

Abstract
We have demonstrated that globular proteins, such as hen egg lysozyme in phosphate buffered saline at room temperature, lose native structural stability and activity when adsorbed onto well-defined homogeneous solid surfaces. This structural loss is evident by α-helix to turns/random during the first 30 min and followed by a slow α-helix to β-sheet transition. Increase in intramolecular and intermolecular β-sheet content suggests conformational rearrangement and aggregation between different protein molecules, respectively. Amide I band attenuated total reflection/Fourier transformed infrared (ATR/FTIR) spectroscopy was used to quantify the secondary structure content of lysozyme adsorbed on six different self-assembled alkanethiol monolayer surfaces with –CH3, –OPh, –CF3, –CN, –OCH3, and –OH exposed functional end groups. Activity measurements of adsorbed lysozyme were in good agreement with the structural perturbations. Both surface chemistry (type of functional groups, wettability) and adsorbate concentration (i.e., lateral interactions) are responsible for the observed structural changes during adsorption. A kinetic model is proposed to describe secondary structural changes that occur in two dynamic phases. The results presented in this article demonstrate the utility of the ATR/FTIR spectroscopic technique for in situ characterization of protein secondary structures during adsorption on flat surfaces.
INTRODUCTION
Protein stability and activity are essential for use in various medical and analytical devices, such as biosensors, biocatalytic chips, biomaterials for implants, drug delivery vehicles, tissue engineering, and affinity chromatography. Unfolding of a native protein is likely the first step in the conversion of a soluble protein into β-sheet-rich structured aggregates such as amyloid and prion deposits (Booth et al., 1997; Kelly, 1998; Dobson, 1999). How proteins unfold and aggregate when adsorbed at a solid-liquid interface in an aqueous environment has been the focus of recent experimental and theoretical studies (Norde and Zoungrana, 1998; Adams et al., 2002; Castells et al., 2002; Latour and Rini, 2002; Buijs et al., 2003; Ostuni et al., 2003; Engel et al., 2004; Sethuraman et al., 2004b). In the adsorbed state, protein residues are involved in nonnative interactions with exposed functional groups at the solid surface interface and hence are likely to be perturbed. Proteins can unfold or adopt a nonnative conformation at hydrophobic interfaces without incurring the energy penalty of exposing the interior hydrophobic residues to water. They are stabilized due to interaction with the exposed hydrophobic groups on the solid surface. Chaperone molecules are known to assist in protein folding and may use this hydrophobic-induced stability to advantage.
Protein aggregation is a well-studied phenomenon in solution (Dong et al., 1995, 2000; Price et al., 1999). Understanding the mechanism of protein aggregation is critical in determining whether higher-ordered aggregate structure can be formed (Stefani and Dobson, 2003). The aggregated state could be an intermediate in a folding pathway leading to highly ordered fibrillar structures (Stefani and Dobson, 2003; Dobson, 2003b; Caughey and Lansbury, 2003). Hence the mechanism of structural changes during protein adsorption is relevant and how these might induce aggregation and protein-protein interactions on the surface leading to the formation of further nonnative structures is worth pursuing.
It is difficult to obtain high-resolution x-ray structural information for the adsorbed proteins. Since the usual method for assessing secondary structure in aqueous solution, circular dichroism, is not easily adaptable to adsorbed proteins on solid surfaces, we have used a quantitative approach based on amide I band attenuated total reflection/Fourier transformed infrared (ATR/FTIR) spectroscopy (Vedantham et al., 2000). FTIR spectroscopy is a surface-sensitive technique that has been used to investigate protein structure. Each secondary structural element (helix, sheet, turns, and random structures) gives rise to characteristic stretching frequencies in the amide I region (1700–1600 cm−1). Hence, quantitative structural information is obtained from the band position and relative band areas. Extent of aggregation is characterized by the appearance of nonnative spectral bands around 1627–1622 cm−1 representative of intermolecular antiparallel β-sheet motifs (Dong et al., 1995, 2000; Zurdo et al., 2001; Sokolowski et al., 2003; Militello et al., 2004). It is difficult to predict the orientation of proteins on surfaces using FTIR; hence, other studies using complementary techniques have been conducted in this regard (Tronin et al., 2002; Daly et al., 2003).
Here we investigate the structural perturbation, aggregation, and activity of hen egg lysozyme on a series of well-defined homogeneous self-assembled monolayer (SAM) surfaces (alkanethiols on gold) with uncharged exposed functional groups –CH3, –OPh, –CF3, –CN, –OCH3, –OH, –CONH2, and –EG3OH surfaces. The surfaces used in this study are SAMs, with the gold layers deposited on “track-etched” poly(carbonate) (PCTE) membranes. Henceforth, these surfaces are referred to as SAM surfaces. The enzyme, hen egg lysozyme, has not been shown to form fibrils in vivo, and only mutants have been associated with amyloid-related diseases (Pepys et al., 1993). Wild-type lysozyme can form fibrils in vitro by incubating in pH 2.0 at 65°C for 2 weeks (Krebs et al., 2000). The formation of fibrils involves a conformation transition from α-helix to β-sheet (Fändrich et al., 2001; Caughey and Lansbury, 2003). Lysozyme is a stable protein and does not aggregate in solution even at higher bulk concentrations (up to 50 mg/ml) because of its four stabilizing disulfide bonds. We have shown earlier that lysozyme undergoes an α-helix to β-sheet transition during adsorption onto a series of heterogeneous polymeric surfaces and that lysozyme unfolds first very fast and later very slowly, i.e., orders of magnitude slower on a fluorinated hydrocarbon surface than in solution (Sethuraman et al., 2004b). For comparison purposes, we also report on lysozyme activity while adsorbed on a series of synthetic polymeric nanoporous surfaces.
KINETIC MODEL
Based on our earlier work with lysozyme adsorption onto polymeric surfaces, we now extend our earlier model to describe the kinetics of secondary structural changes for adsorbed hen egg lysozyme on the SAM surfaces. The protein secondary structure is classified as five independent structures, namely α-helix, intramolecular β-sheet, turns, random, and intermolecular β-sheet. Intermolecular β-sheet structure is a common secondary structural element in the aggregated state (Dong et al., 1995, 2000; Martinez et al., 1996; Kubelka and Keiderling, 2001), these structures are hydrogen-bonded antiparallel β-sheets with strong characteristic bands in the amide I region of the infrared spectrum. Fractional concentration of a secondary structure is the fraction of total amino acids forming a particular secondary structure such as helix, intrasheet, turns, random, or intersheet. Here the model follows the time profile of the fractional concentrations and predicts that α-helices (α) convert to intermediate structural components (I: turns and random) reversibly upon adsorption. The intermediate state could further form irreversibly intramolecular β-sheets (βintra, intra) and intermolecular β-sheet (βinter, inter) contacts between different protein molecules leading to aggregates. Also, we have used time-dependent kinetic rate constants to describe the secondary structural transition kinetics of adsorbed proteins.
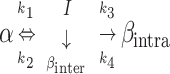 |
(1) |
where
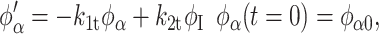 |
(2) |
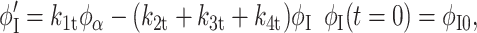 |
(3) |
 |
(4) |
 |
(5) |
where φα, φI, φβintra, and φβinter are fractional concentrations of ordered helix, turns, and random structure (together termed as intermediate), intramolecular β-sheet, and intermolecular β-sheet, respectively. The rate constants, kit = ait + bi, with i = 1, 2, 3, and 4, are assumed to be linearly time-dependent, and ai and bi are constants obtained from the fit. k1, k2, k3, and k4 are rate constants for the loss of fractional α-helix (α), the gain of fractional α-helix from intermediate (I), the loss of intermediate or formation of intramolecular β-sheet (βintra), and the loss of intermediate or formation of intermolecular β-sheet (βinter), respectively. The β-sheet states (intra and inter) are assumed to be irreversible i.e., there is no formation of α-helix from β-sheet secondary structures. φi's are first differentials of the fractional concentrations φi with respect to time, t. φi0 is the initial concentration for each species. The intermediate fractional concentration φI is assumed to be equal to the total of the fractional concentrations of turns and random. Equations 2–5 were solved numerically using the Runge-Kutta method. The kinetic model was fit to the experimental data by using least squares fit. The low amount of protein adsorbed on –CONH2 and –EG3OH SAM surfaces did not allow for high-quality spectral data for structural analysis.
