of the tilt angle of the 1IBN and 1IBO structures (see Fusogenic and nonfusogenic mutants section for a discussion of this point). Each system was then solvated with water, resulting in a total of ∼19,000 atoms (box dimensions were 6.2 × 6.7 × 6.2 nm3) and then subjected to 500 ps of solute-restrained MD simulations to allow the lipid molecules to relax. The final structures were submitted to 5-ns simulation runs. Longer simulations were performed on the HA5 peptide, up to 10 ns, as well as simulations in a different bilayer (palmitoyl-oleoyl-phosphatidylethanolamine) and the average properties (tilting angle, percentage of helical structure) did not change significantly. In the past Brunner (1989) showed by photolabeling techniques that the peptide only penetrates one leaflet of the bilayer; therefore, we decided to start from an asymmetric positioning of the peptide in the membrane. The slight asymmetry of the bilayer seemed not to have any effects on the conformational behavior of the peptide. Given that the average properties for all peptides studied here did not vary significantly after 3 ns time, only 5 ns of simulation data was used for the analysis. This allowed us to maximize the number of mutants that could be studied. MD simulations were performed using GROMACS (Berendsen et al., 1995). The LINCS algorithm was used to constrain all bond lengths within the lipids. A cutoff of 0.9 nm for Coulomb and Lennard-Jones interactions was used and particle mesh Ewald (Essmann et al., 1995) was used to calculate the remaining electrostatic contributions on a grid with 0.12 nm spacing. NPT conditions (i.e., constant number of particles, pressure, and temperature) were used in the simulations. A constant pressure of 1 bar in all three directions was used, with a coupling constant of 1.0 ps (Berendsen et al., 1984). Water, lipids, and protein were coupled separately to a temperature bath at 300 K with a coupling constant of 0.1 ps. The velocity of the atoms was not rescaled during the simulation. The lipid parameters were as in previous MD studies of lipid bilayers (Tieleman et al., 1999) and the GROMOS96 force field (43al) was used for the peptide (van Gunsteren et al., 1996). The SPC water model was used for the solvent (Berendsen et al., 1981). Analysis programs from GROMACS were used, together with our own programs (for tilt angle and secondary-structure calculations). The bilayer thickness was determined as the average difference of the y coordinates (the membrane plane was in xz orientation) between the phosphorus atoms of the upper and lower leaflets. The depth of residues inside the bilayer was determined as the average difference in the y coordinates of each residue's Cα atom and the phosphorus atoms of the upper leaflet.
TABLE 1.
Peptides studied
| Peptide | Sequence | Specification |
|---|---|---|
| HA5 | GLFGAIAGFIENGWEGMIDG | H3 influenza haemagglutinin |
| HA7 | GLFGAIAGFIENGWEGMIDG | H3 influenza haemagglutinin |
| G1L | LLFGAIAGFIENGWEGMIDG | G1L mutation of H3 influenza haemagglutinin |
| ΔG1 | LFGAIAGFIENGWEGMIDG | ΔG1 mutation of H3 influenza haemagglutinin |
| G1E | ELFGAIAGFIENGWEGMIDG | G1E mutation of H3 influenza haemagglutinin |
| G4E | GLFEAIAGFIENGWEGMIDG | G4E mutation of H3 influenza haemagglutinin |
| G4V | GLFVAIAGFIENGWEGMIDG | G4V mutation of H3 influenza haemagglutinin |
| H9 | GLFGAIAGFIEGGWPGLVAG | H9 influenza haemagglutinin |
| H2B | GFFGAIAGFLEGGWEGMIAG | influenza B |
Helix-axis calculation
To analyze the tilting of the FPs within the membrane during the MD trajectories, we selected parts of the structure embedded in the modeled membrane that are known to be helical (residues 3–11), and then fitted the helical axes as accurately as possible. The method used is analogous to that used by (Thomas, 1991) to fit curved helices accurately in a simplified mechanical model of protein folding, a feature implemented in the WHAT IF program as a graphical option (Vriend, 1990). The mathematics is described in (Thomas, 1994), and exploits the special properties of the rarely used Chebyshev polynomials orthogonal on the discrete domain. If the helix (or other secondary-structure element) is optimally fitted by an expansion of arbitrarily high order in these polynomials, the best-fit straight-line approximation is simply given by the first order (linear) part of the expansion. We did not incorporate the computer code into the molecular dynamics program, but instead wrote a stand-alone routine controlled by a perl script.
RESULTS
Sequence plasticity
The first 11 residues of HAFP (GLFGAIAGFIE) are conserved amongst the antigenic subtypes of orthomyxoviruses of influenza A viruses (Nobusawa et al., 1991), suggesting an evolutionary conserved function for this segment. This stretch contains the GxxxG motif known to stabilize helix-helix interactions in both membrane and soluble proteins (Russ and Engelman, 2000; Kleiger et al., 2002). The motif is also present for residues 17–20 (GMIDG), with some sequence variability for different subtypes and influenza B viruses. Helix-helix interactions with the GxxxG motif have been described for dimers, where the glycyl residues of the motif of one helix interact with the glycyl residues of the GxxxG motif of the other helix. This type of interaction allows for close association of the two helices and favors backbone-backbone contacts, as in the case of glycoporin A (MacKenzie et al., 1997). For the HAFP peptide, the presence of this motif could stabilize homotrimer interfaces. From the sequence searching we performed for HAFP, many of the hits involved transport proteins, permease proteins, and amyloid precursors. Of all the significant hits only a few have a structure in the PDB database; for some the structure of a homologous sequence was used. Multiple alignment of the HAFP with the membrane fragment of porin (2OMF), a membrane protein responsible for the passive transport of small hydrophilic molecules, shows a similar pattern of glycine and hydrophobic residues, particularly at the C-terminal part of HAFP. Analogous similarities were found for the amyloid Alzheimer β-peptide (1IYT and 1HZ3) and for the soluble protein phosphofructokinase (6PFK). These sequences form a subgroup in the phylogenetic tree derived from alignment scores. The relation to the other subgroup comprising 1LDF, 1J4N, and 1L7V is less evident, but alignment scores indicate a reasonable similarity. Although sequence similarity of the extracted fragments clearly shows a common pattern within the set, there is no strong evidence for a homologous relationship between any of the fragments. The most surprising fact is that these sequence fragments adopt very different conformations, depending on the local structural environment (segments highlighted in Fig. 1). In apolar or membrane environments, the segments are mostly in helical conformations, with the exception of the porin case. For the soluble proteins (6PFK and 1L7V), the segments mostly adopt a helix-turn-helix structural pattern. By analyzing the CAO scores (Lin et al., 2003), which combine sequence and structure information by reporting the probability of side-chain contact substitution, we observe generally negative pairwise scores between the HAFP and the fragments, meaning that despite the similarity, the HAFP peptide would (in evolutionary terms) probably not replace the fragment sequences. The shortness of the examined sequence does not allow for strict generalizations, but on the basis of these comparisons, we conclude that these similar sequences adopt distinct conformations. However, this does not necessarily imply a common mechanism of membrane insertion. The sequence similarity with the amyloid αβ peptide and with a peptide from the prion sequence have been already pointed out elsewhere (Crescenzi et al., 2002; Del Angel et al., 2002; Forloni et al., 1993). These segments are part of proteins that undergo large conformational transitions, leading to pathogenic forms. Both peptides have been shown to induce channel formation, suggesting that the amino acid composition could be associated with structural mobility and (toxic) pore formation ability. Another common feature is the environmental dependence of the structural forms of these peptides. Typical is the case of the amyloid peptide, for which two completely different structures have been proposed in water/trifluoroethanol solution (Fig. 1, 1HZ3) and in membrane-like environments (Fig. 1, 1IYT) (Crescenzi et al., 2002; Massi et al., 2001). The structural plasticity and environmental adaptation of these fragment sequences are therefore some of the key features required for their functional mechanism to occur. These features would represent a necessary condition for the peptide to be extruded from the proteic environment and, subsequently, to be stably inserted into the viral membrane.