EXPERIMENTAL
Materials
All materials and reagents were used as received. Alkanethiols, 1-undecanethiol and 11-mercapto-1-undecanol were purchased from Sigma-Aldrich Chemicals (Milwaukee, WI). The remaining alkanethiols 11-Phenoxy-1-mercaptoundecane, 1,1,2,2-tetrahydrofluoro-1-dodecanethiol, 11-cyano-1-undecanethiol, 11-methoxy-1-undecanethiol, 1-mercaptoundec-11-yl tri ethylene glycol, 11-mercaptoundecanamide were synthesized according to well-established protocols (Table 1) (Nuzzo et al., 1990; Prime and Whitesides, 1993). Chicken egg white lysozyme (L6876), Micrococcus lysodeikticus cells (ATCC No. 4698, M-3770), acetic acid, Ponceau S, sodium chloride, sodium phosphate monobasic, sodium phosphate dibasic and potassium chloride were obtained from Sigma (St. Louis, MO). PCTE membranes were a gift from Whatman (Clifton, NJ). The membranes used have a nominal pore diameter of 200 nm and contained ∼6 × 108 pores/cm2 of membrane surface area. The PCTE membranes were used as a substrate for the SAMs to increase the surface area, the amount of adsorbed species, and hence the signal/noise ratio. The chemicals SnCl2, AgNO3, Na2SO3, NH4OH, trifluoroacetic acid, formaldehyde, methanol (HPLC grade), and ethanol (HPLC grade) were obtained from Sigma. Commercial gold plating solution of Na3Au(SO3)2 (Oromerse Part B, Technic, Na3Au(SO3)2 concentration is 7.9 × 10−3 M) was used after dilution in water (40 times). Regenerated cellulose (RC), polyethersulfone (PES), and poly(vinyl pyrrilidone)-modified PES (pvp-PES) membranes were gifts from Pall-Fitron (East Hills, NY). Polyvinylidine difluoride (PVDF) and polysulfone (PS) membranes were gifts from Danish Separation Systems, A/S (Nakskov, Denmark), and polytetrafluoroethylene (PTFE) membranes were a gift from W. L. Gore and Associates (Elkton, MD). (Table 2).
TABLE 1.
Roughness-corrected sessile drop contact angles of water on SAMs under cyclooctane and air
| Functional group | Alkanethiol | θCO* (deg) | cos θCO | θair† (deg) | cos θair |
|---|---|---|---|---|---|
| –CH3 | HS(CH2)10CH3 | 160 ± 2 | −0.93 | 108 ± 4 | −0.31 |
| –OPh | HS(CH2)11OPh | 157 ± 2 | −0.92 | 80 ± 4 | 0.17 |
| –CF3 | HS(CH2)2(CF2)9CF3 | 150 ± 3 | −0.86 | 110 ± 4 | −0.34 |
| –CN | HS(CH2)11CN | 141 ± 2 | −0.77 | 58 ± 3 | 0.53 |
| –OCH3 | HS(CH2)11OMe | 110 ± 2 | −0.34 | 75 ± 4 | 0.26 |
| –OH | HS(CH2)11OH | 60 ± 4 | 0.5 | <15 | >0.97 |
| –EG3OH | HS(CH2)11(OCH2CH2)3OH | 55 ± 3 | 0.57 | 35 ± 4 | 0.82 |
| –CONH2 | HS(CH2)10CONH2 | 23 ± 2 | 0.92 | <15 | >0.97 |
Contact angle values are the mean of at least five measurements.
Contact angles of water drops on surfaces under cyclooctane.
Contact angles of air bubbles on surfaces under water.
TABLE 2.
Description and properties of the nanoporous surfaces (membranes)
| Nanoporous surfaces (membrane) | Symbol | MWCO* (kDa) | Contact angle (θ)† | Wettability cos (θ) |
|---|---|---|---|---|
| Poly(tetrafluoroethylene) | PTFE | unknown | 120 ± 3 | 0.499 ± 0.045 |
| Poly(vinylidine difluoride) | PVDF | 20 | 76 ± 2 | 0.243 ± 0.033 |
| Poly(sulfone) | PS | 20 | 70 ± 2 | 0.343 ± 0.033 |
| Poly(ether sulfone) | PES | 10 | 55 ± 2 | 0.574 ± 0.028 |
| Poly(vinyl pyrolidinone)-poly(ether sulfone) | pvp-PES | 10 | 48 ± 2 | 0.669 ± 0.026 |
| Regenerated cellulose | RC | 10 | 27 ± 2 | 0.891 ± 0.016 |
MWCO. Molecular weight cutoff is defined as that molecular weight (in kDa) of a dissolved solute for which 90% of its mass is retained by a membrane during ultrafiltration.
Sessile captive air bubble contact angle. Atomic force microscope measurements and a zig-zag model of the surface were used to correct for roughness effects according to Taniguchi and co-workers (Taniguchi et al., 2001; Taniguchi and Belfort, 2002). Contact angle values are reported as the mean ± SD of 10 measurements.
Preparation of SAM surfaces
The electroless plating procedure, reported by Martin and co-workers (Jirage et al., 1999), was used to deposit the gold onto the PCTE membranes. The temperature during the deposition process was fixed at 2°C, and pH 10 for the gold solution was used. After gold plating, the membranes were thoroughly rinsed with water and were immersed for 12 h in an aqueous solution of 25% HNO3 and then rinsed with water. The gold-coated membranes were then immersed in solutions of the specified alkanethiols in ethanol (5 mM thiol) for 24 h, rinsed with ethanol, and dried under nitrogen (Chun and Stroeve, 2002). The SAM surfaces were then used for protein adsorption experiments. The alkanethiols synthesized and used in this study are listed in Table 1.
Activity measurements
Enzymatic activity of the adsorbed lysozyme was determined by measuring the decrease in turbidity of the Micrococcus lysodeikticus suspension at 450 nm (Bower et al., 1998). The cells were suspended in 10 mM phosphate-buffered saline (PBS), pH 7.4, and incubated at 25°C for 2 h. The absorbance of the mixture was adjusted by dilution with PBS to obtain an optical density of 1.16 ± 0.05 at 450 nm. For each activity assay, the SAM or polymeric surface, with total adsorbed protein amount of 0.8 ± 0.1 μg, was immersed in 3.0 mL of suspension and the decrease in optical density (absorbance) was recorded for different immersion times using a Hitachi U 2000 Double-Beam UV/VIS spectrophotometer (Hitachi Instruments, Danbury, CT). The total surface area of the surface immersed in the 3.0 mL suspension varied with each surface, as the amount of protein adsorbed varied depending on the surface chemistry. The slopes were determined from the initial linear region of each curve. All assays were repeated three times using different samples.
ATR/FTIR spectral analysis algorithm and measurements
A holistic spectral analysis approach (Vedantham et al., 2000) and second-derivative analysis of the FTIR spectra are used here for protein secondary structure estimation. The holistic technique uses amide I band (1700–1600 cm−1) infrared spectra for estimating protein secondary structure. It combines the superposition of reference spectra of pure secondary structural elements with simultaneous aromatic side-chain, water-vapor, and solvent background subtraction. A key element of the technique is the calculation of the reference spectra for ordered helix (called α-helix), sheet (called intramolecular β-sheet), turns, and unordered helix and unordered (called random) structures from a basis set of well-characterized proteins. The spectral analysis technique and data collection were previously described in detail (Vedantham et al., 2000; Sethuraman et al., 2004b). The mean peak positions for Gaussian-Lorentzian band shapes corresponding to these reference spectra and structural motifs were α-helix (1656 cm−1), intramolecular β-sheet (1693 and 1633 cm−1), unordered and unordered helix (1672, 1648, and 1620 cm−1), and turn structures (1720, 1668, 1630, and 1617 cm−1). The deconvoluted spectra for each of these four structural components are easily distinguishable. The fractional concentration for each secondary structure to the total structure was obtained using this algorithm. The estimates for the fractions of turns and unordered helix and random motifs are lumped together because they are the least accurate of the estimates (compared with α-helix and β-sheet). This is possibly because they depend on multiple individual and variable peaks, i.e., α-helix, β-sheet, unordered and unordered helix, and turns structures depend on one, two, three, and four peaks, respectively.
The dominant peaks for the intramolecular-β-sheet motifs at ∼1693 cm−1 and 1633 cm−1 are commonly observed experimentally. There exists the possibility that intermolecular protein contacts could lead to a β-sheet like motif observed at ∼1622 cm−1. The optimization algorithm (used with ATR/FTIR) subsumes this peak as turns and hence we use the second derivative analysis described below to estimate the extent of intermolecular β-sheet content.