FIGURE 1.

Structures of the best hits from sequence searching using HAFP as a template. Alignments with the matched sequences are reported. In case the matched sequence has no known structure, the next best match with a structure is reported in the alignment. Amino acid stretches in the alignment are colored in blue on the structure. Molecules are referred to by their PDB entry codes (see Methods section).
Importance of the starting conformation
A number of biophysical studies have been performed to determine the structure of fusion peptides in a membrane-mimetic environment and to characterize peptide binding to the lipid bilayer. The high hydrophobicity of these peptides impedes measurement of their partitioning between aqueous and membrane phases. To overcome this problem, the insertion of hydrophilic residues into the sequence has allowed the NMR structure determination of a fusogenic homolog of the HA fusion peptide, called E5 (GLFEAIAEFIEGGWEGLIEG), in dodecylphosphocholine and in sodium dodecylsulfate (SDS) micelles (Dubovskii et al., 2000; Hsu et al., 2002). To study the native sequence in an environment similar to the cellular one, a new host-guest fusion peptide system was engineered so as to render the HA fusion peptide completely water-soluble while retaining high affinity for model lipid membranes. The fusion peptide is tethered with a flexible linker to a hydrophilic host peptide that solubilizes the entire system (Han and Tamm, 2000). This has allowed partition experiments of fusion peptides in lipid bilayers (Li et al., 2003) and NMR studies of the structure of the fusion domain in detergent micelles at pH 5.0 and 7.4. Two different conformations for the two pH environments were proposed in that study. We will refer to these as HA5_NMR and HA7_NMR (Han et al., 2001). Based on the experimental observations made, the authors suggest that the secondary structure of the C-terminal segment of HAFP GWEGMIDG (residues 13–20) undergoes large conformational rearrangements upon a change in pH. The first segment is mostly α-helical for both conformers, but the pH 5.0 conformer presents a short stretch of 310 helix from residues 13–18. The NMR study in SDS micelles of the E5 peptide (Hsu et al., 2002) also suggested two different structures at pH 4.3 and 7.3. The low pH structures from all of these studies are similar, with most of the peptide being in a helical conformation, with a hinge in the region around residues Gly12 and Gly13. By comparing the structure of the E5 peptide (Dubovskii et al., 2000) in SDS with HA5_NMR, one can observe that the “V” shape adopted by HA5_NMR (Han et al., 2001) is more rounded for E5 and the peptide assumes a boomerang-shaped conformation with an oblique orientation of the first 11 residues. No hydrophobic pocket due to interaction between Phe9 and Trp12 is formed in the E5 structure, although there is a clear hydrophobic face that faces the membrane. The root-mean-square deviation (RMSD) for the first 11 Cα atoms is 0.96 Å between HA5_NMR and E5. This structural similarity indicates that the orientation and structure of the first 11 residues and of the Glu15 residue are the same. Specifically, Glu 11 and Glu 15 point toward the phosphate groups of the bilayer interface in both structures and the Phe residues are on the opposite side pointing toward the lipid tails in the membrane.
All these considerations seem to imply that the N-terminal 11 residues of the two fusion peptides are in a helical conformation and that residues 11 and 15 should be located at the phosphate-water interface. We therefore constructed two structures starting from the coordinates of HA5_NMR and HA7_NMR, inserted into a preformed POPC lipid bilayer (see Methods section) and simulated each system for 5 ns (the corresponding conformers are referred to as HA5 and HA7). The percentage of secondary-structure elements for the analyzed structures is shown in Fig. 2. Panel a illustrates values for the experimentally determined structures and panel b for the simulated ones averaged over the last 2 ns. The general pattern helix-bend (turn)-helix of the HA5_NMR is maintained for HA5, with a percentage of residues in a helical conformation being slightly higher than for the experimentally determined structures. The HA7 starting structure remains disordered for the segment 12–20, suggesting that if the peptide were to enter the membrane in a partially disordered state, the refolding process would be prevented by the competing favorable interactions with the polar headgroups at the membrane interface. Residues 10 and 11 are more ordered in the simulated peptides, since, as already observed for HA5, the method tends to overestimate the helical content. In Fig. 3, the NMR structures are compared with the simulated ones. In Table 2, some structural parameters for HA5 and HA7 are reported. The helical content measured from the NMR structures HA5_NMR and HA7_NMR are close to the ones measured from the 5-ns trajectories, with the HA5 values higher than the HA7 ones. As already mentioned, the helical contents derived from the simulations are often higher than the measured ones, but the length of the simulation does not allow us to observe statistically relevant unfolding-refolding events. We are not able, on the basis of our simulations, to differentiate, with respect to fusogenic activity, between HA5 and HA7. We have to stress, nevertheless, some contradiction in the literature about the definition of active forms at different pH for the mechanism of insertion. In a pioneering study of the membrane-binding conformational properties of influenza haemagglutinin (Lear and De Grado, 1987) it was clearly shown that the fusion peptide could fuse vesicles at a rate that is independent of pH between 5.0 and 7.0. Later on, other studies (Han et al., 2001; Epand et al., 2001) have stressed the importance of a low pH for the fusogenic activity to occur. Our simulations are performed at constant pH and therefore the only difference between the HA5 and the HA7 conformers is their starting conformation. It would be beyond the scope of our simulations to claim a difference in activity for the same sequence. Under our conditions both conformers adopt a tilted orientation and the HA7 structure results are more disordered and generally less stable, but this could also be due to a less folded starting structure. In principle, an infinitely long simulation should be able to yield the equilibrium conformation of the two conformers. In the past, simulations on peptides and proteins have successfully produced unfolding-refolding events, but these were performed on longer timescales (at least 10 times longer) (Daura et al., 1998, 1999) or with more efficient sampling procedures (Kleinjung et al., 2003). None of these simulations was perfomed in an explicit membrane environment. The analysis of the equilibrium between the two conformers studied here would be interesting subject matter for future studies.