The second-derivative analysis of the FTIR spectra was performed as per the method of Dong et al. (1990, 1995). Omnic software (v. 6.1a) from Nicolet (Madison, WI) was used to subtract the background surface, buffer, and water vapor contributions from the protein-adsorbed SAM/Au/membrane spectra and to calculate the second derivative using a nine-point Savitsky-Golay derivative. The remaining spectrum was imported into Origin (v. 7.0) software (Origin Lab, Northampton, MA) where it was baseline-corrected and area-normalized. All the second-derivative plots were area normalized (1700–1600 cm−1) for comparison. The intermolecular β-sheet fraction obtained from this analysis was defined as the fraction of area under the peak around ∼1627–1622 cm−1 to the total area under the amide I region 1700–1600 cm−1.
All protein adsorbed onto membranes and protein solution spectra were recorded in their respective aqueous (H2O) buffer solution (Vedantham et al., 2000). The spectra were collected with a Nicolet Magna 550 Series II FTIR spectrometer with a horizontal ATR accessory (Spectra Tech, Shellton, CT). The ATR accessory was a trapezoidal germanium crystal (7.0 × 1.0 cm) with ends cut to 45° generating 12 internal reflections, and was mounted onto a sample trough. The spectrometer was equipped with a liquid nitrogen-cooled mercury cadmium telluride detector. To reduce the contributions of water vapor and carbon dioxide, the IR system was continuously purged with air from an FTIR Purge gas generator (Model 74-45, Balston, Haverhill, MA) at 50 standard ft3/min and supplemented with nitrogen gas from the vent of a liquid nitrogen tank. The adsorbed protein spectra were obtained by clamping the wet surface against the crystal. The crystal was then sealed with parafilm to minimize evaporation during acquisition. All ATR-corrected spectra were collected in the 4000−1000 cm−1 range as sets of 1024 time-averaged, double-sided interferograms with Happ-Genzel apodization. Spectral resolution was set at 2 cm−1 with a gain of 8 and an aperture of 40. After each experiment, the exposed surface of the germanium crystal was cleaned via a five-step process (Vedantham et al., 2000): 1), rinsing with DI water; 2), soaking in a 1% (w/w) SDS solution for 10 min; 3), rinsing thoroughly with DI water; 4), rinsing thoroughly with a 50% (w/w) aqueous ethanol solution; and 5), drying with compressed air filtered through cotton to remove oils and particulates. Amide I band signal/noise (S/N) ratios varied from 600 to 150, whereas amide III band S/N ratios varied from 100 to 20. Amide band S/N ratios were calculated as 2.5 times the maximum intensity of the background-subtracted band divided by 3 times the standard deviation of the intensity between 1850 and 2200 cm−1. FTIR spectra of the cleaned crystal surface indicated absence of protein adsorbed on the surface.
Protein adsorption experiments
Before static adsorption, each surface was thoroughly rinsed in DI water. The protein solutions were filtered through a 0.22-μm microfiltration membrane (Express PES; Millipore Corp, Bedford, MA) to remove large protein aggregates from solution before adsorption (Prime and Whitesides, 1993). Then, static adsorption measurements were conducted by immersing the polymeric or SAM surfaces into 5 ml of protein solution containing 16 mg/ml in 10 mM PBS at pH 7.4 and allowed to incubate at 25°C. The protein-adsorbed surfaces were only used for the activity assays. Analyses of the irreversibly adsorbed lysozyme on the SAM surfaces were performed by rinsing membranes with the starting buffer and using ATR/FTIR. For the kinetic experiments, washing of the surfaces and the adsorption procedure were similar to the procedures described above. The SAM surfaces were immersed in the protein solution for time t = 0, 0.5, 1, 5, 10, 24, and 48 h, and then analyzed using ATR/FTIR. All buffers were filtered through 0.22-μm filters before use.
Contact angle
Contact-angle measurements were used to estimate the interfacial energy and wettability of surfaces. The captive bubble method (Taniguchi et al., 2001) was used to measure the contact angle of an air bubble in water and the sessile contact angle was used for a water drop in cyclooctane on solid surfaces (Sigal et al., 1998). For all systems, average values were obtained from multiple contact-angle values (at least five) using an optical system (SIT camera, SIT66, Dage-MTI, Michigan, IN) connected to a video display. Corrections of the contact angles for roughness were obtained using the methods of Taniguchi and co-workers (Taniguchi et al., 2001; Taniguchi and Belfort, 2002). Following Sigal et al. (1998), the wettability is defined as the cosine of the sessile contact angle (−0.97 < cos θ < 0.97).
Dye-binding analysis
For dye-binding analysis (Ulbricht et al., 1996), the SAM/gold membranes with adsorbed lysozyme were immersed for 1 h in a solution of Ponceau S (20 g/l, in water with 30% (w/v) trichloroacetic acid and 30% (w/v) sulfosalicylic acid), then washed three times with water, immersed for 1 h into 5% (v/v) acetic acid and again washed three times. Then, the protein-dye complex was qualitatively eluted with 3 mL of 100 mM NaOH solution (1 h). The surfaces were removed, the solutions were neutralized by addition of 50 ml of 6M HCl, and the absorbance of the red solutions was measured at 515 nm. See Ulbricht et al. (1996) for additional details.
Lysozyme secondary structure
Hen egg white lysozyme comprises two structural domains; an α-domain consisting of four α-helices and a C-terminal 310 helix, and a β-domain consisting of a triple-stranded antiparallel β-sheet, a 310 helix, and a long loop. A short double-stranded antiparallel β-sheet links the two domains, as does one of the four-disulphide bridges. The active site lies between the two domains. The protein is known to be structurally robust in solution (Arai and Norde, 1990). Lysozyme is a component of tears and mucous and prevents bacterial infection. It disrupts bacteria by hydrolyzing the cell walls. The secondary structure contents of lysozyme in PBS buffer such as α-helix, β-sheet, turns, and random structures as determined with ATR/FTIR (Vedantham et al., 2000) are 0.40, 0.07, 0.40, and 0.13, respectively, and with x-ray (Frishman and Argos, 1995) are 0.46, 0.07, 0.42, and 0.05, respectively. Thus, for wild-type lysozyme in free solution, the α–helix content is ∼40% (by ATR/FTIR) and the intra β-sheet content is ∼7%. Lysozyme has a low dipole moment and approaches the surface in a more random orientation (Norde, 2003) relative to proteins with large dipole moments such as ribonuclease A which adsorb in a preferred orientation (Lee and Belfort, 1989).
In this study, porous PCTE membranes are used as the base surface, as they can be adapted for ATR/FTIR measurements in the wet state and also allow larger surface area for protein adsorption. Hard surfaces like glass or silicon cannot be used on the ATR crystal, whereas transmission and grazing-angle modes only allow for dry flat surface measurements.
RESULTS
Surface effects on secondary structure
To probe the secondary structural changes of adsorbed lysozyme, a series of self-assembled monolayer of alkanethiol on Au/PCTE membranes were selected as adsorbents (Fig.1) (Table 1). Following Sigal et al. (1998), we have used cos θ as a measure of the wettability of the surfaces, where θ is the sessile contact angle between a cyclooctane drop and the different surfaces in water and is corrected for roughness (Taniguchi et al., 2001; Taniguchi and Belfort, 2002). First we characterize the lysozyme loading onto different surfaces. In most cases, proteins adsorb with a greater affinity onto hydrophobic surfaces than onto hydrophilic surfaces. The adsorbed amount of lysozyme, estimated using the Ponceau S dye binding technique (Ulbricht et al., 1996), was measured for eight surfaces (Fig. 2). The amount of lysozyme adsorbed, Γads, after 48 h on each of the surfaces decreased with increasing surface wettability (cos θ). Thus, the more hydrophobic surfaces (–CH3, –OPh, –CF3, –CN) adsorbed the highest amount of lysozyme (∼2–3 three calculated adsorbed layers, based on the dimension of lysozyme, 4.5 × 3.0 × 3.0 nm), whereas the most wettable surfaces (–OH,–CONH2, and –EG3OH) adsorbed the least amount of lysozyme (less than an adsorbed monolayer and indistinguishable among these surfaces) during this period. SAM –OPh surface exhibits maximum adsorption, most probably due to additional π-π interactions.
FIGURE 1.

SAM/gold/PCTE surfaces for protein adsorption. Schematic representation of a straight-through pore model of the 200-nm mean pore size PCTE membrane. Hen egg lysozyme was adsorbed onto SAMs of alkanethiols with chosen functional groups (X) on gold, deposited by electroless plating. The protein's secondary structure was interrogated by ATR/FTIR.