FIGURE 2.

Calculated percentages of secondary-structure elements of (a) the experimental structures (1IBN and 1IBO) and (b) the HA5 and HA7 simulations. Percentages of helix structure (expressed as the sum of α and 310) are represented by black bars.
FIGURE 3.
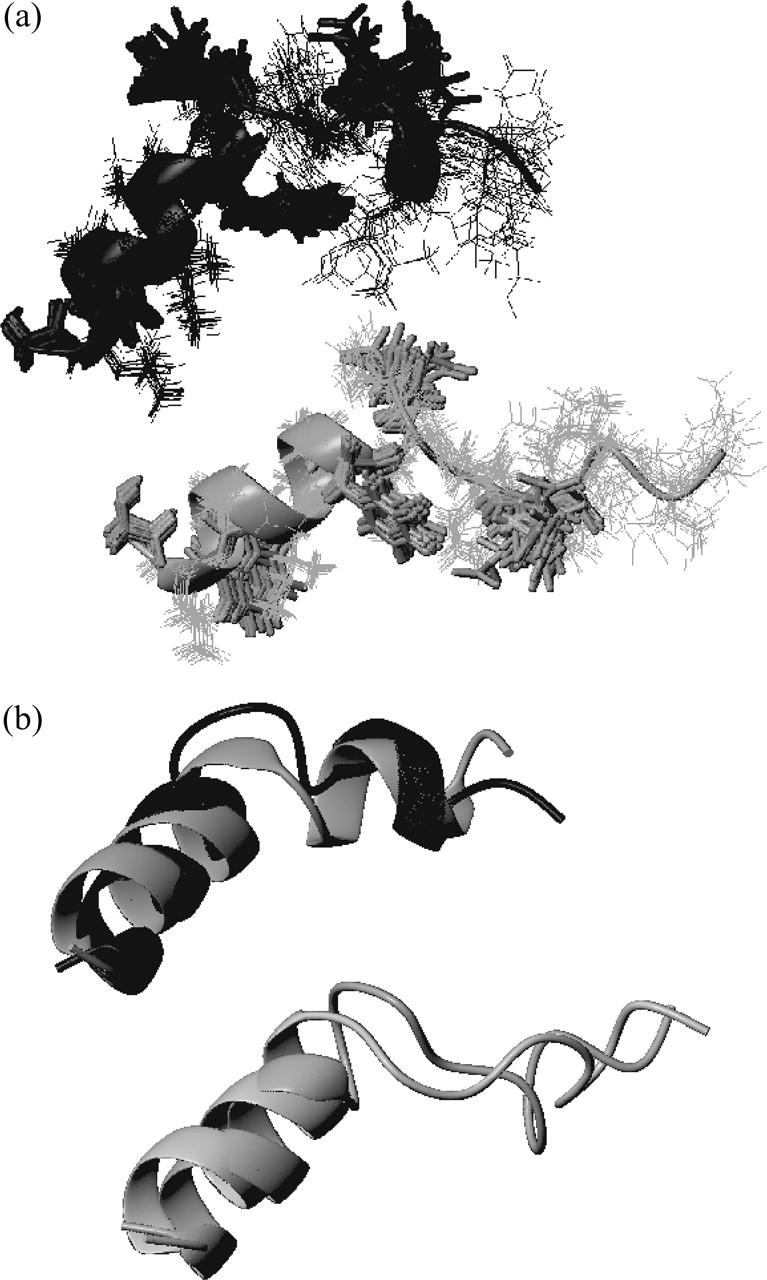
(a) HA5_NMR (black) and HA7_NMR (gray). (b) Superposition of the final HA5 conformer from MD simulations (gray) to HA5_NMR (black). (Bottom) Superposition of the final HA7 conformer from MD simulations (light gray) and of the HA7_NMR structure (dark gray).
TABLE 2.
Relevant structural parameters for HA5 and HA7 in POPC
| Peptide | Hcalc*(%) | HCD†(%) | HNMR‡(%) | Tilt angle(°) | Thickness§ (Å) |
|---|---|---|---|---|---|
| HA5 | 71.0 | 33 | 66.0 | 32.7 | 31.7 |
| HA7 | 47.6 | - | 40.2 | 31.0 | 26.9 |
Helical content (in % over all residues) calculated from the last 2ns of each trajectory.
Helical content measured from CD experiments in POPC.
Helical content calculated from the PDB structures in SDS micelles.
Membrane thickness measured as the average difference in the Y coordinates of the phosphor atoms of the upper and lower leaflets (membrane is in XZ plane).
The bilayer thickness, measured as the average distance between the phosphorus atoms of the upper and lower leaflets of POPC, is also reported in Table 2. These values have to be compared with the thickness of the simulated bilayer of POPC without the presence of peptides (36 Å, data not shown) and with the experimentally measured value (40 Å) (Kinoshita et al., 1998). By comparison with these reference values, we observe a thinning effect of ∼8 Å, which is more pronounced for HA7 than for HA5. This difference may be an artifact since the method used to estimate the membrane thickness is based on average distances, which can significantly oscillate during the simulation, especially in the case of the more disordered HA7. This structure has residue Glu15 pointing toward the hydrophobic face of the membrane (as in the NMR structure). This residue experiences a significant rearrangement that causes a considerable displacement of the phosphate groups in close proximity to Glu15. As a consequence only qualitative observations can be made about the relative thinning effect. Overall, there is a significant change in bilayer thickness for the simulations with peptide over those with POPC alone. In general, the thinning could be related either to the interaction of polar residues in the peptides, which, in the process of insertion into the membrane, would “pull down” the phosphate groups of the upper leaflet, or to the disorder in the hydrophobic tails generated by the peptide. This latter point will be discussed later in more detail.
Fusogenic and nonfusogenic mutants
All the selected sequences have been subjected to 5ns of simulation in POPC and water. The final snapshots from the simulations are shown in Fig. 4. For simplicity only the upper layer of water and phosphorus atoms (yellow spheres) is represented. The panels on the left correspond to fusogenic sequences, whereas the ones on the right correspond to nonfusogenic sequences. It can be easily observed that fusogenic peptides preserve in general the inverse V shape of the starting structure, and all insert with a tilt angle. The nonfusogenic peptides tend to migrate to the interface and, due to their amphiphilic nature, tend to increase their helical content. This result agrees with previous NMR results of fusogenic and nonfusogenic mutants, where the first residue was mutated, in dodecylphosphocholine micelles (Tamm, 2003). The final structures from the MD simulations of all the sequences have been superimposed onto the HA5 starting one. The average RMSD on all Cα is ∼2.5 Å with a better superimposition for the first 11 residues. The average RMSD on all Cα in the NMR bundle structures from Han et al. (2001) is at most ∼2.2 Å; therefore it can be assumed that the mobility of the structures in the lipid environment has not influenced the structure substantially.
FIGURE 4.
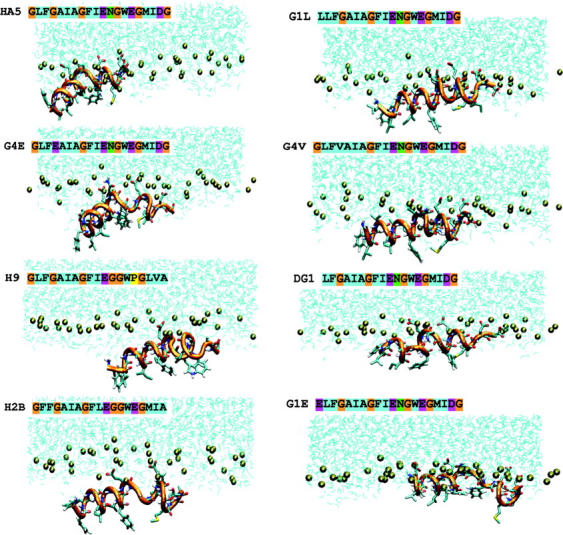
Snapshots of the final conformations of the fusogenic sequences (left panel) and of the fusion-impaired sequences (right panel). The phosphor atoms are represented by brown spheres. The lipid tails are omitted for clarity.
The inverse V shape is retained for the mutant G1S, which has been shown to promote hemifusion in full-length HA (Qiao et al., 1999). The G1V mutant is a controversial case, because it has been suggested that it orients approximately parallel to the membrane surface in the study of Tamm (2003), but other studies have found this peptide to insert perpendicularly to the membrane surface (Epand et al., 2001). Therefore we cannot conclusively compare our G1L peptide results with experimental data on G1V.
The peptide H2B inserts most deeply into the membrane. This could be attributed to the presence of two phenylalanines at the N-terminal region of the sequence, which increases the hydrophobic moment of the inserting helix and stabilizes interactions with the lipid chains. In Fig. 5, the tilt angles of the HA5 and HA7 and G4E peptides and of the fusion-impaired mutants G1L, ΔG1, and G4V are reported. The experimental values reported refer to two different experimental immersion depth measurements for HAFP, obtained from EPR data on singly spin-labeled peptides (Macosko et al., 1997) (exp1) and by mapping NMR data onto the best-fit EPR data (Han et al., 2001) (exp2). The fusogenic peptides equilibrate their tilt angles after ∼3 ns to values very close to the experimental ones (between ∼25° and ∼38°). It is important to stress that tilt-angle values stabilize after ∼3–4 ns, which is another reason why all our simulations were performed for at least 5 ns. Recent work demonstrated that to study the mechanism of insertion of peptides into a lipid bilayer, 30 ns of simulation are necessary (Shepherd et al., 2003), but the purpose of our study is the comparison between different systems of peptides already inserted into the bilayer, and the equilibration of the tilt angle supports our choice of 5 ns. While we were completing this work, a study on the bilayer conformation of the native sequence of the fusion peptide appeared (Huang et al., 2004), where the peptide has been simulated in two different protonation states for the N-termini for 19 ns. The final structure and position of the peptide in this study is, as far as we can judge, not very different from ours; therefore the shorter simulation time chosen should not influence dramatically the structure and positioning of the peptide, and our comparison across mutants should be not substantially affected.
FIGURE 5.

Time evolution of the angle between the helix axis (calculated between residues 3 and 11) and the plane of the membrane for the HA5, HA7, and G4E (dark circles, squares, and triangles, respectively), and for G1L, G4V, and ΔG1 (shaded circles, squares, and triangles, respectively).
Tilt angles for the nonfusogenic mutants are significantly smaller in comparison to the fusogenic ones and are, in general, oriented such that their long axis is perpendicular to the membrane normal.
To describe the insertion mode of the peptide, we report in Fig. 6 the depth of insertion of the 11 N-terminal residues. The reason we focused on these residues is that in all sequences this segment is mostly in helical conformation. We can see that the fusion-impaired mutant G1L is less deeply inserted. This result has been confirmed in experimental data, which has shown that for G1S, G1E, and G1L the depth of insertion and lipid perturbation were lower than for the native sequence (Wu et al., 2003). From our simulations, we can derive that the tilted insertion is mainly due to residues 3, 5, and 6. The most deeply inserted, Ile6, is located at ∼9 Å depth, in good agreement with experimental data (Macosko et al., 1997; Zhou et al., 2000; Han et al., 2001). By inserting deeper into the bilayer and in a tilted way, the fusogenic peptides may destabilize the membrane, promoting membrane fusion. The tilted insertion and perturbation of the membrane has been already illustrated in other computational studies (Lins et al., 2001).