FIGURE 2.

Amount of lysozyme adsorbed for 48 h onto different SAM surfaces versus surface wettability (cos θco, where θco is the sessile contact angle of a cyclooctane drop placed onto the SAM in water).
The structural stability of lysozyme can be determined through its catalytic activity. The activity of adsorbed lysozyme after 48 h was assayed on different SAM surfaces (Table 1) and a series of synthetic polymeric nanoporous surfaces (Table 2) by measuring the initial slope of the decrease in turbidity of an M. lysodeikticus suspension (Fig. 3 a, inset). Activity is plotted against wettability for each SAM surface in Fig. 3 a. Except for the abnormally low activity of adsorbed lysozyme on the –CN surface, activity increases with increasing wettability. Lysozyme displays nearly native activity on the hydrophilic surfaces (–OH,–CONH2, and –EG3OH). As can be seen in Fig. 3 b, lysozyme loses most of its activity while adsorbed onto the –CH3 and –CN surfaces and this decreases with increasing surface coverage. For the polymeric surfaces, lysozyme loses 20% of its native activity while adsorbed onto regenerated cellulose, a well-known hydrophilic surface, and exhibits almost complete loss of activity on PTFE. The results imply that, in addition to surface chemistry, protein-protein lateral interactions need to be considered.
FIGURE 3.

Activity of adsorbed lysozyme at 16 mg/ml in PBS buffer, pH 7.4 and 25°C after 48 h. (a) Lysozyme activity (solid diamonds) (initial linear slopes, see inset) versus wettability of the SAM surfaces. The decrease in turbidity of the Micrococcus suspension (at 450 nm) was monitored to measure the activity of lysozyme in solution and after adsorption onto the eight different SAM surfaces. The solid line is a third-order polynomial fit (y = 34.171x3 – 35.631x2 + 14.081x + 86.926, R2 = 0.970) for the activity excluding the –CN data point. (Inset) Initial linear region of lysozyme activity on different SAM surfaces. The solid lines are linear fits (y = ax + b) for the activity on the different surfaces. The values of a, b, and R2 for lysozyme in free solution and adsorbed onto –CONH2, –EG3OH, –OH, –OCH3, –CF3, –OPh, –CH3, and –CN surfaces are (−0.0038, 1.159, 0.999), (−0.0036, 1.145, 0.975), (−0.0035, 1.161, 0.995), (−0.0035, 1.155, 0.967), (−0.0029, 1.160, 0.988), (−0.0015, 1.154, 0.975), (−0.0013, 1.158, 0.988), (−0.0003, 1.161, 0.989), and (−0.0001, 1.160, 0.967), respectively. (b) Lysozyme activity adsorbed onto eight SAM (solid diamonds) and six polymeric (solid squares) surfaces versus the amount of lysozyme adsorbed. The dashed vertical line indicates flat-on close-packed monolayer coverage (estimated using lysozyme dimensions of 4.5 × 3.0 × 3.0 nm) at 3.6 mg/m2.
To investigate the secondary structure of the adsorbed protein, we have analyzed the data using two different techniques, 1), second-derivative analysis to locate peaks due to secondary structural components (Stefani and Dobson, 2003); and 2), a holistic-optimization ATR/FTIR (Vedantham et al., 2000) approach to calculate secondary structure fractions of the adsorbed proteins. All experiments were conducted in 1H2O solutions. In Fig. 4 a, raw spectra for the amide I region for lysozyme in solution and adsorbed on each of the six different SAM surfaces are presented. The second-derivative analysis (Fig. 4 b) indicates the presence of nonnative intramolecular antiparallel β-sheet structures of lysozyme on two hydrophobic surfaces (1633 cm−1 for –CH3; 1694 cm−1 for –OCH3) after adsorbing for 48 h. Lysozyme in free solution and on the –OH surface show similar second-derivative peaks, confirming that the hydrophilic surface, which adsorbs the least amount of protein (Fig. 2), does not strongly perturb the protein structure. The intensity of the second-derivative peaks around 1656 cm−1 in Fig. 4 b for lysozyme adsorbed on –OCH3 and –CH3 are lower than that in free solution and both peaks are also shifted to lower wavenumbers. This indicates a decrease in helical content and a shift toward random structure (∼1650–1640 cm−1). The peaks at 1627 cm−1 and 1622 cm−1 indicate the formation of intermolecular contacts (aggregation) on the hydrophobic surfaces (–OCH3 and –CH3, respectively). In solution, even after 48 h, lysozyme remained a monomer as confirmed by dynamic light-scattering measurements (data not shown). Intermolecular sheet fraction on different surfaces (lysozyme aggregation), obtained from the area under the second-derivative peak (∼1627–1622 cm−1), is plotted in Fig. 5. The wavenumber location for the second-derivative peaks does not occur at the same value for lysozyme adsorbed on different surfaces, as IR absorbances are affected by the local environment of the different structural groups. As expected, the extent of aggregation decreased with increasing wettability (Fig. 5), also indicating that surfaces that strongly perturb the native protein structure also aid in protein aggregation. Also, aggregation increases as more adsorbed protein molecules crowd onto the hydrophobic surfaces (Fig. 5, inset). The kinetics of aggregation on the five different SAM surfaces is described below.
FIGURE 4.

ATR/FTIR spectroscopy of hen egg lysozyme in PBS and adsorbed from PBS at 25°C for 48 h. (a) Superimposed ATR/FTIR spectra (absorbance scale arbitrary) for lysozyme in free solution (PBS) (#1) and lysozyme adsorbed for 48 h from PBS onto six different surfaces (–OH, –OCH3, –CN, –CF3, –OPh, and –CH3, numbers 2–7, respectively; see Table 1). (b) Superimposed second derivative of ATR/FTIR spectra (all spectra were area normalized from 1700−1600 cm−1) of lysozyme in free solution (thin solid line) and adsorbed onto –OH (dot-dashed line), –OCH3 (shaded line), and –CH3 (thick solid line) after 48 h. The initial free-solution lysozyme concentration was 16 mg/ml in PBS at 25°C for all experiments.
FIGURE 5.

Fraction of intermolecular β-sheet of adsorbed lysozyme obtained from the second derivative peak area (solid circles) (1627–1622 cm−1) for lysozyme adsorbed onto six different SAM surfaces versus the wettability at 16 mg/ml in PBS buffer, pH 7.4, and 25°C after 48 h. (Inset) Fraction of second-derivative peak area (solid squares) (1627–1622 cm−1) for lysozyme adsorbed onto six different SAM surfaces versus the amount of lysozyme adsorbed.
Secondary structure transition kinetics
Adsorption of proteins from aqueous solution onto a solid surface is the net result of hydrophobic, steric, and electrostatic interactions between the protein residues, surface functional groups, and water. Rearrangements of the adsorbed protein structure are the result of a delicate balance between these interactions. Exploring the mechanism of adsorbed protein unfolding is important for understanding the principles of protein stability. The results using the ATR/FTIR algorithm and second-derivative analysis to estimate the secondary structural components of lysozyme in Fig. 6 show the kinetics of a secondary structural conformational transition from α-helix to inter and intra β-sheet and turns/random for lysozyme. From the data presented in Fig. 6, it is clear that the unfolding of adsorbed lysozyme is a complicated process containing a thermodynamically stable intermediate. As reported previously for adsorbed lysozyme, there are two unfolding regimes on SAM surfaces: 1), short time period (0–1 h), where there is a loss of alpha helix and gain in turns and random content, without change in the β-sheet content; and 2), long time periods (1–48 h), which includes gain in (intra and inter) β-sheet content. The kinetic modeling presented here follows the kinetics of change for the long time period (1–48 h). The boundary value problem (Eqs. 2–5) was solved numerically and is displayed in Fig. 6. The initial values, i.e., at t = 0 for the model fit, are the secondary structure fractional concentrations of the adsorbed species and are chosen as the first measurable data point, t = 30 min. The initial values (starting at 30 min into the kinetic experiment) of φα0, φI0, φβinter0, and φβintra0 for –CH3, –OPh, –CF3, –CN, –OCH3 were (0.22, 0.69, 0.07, 0.02), (0.22, 0.69, 0.07, 0.02), (0.24, 0.68, 0.06, 0.02), (0.23, 0.68, 0.07, 0.02), and (0.32, 0.60, 0.07, 0.01), respectively. The transition from the native solution structure to t = 30 min on the surface is difficult to measure, and hence, this transition has not been modeled. The data for the intermolecular β-sheet (φβinter) was obtained from the second-derivative analysis, and data for the helix (φα), intramolecular β-sheet (φβintra), and intermediate (turns/random) (φI) was obtained from the ATR/FTIR spectral analysis algorithm. Since the fraction of turns/random also includes intermolecular β-sheet (see ATR/FTIR spectral analysis algorithm and measurements), the latter fraction was subtracted to obtain pure turns/random content. Details of the structural changes during the initial time period (<30 min) were difficult to observe due to the low amount of adsorbed protein on the surface (low S/N ratio). However, after 1 h, one could clearly observe that the fractional α-helix content had dropped precipitously from 0.4 to ∼0.23 for a loss of ∼43%, whereas the fractional intermediate (turns and random) content had increased by a similar amount from 0.53 to ∼0.70 for a gain of ∼32% and the intramolecular β-sheet fractional content did not perceptibly change (it remained at 0.07). The expressions for the rate constants k1, k2, k3, and k4, from the model fit are presented in the legend of Fig. 6 as a function of time for the different surfaces. The model fits the data well, especially since it simultaneously predicts four fractional concentrations for five different surfaces. The 2-norm squared of the residual for the –CH3, –OPh, –CF3, –CN, and –OCH3 SAM surfaces was 2.60 × 10−3, 8.50 × 10−3, 1.46 × 10−3, 2.50 × 10−3, and 1.50 × 10−3, respectively.