FIGURE 6.

Depth of insertion of the first 11 residues of the HA5 fusion peptide. The experimental values mentioned are extracted from Fig. 5 of Han et al, (2001).
To better investigate the process of insertion, we also performed simulations with charged N- and C-termini for the native sequence (referred to as HA_c). The charged amino group is initially deeply inserted into the membraneous hydrophobic phase. Within the first 60 ps of simulations (see Fig. 7) a shell of solvation has already formed around the amino terminus and remains stable for the remainder of the simulation. The first three residues assume a “hook-like” conformation and the rest of the segment 4–11 is in helical conformation with a tilt angle of insertion of 28.4°. The calculated radial distribution function shows an average number of two or three molecules of water solvating the N-terminus (data not shown). This simulation result could explain at a molecular level the experimentally observed high pKa value for this amino group (Zhou et al., 2000), because the base strength increases if the protonated form is stabilized. In this study the authors conclude that the N-terminus of the peptide is close to the aqueous phase and protonated. Our simulations allow us to follow the inverse process with the amino terminus reaching out to the polar phase, yielding a stable solvated state. We have analyzed in detail the differences between charged and uncharged terminal groups for the native sequence of the influenza haemagglutinin in the conformations HA5 and HA7 and discussed them in a previous article (Vaccaro et al., 2004). During the simulation with charged termini we observe a measurable penetration of water into the apolar lipidic environment. In this study, we have therefore chosen to use a neutral N-terminus, as a description of a protonation/deprotonation mechanism is not feasible at this point in time. Experimental calorimetric data on peptides with a free N-terminal amino group have been performed by Seelig and co-workers (Wieprecht and Seelig, 2002) and it has been pointed out that protonation/deprotonation of the N-terminus is the most likely reaction accompanying binding of peptides to membranes at pH 7. An accurate description of the different equilibria in dependence of the specific media (membrane interface-bulk solution) in which the protonated peptide is located, is given for lipid binding of somatostatin analogs (Seelig et al., 1993), but no study of this type has been performed so far on the influenza haemagglutinin analogs. Considering that in the past a number of studies on the fusion activity of synthetic peptide analogs of the HA fusion peptide have been performed on acetylated N-termini peptides (Murata et al., 1987; Bailey et al., 1997; Zhelev et al., 2001), we estimate that the approximation made by using neutral termini in the analysis of fusogenic or nonfusogenic behavior is equally valid for simulation and experiment.
FIGURE 7.

Time evolution of the solvation process of the charged N-terminus (circled) for the HA5_c simulation. After 60 ps two molecules of water are constantly solvating the charged group throughout the simulated time.
In Fig. 8, calculated order parameters (SCH) from the simulations are reported for equilibrated POPC alone (upper bilayer) and for the POPC upper leaflet in the presence of the fusogenic HA5 and the nonfusogenic G4V peptides. Black lines refer to the palmitic chains and red lines to the oleyl chains; experimental values (Seelig and Seelig, 1977, 1980) are reported in filled circles. For POPC alone, calculated values are slightly higher in magnitude (indicating more order in the system) than experimental values, especially for the oleyl chain. Our reference state in the following comparison will be the order parameters from simulations of POPC alone, this being the starting point before the insertion of the peptide. As evidenced from the lower SCH (magnitude) values, the peptide insertion increases the amount of disorder in the leaflet. It is interesting to note that this phenomenon is more pronounced for the fusogenic peptide HA5, especially the SCHs corresponding to the oleyl chains, which are the more hindered ones. The nonfusogenic mutant G4V perturbs the palmitic chains and, less significantly, the oleyl chain. Localized destabilization of the lipid bilayer by the fusogenic HA peptide have been proposed also from the results of micropipette aspiration experiments of single vescicles to study the mechanical properties of the lipid molecules (Lau et al., 2004), and oligomerization of FP was suggested to enhance localized destabilization of lipid bilayers.
FIGURE 8.

Calculated order parameters for the simulated POPC (upper panel), POPC + HA5 peptide (middle panel), and POPC + G4V peptide (bottom panel).
G1E
The mutant G1E has been the most difficult to interpret. The initial structure imposed on our mutants was chosen on the basis of experimental data from the native fusogenic peptide. To study the changes in the tilt angle during the course of the simulation, an artificial initial tilt of ∼50° was imposed. For G1E, the presence of the glutamate residue (which is protonated because it is positioned deep in the apolar lipidic phase) strongly stabilizes the helix dipole moment and therefore the process of migration is very slow. We have extended this simulation to 10 ns and as can be seen in Fig. 9 the tilt angle still oscillates around values larger than the ones observed for the other studied peptides. At the end of the 10-ns simulation the Glu residue is still in oblique orientation (Fig. 9), because water molecules and phosphate groups have been dragged down toward the apolar phase, attracted by the polar field of the Glu residue, as shown in the right panel of Fig. 9. To speed up a process that would require a very long simulation time to be observed, we started from a configuration with a tilt corresponding to the minimum value observed during the 10-ns period and performed an additional 5 ns of simulation. As expected for a nonfusogenic peptide, under these conditions G1E migrated toward the interface and reached the conformation shown in Fig. 4. Although these conditions are not exactly the same as those used for the other peptides, it should be noted that we are trying to simulate a process that is opposite to the one naturally occurring during the insertion of the peptides into the membrane. In reality the peptide would have to pass through the polar phase and then reach the apolar lipidic tails. During this process a charged residue would presumably be trapped by favorable interactions with the aqueous phase and it would not tilt into the membrane.
FIGURE 9.

Evolution of the angle between the helix axis (calculated between residues 3 and 11) and the membrane plane for G1E. In the lower panel the specific interactions of water molecules and phosphate groups with residue Glu 1 are shown.
CONCLUSIONS
This work describes simulations at a molecular level of the relative positioning of fusogenic and nonfusogenic peptide analogs of the influenza haemagglutinin fusion peptide in lipid bilayers. The sequence pattern characteristic of these peptides has been found to be extremely plastic and able to adopt different conformations as a function of the different environments. Similarity with the amyloid αβ peptide and with a peptide from the prion protein was detected, and the structural plasticity of the FP could be a characteristic required for proteins that undergo large conformational transitions, leading in some cases to a pathogenic form.