FIGURE 6.

Fraction of secondary structure components of adsorbed native lysozyme versus time. Ordered helix (α-helix, solid squares), intermolecular sheet (β-sheet, solid diamonds), intramolecular sheet (β-sheet, solid circles), and turns/random (solid triangles) for lysozyme adsorbed onto different SAM surfaces in PBS buffer at pH 7.4 and 25°C for 48 h. The four solid lines in each figure are the model fits that use the experimental value at t = 30 min as the initial value (i.e., t = 0). The rate constants for the best fit were (a) CH3: k1 = 2.17 × 10−3t + 0.453; k2 = −0.36 × 10−3t + 0.143; k3 = −4.27 × 10−4t + 0.014; k4 = 0.31 × 10−3t + 0.010; and R2 = 0.997. (b) OPh: k1 = 41.6 × 10−3t + 0.443; k2 = 10.3 × 10−3t + 0.176; k3 = −7.70 × 10−4t + 0.025; k4 = −0.44 × 10−3t + 0.014; and R2 = 0.989. (c) CF3: k1 = 34.2 × 10−3t + 0.390; k2 = 5.54 × 10−3t + 0.135; k3 = -9.70 × 10−4t + 0.028; k4 = −0.26 × 10−3t + 0.008; and R2 = 0.960. (d) CN: k1 = 32.7 × 10−3t + 0.356; k2 = 2.28 × 10−3t + 0.120; k3 = -4.79 × 10−4t + 0.015; k4 = −0.17 × 10−3t + 0.006; and R2 = 0.992. (e) OCH3: k1 = 2.6 × 10−5t + 0.394; k2 = −0.70 × 10−4t + 0.175; k3 = −1.72 × 10−4t + 0.005; k4 = −1.17 × 10−4t + 0.004; and R2 = 0.998. Time, t, is in h.
DISCUSSION
The helix-coil transition (α→I) for small peptides in solution is very fast and of the order of 20–180 ns, according to the modeling and experimental literature (Munoz et al., 1997; Williams et al., 1996; Eaton et al., 2000; Hummer et al., 2001; Kauffmann et al., 2001), whereas the folding and unfolding of β-hairpins take ∼6 μs (Munoz et al., 1997). Williams et al. (1996), found that “the characteristic timescale of helix formation would appear to be some three orders of magnitude faster than the characteristic timescale of intramolecular tertiary contact formation” (in free solution). Restraining a protein on a surface should slow down or restrict the formation rate of β-sheet structures as observed here. Thus, during the adsorption of native lysozyme on the hydrophobic SAM surfaces, 1), a large fraction of the α-helices transform into turns and random structures within 1 h with minimum β-sheet formation; 2), then, after 1 h, there is a direct slow transition from α-helix to intramolecular β-sheet; and 3), the fraction of intermolecular sheet increases with time (1–48 h).
Insight could be gained on the behavior of lysozyme during the adsorption process by recent experimental and theoretical work on its folding (Matagne and Dobson, 1998) and unfolding (Williams et al., 1997) in solution. Williams et al. (1997) have shown using a molecular-dynamics procedure in which water molecules are artificially inserted into cavities during unfolding, that the first rate-limiting transition corresponds to the “disruption of the tertiary contacts within the protein, which decouples its domains” and that “subsequently, the helical domain slowly loses its compactness and the helices fluctuate rapidly. The protein then adopts a ‘molten globule-like’ structure in which the native β-sheet is essentially in tact.” We observe a loss of helix during the first hour of adsorption (or at very low surface concentrations) from 40% in solution to 30%, 21%, 21%, 21%, and 21% when adsorbed onto –OCH3, –CH3, –OPh, –CF3, and –CN SAM surfaces, respectively, for 1 h (Fig. 7 a), while maintaining the intramolecular β-sheet structure, which changed from 7% native to 7%, 8%, 9%, 8%, and 7%, respectively, on the same surfaces. This result is similar to that predicted by Williams et al. (1997) where they state that “only some of the secondary structure is completely stable (for their case 90% of the β-sheet and 40% of the helix).” We then observe a slow increase in the intramolecular β-sheet and intermolecular β-sheet content (1–48 h). Thus, our early phase results are consistent with the predicted behavior from molecular modeling.
FIGURE 7.

Fraction of secondary structure components of adsorbed native lysozyme versus time at 16 mg/ml in PBS buffer, pH 7.4 and 25°C for –OH (−), –OCH3 (solid circles), –CN (solid squares), –CF3 (solid triangles), –OPh (solid diamonds), and –CH3 (X) SAM surfaces. (a) Helix fraction versus time for lysozyme adsorbed onto the six different SAM surfaces. (b) Intramolecular sheet fraction versus time for lysozyme adsorbed onto the six different SAM surfaces. (c) Intermolecular sheet fraction versus time for lysozyme adsorbed onto the six different SAM surfaces. The horizontal dashed line indicates the helix and intramolecular sheet fraction in free solution (intermolecular is absent), respectively, for native lysozyme.
The kinetic model presented here follows the rate of secondary structure change of an adsorbed protein on the SAM surfaces for long time periods (1–48 h). The α-helix content of native lysozyme dropped from its native structure value of 40% in solution to 32%, 26%, 15%, 14%, 12%, and 9%, when adsorbed onto –OH, –OCH3, –CH3, –OPh, –CF3, and –CN, respectively, for 48 h (Fig. 7 a). Also during this period, a concomitant increase occurred in the intramolecular β-sheet content, from 7% for the native lysozyme in free solution to 8%, 11%, 19%, 25%, 24%, and 18% (Fig. 7 b). Since protein aggregates formed on a surface could get desorbed and form seeds for further aggregation and adsorption (Tzannis et al., 1996), it is instructive to determine what condition the lysozyme was in (native or aggregated) during the adsorption process on the SAM surfaces. The intermolecular β-sheet content increased from 0% for the native lysozyme to 3%, 5%, 10%, 12%, 8%, and 8% (Fig. 7 c) for the same surfaces, respectively. Lysozyme aggregation on these surfaces reached 70% of its maximum within 5 h. Not all the loss in α-helix content was converted to β-sheet structure, since the remaining secondary structural components (turns and random) also changed from 53% to 55%, 48%, 56%, 65%, and 58%, respectively. Notice that for the polar surface, –OH, the secondary structural content of adsorbed native lysozyme was close to that in free solution (dashed lines in Fig. 7, a and b) and thus lysozyme was only weakly perturbed from its native structure, confirming the results from the activity tests. Sufficiently high S/N ratio of the protein spectra was only obtained on the –OH surface after 48 h adsorption, hence, the three data points for this surface in Fig. 7. The rate of loss of the helical structure and gain in intermolecular and intramolecular β-sheet was the highest for lysozyme on the –OPh SAM surface and the lowest for the least hydrophobic surface (–OCH3). The behavior on the –OPh surface is due to a greater attractive interaction between the protein and –OPh SAM surface. Direct intermolecular adhesion measurements between lysozyme and different functionalized SAM surfaces, using a modified atomic force microscope cantilever, show that “pull-off” or adhesive forces are substantial (∼10 ± 3 mN/m) between lysozyme and hydrophobic surfaces (–CH3, –OPh, –CN, –OCH3, and –CF3), low (∼2.3 ± 0.4 mN/m) between lysozyme and hydrophilic surfaces (–OH, –CONH2, and –ethylene glycol), and exceedingly low between lysozyme and lysozyme (∼0.5 ± 0.2 mN/m) (Sethuraman et al., 2004a). Importantly, it was found that the “pull-off” for lysozyme was the highest on the –OPh surface (13.6 ± 3.1 mN/m). From the model, we also find that the values of the rate constants are in the order k1 > k2 > k3 > k4 for lysozyme, at all times, on all the SAM surfaces. The largest rate constant, k1, is in agreement with the results of Williams et al. (1997), where the helix domain is predicted to be unstable and is the first to unfold, reversibly. The formation of intramolecular β-sheet is a slower process; hence the value of the rate constant k2 is an order of magnitude smaller than that of k1. As the protein surface concentration increases, i.e., more molecules crowd onto the surface, they tend to form intermolecular contacts (aggregates) with a rate constant k4. The value of k4 was found to be the same order of magnitude as that of k3 and smaller than k1 and k2. We assume, in this model, that the structural changes to intramolecular and intermolecular β-sheets are irreversible processes.
From the experimental observations, it is clear that the structural perturbation of lysozyme is the direct result of its interaction with the hydrophobic surface and also the lateral interactions between adjacent lysozyme molecules on the surface. The lateral interactions between native lysozyme molecules in free solution is minimal as their native secondary structure does not change over >70 h (data not shown). To normalize the results, the structural fractional components, fi, which were estimated using the ATR/FTIR spectral analysis algorithm, are plotted against the adsorbed native lysozyme concentration, Γads, in Fig. 8 for the six different SAM surfaces and hence wettabilities (on each surface after 48 h). Since the fractions were estimated using the algorithm, turns, random, and intermolecular sheet are combined as a single fraction. These results are compared with those observed previously for lysozyme adsorbed onto a series of nanoporous surfaces (Sethuraman et al., 2004b), i.e., 1), six different nanoporous polymeric surfaces (Table 2) and hence wettabilities (on each surface after 300 min); 2), nine different lysozyme bulk concentrations, Cs, (on PTFE for 300 min); and 3), 17 different time points (on PTFE at 16 mg/ml). As Γads increases, a secondary structural conformational transition from α-helix to intra and inter β-sheet (and turns/random) is similarly observed, confirming the importance of lateral interactions between adsorbed proteins. The calculated monolayer coverage (dashed vertical line) is shown.
FIGURE 8.

Fraction of secondary structure components (helix, intramolecular β-sheet and turns/random/intermolecular sheet) of adsorbed native lysozyme versus amount of lysozyme adsorbed for 1), six different SAM surfaces at 16 mg/ml (on each surface after 48 h) (solid circles, squares, and triangles); 2), six different polymeric surfaces and hence wettabilities (on each surface after 300 min) (open circles, squares, and triangles); 3), nine different lysozyme bulk concentrations, Cs (on PTFE for 300 min) (shaded circles, squares, and triangles); and 4), 17 different time points (on PTFE at 16 mg/ml) (hatched circles, squares, and triangles). Ordered helix (α-helix, solid, open, shaded, and hatched circles), intramolecular sheet (β-sheet, solid, open, shaded, and hatched squares), and turns/random/intermolecular sheet (open, solid, shaded, and hatched triangles). All of the fractions in this figure were estimated using the ATR/FTIR spectral analysis algorithm. Three horizontal full arrows identify the fractional structural components in free solution for the native lysozyme. The dashed vertical line indicates the flat-on close-packed monolayer coverage (estimated using lysozyme dimensions of 4.5 × 3.0 × 3.0 nm) at 3.6 mg/m2. All measurements are in PBS buffer at pH 7.4 and 25°C.
CONCLUSIONS
This work demonstrates that hen egg lysozyme undergoes structural changes during adsorption onto a series of well-defined homogeneous surfaces. ATR/FTIR was used to follow the secondary structural transition and aggregation on six SAM surfaces (–CH3, –OPh, –CF3, –CN, –OCH3, and –OH). Activity measurements of adsorbed protein were in good agreement with the structural perturbations. The main conclusions of the study are:
A large amount of protein adsorbed on the hydrophobic surfaces (–CH3, –OPh, –CF3, –CN, and –OCH3, ∼2.5–4.5 mg/m2) and a small amount adsorbed on the hydrophilic surfaces (–OH, –EG3OH, and –OCH3, ∼0.1–0.3 mg/m2).
Lysozyme activity dropped with decreasing surface wettability. It showed the least activity on –CN (3% of activity in free solution) and –CH3 (7%) SAM surfaces and the most activity on –CONH2 (95%), EG3OH (92%), and OH (90%) SAM surfaces.
After 48 h of adsorption from PBS, lysozyme exhibited a drop in helical content from 40% in bulk solution to ∼26% on the hydrophobic surfaces. A simultaneous increase in intramolecular β-sheet from 7% to a maximum of 25% occurred on hydrophobic surfaces. There appeared to be a direct conversion of α-helix to β-sheet content (and turns/random) on the different SAM surfaces (Fig. 5).
–CN and –OPh surfaces displayed the largest perturbation of lysozyme for α-helix (Δα = −31%) and β-sheet (Δβ = 18%) structures, respectively.
Lysozyme formed intermolecular β-sheet contacts in the adsorbed state, and the aggregation increased with decreased surface wettability and increased adsorbed protein concentration.
Activity changes (Fig. 3) and fractional conversion of secondary structures (Fig. 8) during adsorption on homogeneous SAMs was qualitatively similar to that on heterogeneous polymer surfaces. An important difference was that the activity of lysozyme adsorbed on regenerated cellulose was well below that for the polar SAMs. These results suggest that synthetic membrane surfaces should emulate homogeneous surfaces to maximally reduce protein adsorption.
An extended kinetic model based on an intermediate species, and which allows for the formation of intramolecular and intermolecular β–sheet, is proposed to describe the secondary structural changes upon adsorption.
Therefore, besides mutagenesis and excursions in pH and temperature, solid surfaces, depending on their surface chemistry and wettability, can perturb the secondary structure of proteins, leading to loss of activity, loss in helical structure, and appearance of alternate structures rich in β-sheets (inter- and intramolecular).
Previous work on surface-induced structural changes using ATR/FTIR (Cheng et al., 1994; Green et al., 1999; Sane et al., 1999) and CD (Giacomelli and Norde, 2003) reports β-sheet-rich structure for globular proteins at interfaces. In earlier work with lysozyme adsorption on hydrophobic chromatography media, using a similar deconvolution method with Raman spectroscopy, β-sheet content increased on adsorption and the greater the loading, the greater was the increase in β-sheet content (Sane et al., 1999). Giacomelli and Norde (2003) have reported a similar result (α-helix to β-sheet transition) with increased loading for a truncated amyloid β-peptide adsorbed onto 100 nm negatively charged PTFE particles using CD. This complements our observations that as the surface becomes more crowded, lateral interactions are also important and it could promote intermolecular β-sheet contacts on the surface. They also report on an increase in helical structures at low surface coverage (Maste et al., 1996; Norde and Zoungrana, 1998). We, however, have not observed any such increase in helical fraction for adsorbed lysozyme. To determine the detailed molecular conformational changes and identify exactly which amino acid and structural motifs are affected by the adsorption, measurements using hydrogen/deuterium exchange and 2-D NMR spectroscopy are needed. (Buijs et al., 2003; Engel et al., 2004).
The mechanism of formation of intermolecular β-sheet is also important in the context of formation of insoluble aggregates related to amyloid- and prion-induced diseases (Dobson, 1999). An important question regarding fibril self-assembly is whether they occur by docking of preformed β-structure or by association of unfolded polypeptides or by both mechanisms. Fändrich and co-workers (2003) have shown that fibril formation for the protein apomyoglobin (a helical protein, no sheet) depends on disordered polypeptide segments and unfavorable conditions for protein folding. This could suggest a similar mechanism for aggregation on a surface, since we have previously shown that the helical and random content normalized by molecular weight correlated well with protein adhesion for seven different globular proteins (Sethuraman et al., 2004a).