We suggest that the amino acid composition could be associated with structural mobility and pore-forming ability. The amphiphilic character of the sequence and the glycine pattern favor helical structures in hydrophobic environments. Our simulated peptides confirm previous structural observations of highly amphipathic conformations adopting an inverse V-shaped structure. One of the most critical parameters in exhibiting fusogenic behavior may be the insertion at an angle to the membrane to perturb the bilayer. In particular, the presence of a fairly stable helix spanning the first 11 N-terminal residues seems necessary to stabilize the tilt angle to a value of ∼30°. This tilted insertion is mainly due to residues 3, 5, and 6, in line with experimental EPR measurements (Macosko et al., 1997; Han et al., 2001), and especially good agreement is found for residue 6, which is inserted to a depth of 9 Å. Lower order parameter values (in magnitude) are observed for fusogenic peptides versus nonfusogenic ones, indicating that the tilted insertion increases the amount of disorder in the leaflet. The C-terminal segment can be partially unfolded without modifying the tilt of the first segment. The lipid bilayer is therefore perturbed by the presence of the FPs and a thinning of ∼8 Å is observed after 5 ns of simulation for either HA5 and HA7, suggesting that the most important factors determining fusogenicity may be found in the interactions of the first 11 residues with the bilayer and not in the degree of secondary structure of the C-terminal segment. This may indicate that the fusogenic activity of a peptide is correlated with the amino acid composition of the first 11 residues. The nonfusogenic mutants tend to migrate to the polar interface and lose the V-shaped structure, in agreement with Fourier transformed infrared spectroscopy data (Han et al., 2001). It is important to recall that fusion peptides are only simplified models of the entire system and therefore cannot entirely describe the complexity of the process. More structural studies are needed to fully understand the mechanism of entry of enveloped viruses into the host membrane.
Acknowledgments
F.F. thanks Ramla Ali for collaboration in initiating the project.
L.V. acknowledges a grant from the University of Salerno, Italy. S.K.S. thanks the Natural Sciences and Engineering Research Council of Canada for a University Faculty Award. This work was supported by the Medical Research Council.
References
- Bailey, A. L., M. A. Monck, and P. R. Cullis. 1997. ph-induced destabilization of lipid bilayers by a lipopeptide derived from influenza hemagglutinin. Biochim. Biophys. Acta. 1324:232–244. [DOI] [PubMed] [Google Scholar]
- Benchor, D., and N. Ben-Tal. 2001. Implicit solvent model studies of the interactions of the influenza hemagglutinin fusion peptide with lipid bilayers. Biophys. J. 80:643–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson, D. A., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., B. A. Rapp, and D. L. Wheeler. 2002. GenBank. Nucleic Acids Res. 30:17–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen, H., J. Postma, W. van Gunsteren, A. DiNola, and J. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- Berendsen, H., J. Postma, W. van Gunsteren, and J. Hermans. 1981. Interaction models for water in relation to protein hydration. In Intermolecular Forces. B. Pullman, editor. Reidel, Dordrecht, The Netherlands.
- Berendsen, H., D. van der Spoel, and R. van Drunen. 1995. Gromacs: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 95:43–56. [Google Scholar]
- Bizebard, T., B. Gigant, P. Rigolet, B. Rasmussen, O. Diat, P. Boseke, S. A. Wharton, J. J. Skehel, and M. Knossow. 1995. Structure of influenza haemagglutinin complexed with a neutralizing antibody. Nature. 376:92–94. [DOI] [PubMed] [Google Scholar]
- Boeckmann, B., A. Bairoch, R. Apweiler, M. C. Blatter, A. Estreicher, E. Gasteiger, M. J. Martin, K. Michoud, C. O'Donovan, I. Phan, S. Pilbout, and M. Schneider. 2003. The SWISS-PROT protein knowledge base and its supplement TrEMBL in 2003. Nucleic Acids Res. 31:365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw, J. P., M. J. M. Darkes, T. A. Harroun, J. Katsaras, and R. M. Epand. 1998. Structure of the haemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell. 95:409–417. [DOI] [PubMed] [Google Scholar]
- Brunner, J. 1989. Testing topological models for the membrane penetration of the fusion peptide of influenza virus hemagglutinin. FEBS Lett. 257:369–372. [DOI] [PubMed] [Google Scholar]
- Bullough, P. A., F. M. Hughson, J. J. Skehel, and D. C. Wiley. 1994. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 371:37–43. [DOI] [PubMed] [Google Scholar]
- Chen, J., K. H. Lee, D. A. Steinhauer, D. J. Stevens, J. J. Skehel, and D. C. Wiley. 1998. Structure of the haemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell. 95:409–417. [DOI] [PubMed] [Google Scholar]
- Crescenzi, O., Tomaselli, S., Guerrini, R., Salvadori, S., D'Ursi, A. M., Temussi, P. A., and D. Picone. 2002. Solution structure of the alzheimer amyloid β-peptide in an apolar microenvironment. Eur. J. Biochem. 269:5642–5648. [DOI] [PubMed] [Google Scholar]
- Cross, K. J., S. A. Wharton, J. J. Skehel, D. C. Wiley, and D. A. Steinhauer. 2001. Studies on influenza haemagglutinin fusion peptide mutants generated by reverse genetics. EMBO J. 20:4432–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daura, X., B. Jaun, W. F. van Gunsteren, and A. E. Mark. 1998. Reversible peptide folding in solution by molecular dynamics simulations. J. Mol. Biol. 280:925–932. [DOI] [PubMed] [Google Scholar]
- Daura, X., W. F. van Gunsteren, and A. E. Mark. 1999. Folding-unfolding thermodynamics of a β-heptapeptide from equilibrium simulations. Proteins. 34:269–280. [DOI] [PubMed] [Google Scholar]