The extent of structural perturbation and aggregation could also provide insights for the mechanism of unfolding on the surface and how it might induce formation of highly structured aggregates. Recent studies have indicated that amyloid fibril formation from native proteins occurs via partially folded intermediates that self-assemble to form fibrils (Cohen and Kelly, 2003; Dobson, 2003a). Formation of intermediates is critical as these are capable of strong intermolecular interactions due to solvent exposure of hydrophobic residues which are otherwise buried in the native fold. This has led Dobson (1999, 2003a) to propose that all polypeptide chains can, in principle, form amyloid aggregates under appropriate conditions. It is interesting to compare the results of protein unfolding on surfaces to those of fibril self-assembly in solution, since the kinetic model presented here is also based on a thermodynamically stable intermediate and predicts that there is an increase in the intramolecular β-sheet component. Hen egg-white lysozyme forms fibrillar aggregates in vitro by incubating the protein at pH 2.0 at elevated temperatures (65°C) for 2 weeks (Krebs et al., 2000). It has been recently shown that reduction of the disulfide bonds in lysozyme causes it to form fibrils more readily (Cao et al., 2004). Protein aggregation into amyloid fibrils is a complex phenomenon that could be influenced by many structural and physiochemical properties of the polypeptide chain. It has also been reported that the local unfolding of residues in the β-domain and the C-helix triggers the formation of amyloid fibrils for this protein (Frare et al., 2004).
In our study, the adsorbed proteins on these SAM surfaces were assayed for the presence of fibrils using Congo red dye without success. The spectral analysis confirms the presence of remaining helical content for lysozyme even in the adsorbed state, indicating residual native structure in the aggregated/adsorbed state and that further perturbation is required to form large fibrillar-like aggregates. Longer time periods >48 h could possibly induce formation of fibrils on these surfaces. This is currently being evaluated.
Acknowledgments
We thank Todd Przybycien for the ATR/FTIR spectral analysis algorithm and advice on its use, Helen Evans, Whatman Inc., and Osmonics Inc. (now GE Osmonics, Inc.) for donating the PCTE membranes, and Charles Martin and Pieter Stroeve for suggestions with the electroless gold plating technique. We also thank Ole J. Olsen, Danish Separation Systems A/S (now Alpha Laval AB), Bill Short and Monty Carlisle, W. L. Gore and Assoc., Inc., and Barry Breslau and Michael Heath, Pall-Filtron Corp. (now Pall Corp.), for donating synthetic membranes.
We acknowledge the support of the US Department of Energy (grant No. DE-FG02-90ER14114) and the National Science Foundation (grant No. CTS-94-00610).
References
- Adams, S., A. M. Higgins, and R. A. L. Jones. 2002. Surface-mediated folding and misfolding of proteins at lipid/water interfaces. Langmuir. 18:4854–4861. [Google Scholar]
- Arai, T., and W. Norde. 1990. The behaviour of some model proteins at solid-liquid interfaces 1. Adsorption from single protein solutions. Colloids Surf. 51:1–15. [Google Scholar]
- Booth, D. R., M. Sunde, V. Bellotti, C. V. Robinson, W. L. Hutchinson, P. E. Fraser, P. N. Hawkins, C. M. Dobson, S. E. Radford, C. C. Blake, and M. B. Pepys. 1997. Instability, unfolding and aggregation of human lysozyme variants underlying amyloid fibrillogenesis. Nature. 385:787–793. [DOI] [PubMed] [Google Scholar]
- Bower, C. K., Q. Xu, and J. McGuire. 1998. Activity losses among T4 lysozyme variants after adsorption to colloidal silica. Biotechnol. Bioeng. 58:658–662. [DOI] [PubMed] [Google Scholar]
- Buijs, J., M. Ramström, M. Danfelter, H. Larsericsdotter, P. Håkansson, and S. Oscarsson. 2003. Localized changes in the structural stability of myoglobin upon adsorption onto silica particles, as studied with hydrogen/deuterium exchange mass spectrometry. J. Colloid Interface Sci. 263:441–448. [DOI] [PubMed] [Google Scholar]
- Cao, A., D. Hu, and L. Lai. 2004. Formation of amyloid fibrils from fully reduced hen egg white lysozyme. Protein Sci. 13:319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castells, V., S. Yang, and P. R. Tassel. 2002. Surface-induced conformational changes in lattice model proteins by Monte Carlo simulation. Phys. Rev. E. 65:0319121–0319128. [DOI] [PubMed] [Google Scholar]
- Caughey, B., and P. T. Lansbury. 2003. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 26:267–298. [DOI] [PubMed] [Google Scholar]
- Cheng, S.-S., K. K. Chittur, C. N. Sukenik, L. A. Culp, and K. Lewandowska. 1994. The conformation of fibronectin on self-assembled monolayers with different surface composition: An FTIR/ATR study. J. Colloid Interface Sci. 162:135–143. [Google Scholar]
- Chun, K.-Y., and P. Stroeve. 2002. Protein transport in nanoporous membranes modified with self-assembled monolayers of functionalized thiols. Langmuir. 8:4653–4658. [Google Scholar]
- Cohen, F. E., and J. W. Kelly. 2003. Therapeutic approaches to protein-misfolding diseases. Nature. 426:905–909. [DOI] [PubMed] [Google Scholar]
- Daly, S. M., T. M. Przybycien, and R. D. Tilton. 2003. Coverage-dependent orientation of lysozyme adsorbed on silica. Langmuir. 19:3848–3857. [Google Scholar]
- Dobson, C. M. 1999. Protein misfolding, evolution and disease. Trends Biochem. Sci. 24:329–332. [DOI] [PubMed] [Google Scholar]
- Dobson, C. M. 2003a. Protein folding and disease: view from the first Horizon symposium. Nat. Rev. Drug Discov. 2:154–160. [DOI] [PubMed] [Google Scholar]
- Dobson, C. M. 2003b. Protein folding and misfolding. Nature. 426:884–890. [DOI] [PubMed] [Google Scholar]
- Dong, A., P. Huang, and W. S. Caughey. 1990. Protein secondary structures in water from second-derivative amide I infrared spectra. Biochemistry. 29:3303–3308. [DOI] [PubMed] [Google Scholar]
- Dong, A., S. J. Prestrelski, S. D. Allison, and J. F. Carpenter. 1995. Infrared spectroscopic studies of lyophilization- and temperature-induced protein aggregation. J. Pharm. Sci. 84:415–424. [DOI] [PubMed] [Google Scholar]
- Dong, A., T. W. Randolph, and J. F. Carpenter. 2000. Entrapping intermediates of thermal aggregation in alpha-helical proteins with low concentration of guanidine hydrochloride. J. Biol. Chem. 275:27689–27693. [DOI] [PubMed] [Google Scholar]
- Eaton, W. A., V. Munoz, S. J. Hagen, G. S. Jas, L. J. Lapidus, E. R. Henry, and J. Hofrichter. 2000. Fast kinetics and mechanisms in protein folding. Annu. Rev. Biophys. Biomol. Struct. 29:327–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel, M. F. M., A. J. W. G. Visser, and C. P. M. van Mierlo. 2004. Conformation and orientation of a protein folding intermediate trapped by adsorption. Proc. Natl. Acad. Sci. USA. 101:11316–11321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fändrich, M., M. A. Fletcher, and C. M. Dobson. 2001. Amyloid fibrils from muscle myoglobin. Nature. 410:165–166. [DOI] [PubMed] [Google Scholar]
- Fändrich, M., V. Forge, K. Buder, M. Kittler, C. M. Dobson, and S. Diekmann. 2003. Myoglobin forms amyloid fibrils by association of unfolded polypeptide segments. Proc. Natl. Acad. Sci. USA. 100:15463–15468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frare, E., P. Polverino De Laureto, J. Zurdo, C. M. Dobson, and A. Fontana. 2004. A highly amyloidogenic region of hen lysozyme. J. Mol. Biol. 340:1153–1165. [DOI] [PubMed] [Google Scholar]
- Frishman, D., and P. Argos. 1995. Knowledge-based protein secondary structure assignment. Proteins. 23:566–579. [DOI] [PubMed] [Google Scholar]
- Giacomelli, C. E., and W. Norde. 2003. Influence of hydrophobic Teflon particles on the structure of amyloid β-peptide. Biomacromolecules. 4:1719–1726. [DOI] [PubMed] [Google Scholar]