- Del Angel, V. D., F. Dupuis, J. P. Mornon, and I. Callebaut. 2002. Viral fusion peptides and identification of membrane-interacting segments. Biochem. Biophys. Res. Commun. 293:1153–1160. [DOI] [PubMed] [Google Scholar]
- Dubovskii, P. V., H. Li, S. Takahashi, A. S. Arseniev, and K. Akasaka. 2000. Structure of an analog of fusion peptide from hemagglutinin. Protein Sci. 9:786–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremov, R. G., D. E. Nolde, P. E. Volynsky, A. A. Chernyavsky, P. V. Dubovskii, and A. S. Arseniev. 1999. Factors important for fusogenic activity of peptides: molecular modeling study of analogs of fusion peptide of influenza virus hemagglutinin. FEBS Lett. 462:205–210. [DOI] [PubMed] [Google Scholar]
- Epand, R. M. 2003. Fusion peptides and the mechanism of viral fusion. Biochim. Biophys. Acta. 1614:116–121. [DOI] [PubMed] [Google Scholar]
- Epand, R. M., R. F. Epand, I. Martin, and J. M. Ruysschaert. 2001. Membrane interactions of mutated forms of the influenza fusion peptide. Biochemistry. 40:8800–8807. [DOI] [PubMed] [Google Scholar]
- Essmann, U., L. Perera, M. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- Faraldo-Gomez, J. D., G. R. Smith, and M. S. Sansom. 2002. Setting up and optimization of membrane protein simulations. Eur. Biophys. J. 31:217–227. [DOI] [PubMed] [Google Scholar]
- Forloni, G., N. Angeretti, R. Chiesa, E. Monzani, M. Salmona, O. Bugiani, and F. Tavaglini. 1993. Neurotoxicity of a prion protein fragment. Nature. 632:543–546. [DOI] [PubMed] [Google Scholar]
- Gething, M. J., R. W. Doms, D. York, and J. M. White. 1986. Studies on the mechanism of membrane fusion: site-specific mutagenesis of the hemagglutinin of influenza virus. J. Cell Biol. 102:11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves, P. N., J. L. Schulman, J. F. Young, and P. Palese. 1983. Preparation of influenza virus subviral particles lacking the HA1 subunit of hemagglutinin: unmasking of cross-reactive ha2 determinants. Virology. 126:106–116. [DOI] [PubMed] [Google Scholar]
- Han, X., J. H. Bushweller, D. S. Cafiso, and L. Tamm. 2001. Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nat. Struct. Biol. 8:715–720. [DOI] [PubMed] [Google Scholar]
- Han, X., and L. Tamm. 2000. A host-guest system to study structure-function relationships of membrane fusion peptides. Proc. Natl. Acad. Sci. USA. 97:13097–13102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff, S., and J. G. Henikoff. 1992. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA. 89:10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heringa, J. 1999. Two strategies for sequence comparison: profile-preprocessed and secondary structure-induced multiple alignment. Comput. Chem. 23:341–364. [DOI] [PubMed] [Google Scholar]
- Heringa, J. 2002. Local weighting schemes of protein multiple sequence alignment. Comput. Chem. 26:459–477. [DOI] [PubMed] [Google Scholar]
- Hsu, C. H., S. H. Wu, D. K. Chang, and C. Chen. 2002. Structural characterizations of fusion peptide analogs of influenza virus hemagglutinin. J. Biol. Chem. 277:22725–22733. [DOI] [PubMed] [Google Scholar]
- Huang, Q., C. L. Chen, and A. Hermann. 2004. Bilayer conformation of fusion peptide influenza virus hemagglutinin: a molecular dynamics study. Biophys. J. 87:14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath, S., and T. C. Wong. 2002. Membrane structure of the human immunodeficiency virus gp41 fusion domain by molecular dynamics simulations. Biophys. J. 83:135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita, K., S. Furuike, and M. Yamazaki. 1998. Intermembrane distance in multilamellar vesicles of phosphatidylcholine depends on the interaction free energy between solvents and the hydrophilic segments of the membrane surface. Biophys. Chem. 74:237–249. [DOI] [PubMed] [Google Scholar]
- Kleiger, G., R. Grothe, P. Mallick, and D. Eisenberg. 2002. The GXXXG and AXXXA: common α-helical interaction motifs in proteins, particularly in extremophiles. Biochemistry. 41:5990–5997. [DOI] [PubMed] [Google Scholar]
- Kleinjung, J., F. Fraternali, S. R. Martin, and P. M. Bayley. 2003. Thermal unfolding simulations of apo-calmodulin using leap-dynamics. Proteins. 50:648–656. [DOI] [PubMed] [Google Scholar]
- Lau, W. L., D. Ege, J. D. Lear, D. Hammer, and W. F. De Grado. 2004. Oligomerization of fusogenic peptides promotes membrane fusion by enhancing membrane destabilization. Biophys. J. 86:272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lear, J. D., and W. F. De Grado. 1987. Membrane binding and conformational properties of peptide representing the amino terminus of influenza virus HA2. J. Biol. Chem. 262:6500–6505. [PubMed] [Google Scholar]
- Li, Y., X. Han, and L. Tamm. 2003. Thermodynamics of fusion peptide-membrane interactions. Biochemistry. 42:7245–7251. [DOI] [PubMed] [Google Scholar]
- Lin, K. X., J. Kleinjung, W. R. Taylor, and J. Heringa. 2003. Testing homology with CAO: a contact-based Markov model of protein evolution. Comput. Biol. Chem. 27:93–102. [DOI] [PubMed] [Google Scholar]
- Lins, L., Charloteaux, B., Thomas, A., and R. Brasseur. 2001. Computational study of lipid-destabilising protein fragments: towards a comprehensive view of tilted peptides. Proteins. 44:435–447. [DOI] [PubMed] [Google Scholar]
- MacKenzie, K. R., J. H. Prestegard, and D. M. Engelman. 1997. A transmembrane helix dimer:structure and implications. Science. 276:131–133. [DOI] [PubMed] [Google Scholar]
- Macosko, J. C., C. Kim, and Y. Shin. 1997. The membrane topology of the fusion peptide region of influenza hemagglutinin determined by spin-labeling EPR. J. Mol. Biol. 267:1139–1148. [DOI] [PubMed] [Google Scholar]
- Massi, F., J. W. Peng, J. P. Lee, and J. E. Straub. 2001. Simulation study of the structure and dynamics of the Alzheimer's amyloid peptide congener in solution. Biophys. J. 80:31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata, M., Y. Sugahara, S. Takahashi, and S. Ohnishi. 1987. pH-dependent membrane fusion activity of a synthetic twenty amino acid peptide with the same sequence as that of the hydrophobic segment of influenza virus hemagglutinin. J. Biochem. 102:957–962. [DOI] [PubMed] [Google Scholar]
- Nobusawa, E., T. Aoyama, H. Kato, Y. Suzuki, Y. Tateno, and K. Nakajima. 1991. Comparison of complete amino acid sequences and receptor-binding properties among 13 serotypes of hemgglutinins of influenza A viruses. Virology. 182:475–485. [DOI] [PubMed] [Google Scholar]
- Qiao, H., R. T. Armstrong, G. B. Melikyan, F. S. Cohen, and J. M. White. 1999. A specific point mutant at position 1 of the influenza hemagglutinin fusion peptide displays a hemifusion phenotype. Mol. Biol. Cell. 8:2759–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ, W. P., and D. M. Engelman. 2000. The GxxxG motif: A framework for transmembrane helix-helix association. J. Mol. Biol. 296:911–919. [DOI] [PubMed] [Google Scholar]
- Seelig, A., and J. Seelig. 1977. Effect of a single cis double bond on the structure of a phospholipid bilayer. Biochemistry. 16:45–50. [DOI] [PubMed] [Google Scholar]
- Seelig, A., and J. Seelig. 1980. Lipid conformation in model membranes and biological membranes. Q. Rev. Biophys. 13:19–61. [DOI] [PubMed] [Google Scholar]
- Seelig, J., S. Nebel, P. Ganz, and C. Bruns. 1993. Electrostatic and nonpolar peptide-membrane interactions. lipid binding and functional properties of somatostatin analogues of charge z=+1 to z=+3. Biochemistry. 32:9714–9721. [DOI] [PubMed] [Google Scholar]
- Shepherd, C. M., H. J. Vogel, and D. P. Tieleman. 2003. Interactions of the designed antimicrobial peptide mb21 and truncated dermaseptin s3 with lipid bilayers: molecular-dynamics simulations. Biochem. J. 370:233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel, J. J., P. M. Bayley, E. B. Brown, S. R. Martin, M. D. Waterfield, J. M. White, I. A. Wilson, and D. C. Wiley. 1982. Changes in the conformation of influenza virus hemmagglutinin at the pH optimum of virus-mediated membrane fusion. Proc. Natl. Acad. Sci. USA. 79:968–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel, J. J., and D. C. Wiley. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69:531–569. [DOI] [PubMed] [Google Scholar]
- Smith, T. F., and M. S. Waterman. 1981. Identification of common molecular subsequences. J. Mol. Biol. 147:195–197. [DOI] [PubMed] [Google Scholar]
- Steinhauer, D. A., S. A. Wharton, J. J. Skehel, and D. C. Wiley. 1995. Studies of the membrane fusion activities of fusion peptide mutants of influenza virus haemagglutinin. J. Virol. 69:6643–6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamm, L. 2003. Th. Biochim. Biophys. Acta. 1614:14–23. [DOI] [PubMed] [Google Scholar]
- Taylor, W. R. 1998. Dynamic sequence databank searching with templates and multiple alignment. J. Mol. Biol. 280:375–406. [DOI] [PubMed] [Google Scholar]
- Thomas, D. J. 1991. A simplified mechanical model of proteins tested on the globin fold. J. Mol. Biol. 222:805–817. [DOI] [PubMed] [Google Scholar]
- Thomas, D. J. 1994. The graduation of secondary structure elements. J. Mol. Graph. 12:146–152. [DOI] [PubMed] [Google Scholar]
- Tieleman, D., M. Sansom, and H. Berendsen. 1999. Alamethicin helices in a bilayer and in solution: Molecular dynamics simulations. Biophys. J. 76:40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccaro, L., K. J. Cross, S. A. Wharton, J. J. Skehel, and F. Fraternali. 2004. Aspects of the fusogenic activity of influenza haemmagglutinin peptides by molecular dynamics simulations. In Viral Membrane Proteins: Structure, Function and Drug Design. W. B. Fischer, editor, Kluwer Academic Plenum, Dordrecht, The Netherlands.
- van Gunsteren, W., S. Billeter, A. Eising, P. Hünenberger, P. Krüger, A. Mark, W. Scott, and I. Tironi. 1996. Biomolecular Simulations: The GROMOS96 Manual and User Guide. BIOMOS b.v., Zürich, Groningen.,
- Vriend, G. 1990. WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 8:52–55. [DOI] [PubMed] [Google Scholar]
- Wharton, S. A., S. R. Martin, R. W. Ruigrok, J. J. Skehel, and D. C. Wiley. 1988. Membrane fusion by peptide analogues of influenza virus haemagglutinin fusion. J. Gen. Virol. 69:1847–1857. [DOI] [PubMed] [Google Scholar]
- Wieprecht, T., and J. Seelig. 2002. Isothermal titration calorimetry for studying interactions between peptides and lipid membranes. In Peptide-Lipid Interactions. S. A. Simon and T. J. McIntosh, editors. Academic Press, New York. 31–56.
- Wilson, I. A., J. J. Skehel, and D. C. Wiley. 1981. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3.0 Å resolution. Nature. 289:366–373. [DOI] [PubMed] [Google Scholar]
- Wong, T. C. 2003. Membrane structure of the human immunodeficiency virus gp41 fusion peptide by molecular dynamics simulations. II The glycine mutants. Biochim. Biophys. Acta. 1609:45–54. [DOI] [PubMed] [Google Scholar]
- Wu, C. W., S. F. Cheng, W. N. Huang, V. D. Trivedi, B. Veeramuthu, B. K. Assen, W. G Wu, and D. K. Chang. 2003. Effects of the alterations of the amino-termina; glycine of influenza hemagglutinin fusion peptide on its structure, organization and membrane interactions. Biochim. Biophys. Acta, 1612:41–51. [DOI] [PubMed] [Google Scholar]
- Zhelev, D. V., N. Stoicheva, P. Scherrer, and D. Needham. 2001. Interaction of synthetic HA2 influenza fusion peptide analog with model membranes. Biophys. J. 81:285–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Z., J. C. Macosko, D. W. Hughes, B. G. Sayer, J. Hawes, and R. M. Epand. 2000. 15N NMR study of the ionization properties of the influenza virus fusion peptide in zwitterionic phospholipid dispersions. Biophys. J. 78:2418–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]