- Green, R. J., I. Hopkinson, and R. A. L. Jones. 1999. Unfolding and intermolecular association in globular proteins adsorbed at interfaces. Langmuir. 15:5102–5110. [Google Scholar]
- Hummer, G., A. E. Garcia, and S. Garde. 2001. Helix nucleation kinetics from molecular simulations in explicit solvent. Proteins. 42:77–84. [PubMed] [Google Scholar]
- Jirage, K. B., J. C. Hulteen, and C. R. Martin. 1999. Effect of thiol chemisorption on the transport properties of gold nanotubule membranes. Anal. Chem. 71:4913–4918. [DOI] [PubMed] [Google Scholar]
- Kauffmann, E., N. C. Darnton, R. H. Austin, C. Batt, and K. Gerwert. 2001. Lifetimes of intermediates in the β-sheet to α-helix transition of β-lactoglobulin by using a diffusional IR mixer. Proc. Natl. Acad. Sci. USA. 98:6646–6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, J. W. 1998. The alternate conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr. Opin. Struct. Biol. 8:101–106. [DOI] [PubMed] [Google Scholar]
- Krebs, M. R., D. K. Wilkins, E. W. Chung, M. C. Pitkeathly, A. K. Chamberlain, J. Zurdo, C. V. Robinson, and C. M. Dobson. 2000. Formation and seeding of amyloid fibrils from wild-type hen lysozyme and a peptide fragment from the beta-domain. J. Mol. Biol. 300:541–549. [DOI] [PubMed] [Google Scholar]
- Kubelka, J., and T. A. Keiderling. 2001. Differentiation of β-sheet-forming structures: ab initio-based simulation of IR absorption and vibrational CD for model peptide and protein β-sheets. J. Am. Chem. Soc. 123:12048–12058. [DOI] [PubMed] [Google Scholar]
- Latour, R. A., Jr., and C. J. Rini. 2002. Theoretical analysis of adsorption thermodynamics for hydrophobic peptide residues on SAM surfaces of varying functionality. J. Biomed. Mater. Res. 60:564–577. [DOI] [PubMed] [Google Scholar]
- Lee, C. −S., and G. Belfort. 1989. Changing activity of ribonuclease A during adsorption: a molecular explanation. Proc. Natl. Acad. Sci. USA. 86:8392–8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez, A., J. Haavik, T. Flatmark, J. L. R. Arrondo, and A. Muga. 1996. Conformational properties and stability of tyrosine hydroxylase studied by infrared spectroscopy. Effect of iron/catecholamine binding and phosphorylation. J. Biol. Chem. 271:19737–19742. [DOI] [PubMed] [Google Scholar]
- Maste, M. C. L., E. H. W. Pap, A. Van Hoek, W. Norde, and A. J. W. G. Visser. 1996. Spectroscopic investigation of the structure of a protein adsorbed on a hydrophobic Latex. J. Colloid Interface Sci. 180:632–633. [Google Scholar]
- Matagne, A., and C. M. Dobson. 1998. The folding process of hen lysozyme: a perspective from the ‘new view’. Cell. Mol. Life Sci. 54:363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Militello, V., C. Casarino, A. Emanuele, A. Giostra, F. Pullara, and M. Leone. 2004. Aggregation kinetics of bovine serum albumin studied by FTIR spectroscopy and light scattering. Biophys. Chem. 107:175–187. [DOI] [PubMed] [Google Scholar]
- Munoz, V., P. A. Thompson, J. Hofrichter, and W. A. Eaton. 1997. Folding dynamics and mechanism of β-hairpin formation. Nature. 390:196–199. [DOI] [PubMed] [Google Scholar]
- Norde, W. 2003. Driving forces for protein adsorption at solid surfaces. In Biopolymers at Interfaces. M. Malmsten, editor. Marcel Dekker, New York. 21–44.
- Norde, W., and T. Zoungrana, T. 1998. Surface-induced changes in the structure and activity of enzymes physically immobilized at solid/liquid interfaces. Biotechnol. Appl. Biochem. 28:133–143. [PubMed] [Google Scholar]
- Nuzzo, R. G., L. H. Dubois, and D. L. Allara. 1990. Fundamental studies of microscopic wetting on organic surfaces. 1. Formation and structural characterization of a self-consistent series of polyfunctional organic monolayers. J. Am. Chem. Soc. 112:558–569. [Google Scholar]
- Ostuni, E., B. A. Grzybowski, M. Mrksich, C. S. Roberts, and G. M. Whitesides. 2003. Adsorption of proteins to hydrophobic sites on mixed self-assembled monolayers. Langmuir. 19:1861–1872. [Google Scholar]
- Pepys, M. B., P. N. Hawkins, D. R. Booth, D. M. Vigushin, G. A. Tennent, A. K. Soutar, N. Totty, O. Nguyen, C. C. Blake, C. J. Terry, T. G. Feest, A. M. Zalin, and J. J. Hsuan. 1993. Human lysozyme gene mutations cause hereditary systemic amyloidosis. Nature. 362:553–557. [DOI] [PubMed] [Google Scholar]
- Price, W. S., F. Tsuchiya, and Y. Arata. 1999. Lysozyme aggregation and solution properties studied using PGSE NMR diffusion measurements. J. Am. Chem. Soc. 121:11503–11512. [Google Scholar]
- Prime, K. L., and G. M. Whitesides. 1993. Adsorption of proteins onto surfaces containing end-attached oligo(ethylene oxide): a model system using self-assembled monolayer. J. Am. Chem. Soc. 115:10714–10721. [Google Scholar]
- Sane, S. U., S. M. Cramer, and T. M. Przybycien. 1999. Protein structure perturbations on chromatographic surfaces. J. Chromatogr. A. 849:149–159. [Google Scholar]
- Sethuraman, A., M. Han, R. S. Kane, and G. Belfort. 2004a. Effect of surface wettability on the adhesion of proteins. Langmuir. 20:7779–7788. [DOI] [PubMed] [Google Scholar]
- Sethuraman, A., G. Vedantham, T. Imoto, T. Przybycien, and G. Belfort. 2004b. Protein unfolding at interfaces: slow dynamics of α-helix to β-sheet transition. Proteins. 56:669–678. [DOI] [PubMed] [Google Scholar]
- Sigal, M., M. Mrksich, and G. M. Whitesides. 1998. Effect of surface wettability on the adsorption of proteins and detergents. J. Am. Chem. Soc. 120:3464–3473. [Google Scholar]
- Sokolowski, F., A. J. Modler, R. Masuch, D. Zirwer, M. Baier, G. Lutsch, D. A. Moss, K. Gast, and D. Naumann. 2003. Formation of critical oligomers is a key event during conformational transition of recombinant syrian hamster prion protein. J. Biol. Chem. 278:40481–40492. [DOI] [PubMed] [Google Scholar]
- Stefani, M., and C. M. Dobson. 2003. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding disease and biological evolution. J. Mol. Med. 81:678–699. [DOI] [PubMed] [Google Scholar]
- Taniguchi, M., and G. Belfort. 2002. Correcting for surface roughness: advancing and receding contact angles. Langmuir. 18:6465–6467. [Google Scholar]
- Taniguchi, M., J. P. Pieracci, and G. Belfort. 2001. Effect of undulations on surface energy: a quantitative assessment. Langmuir. 17:4312–4315. [Google Scholar]
- Tronin, A., A. M. Edwards, W. W. Wright, J. M. Vanderkooi, and J. K. Blasie. 2002. Orientation distributions for cytochrome c on polar and nonpolar interfaces by total internal reflection fluorescence. Biophys. J. 82:996–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzannis, S. T., W. J. M. Hrushesky, P. A. Wood, and T. M. Przybycien. 1996. Irreversible inactivation of interleukin 2 in a pump-based delivery environment. Proc. Natl. Acad. Sci. USA. 93:5460–5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbricht, M., H. Matuschewski, A. Oechel, and H.−G. Hicke. 1996. Photo-induced graft polymerization surface modifications for the preparation of hydrophilic and low-protein-adsorbing ultrafiltration membranes. J. Membr. Sci. 115:31–47. [Google Scholar]
- Vedantham, G., H. G. Sparks, S. U. Sane, S. Tzannis, and T. M. Przybycien. 2000. A holistic approach for protein secondary structure estimation from infrared spectra in H2O solutions. Anal. Biochem. 285:33–49. [DOI] [PubMed] [Google Scholar]
- Williams, M. A., J. M. Thornton, and J. M. Goodfellow. 1997. Modelling protein unfolding: hen egg-white lysozyme. Protein Eng. 10:895–903. [DOI] [PubMed] [Google Scholar]
- Williams, S., T. P. Causgrove, R. Gilmanshin, K. S. Fang, R. H. Callender, W. H. Woodruff, and R. B. Dyer. 1996. Fast events in protein folding: helix melting and formation in a small peptide. Biochemistry. 35:691–697. [DOI] [PubMed] [Google Scholar]
- Zurdo, J., J. I. Guijarro, J. L. Jimenez, H. R. Saibil, and C. M. Dobson. 2001. Dependence on solution conditions of aggregation and amyloid formation by an SH3 domain. J. Mol. Biol. 311:325–340. [DOI] [PubMed] [Google Scholar]