

Abstract
EET in reconstituted Lhca4, a peripheral light-harvesting complex from Photosystem I of Arabidopsis thaliana, containing 10 chlorophylls and 2 carotenoids, was studied at room temperature by femtosecond transient absorption spectroscopy. Two spectral forms of Lut were observed in the sites L1 and L2, characterized by significantly different interactions with nearby chlorophyll a molecules. A favorable interpretation of these differences is that the efficiency of EET to Chls is about two times lower from the “blue” Lut in the site L1 than from the “red” Lut in the site L2 due to fast IC in the former case. A major part of the energy absorbed by the “red” Lut, ∼60%–70%, is transferred to Chls on a sub-100-fs timescale from the state S2 but, in addition, minor EET from the hot S1 state within 400–500 fs is also observed. EET from the S1 state to chlorophylls occurs also within 2–3 ps and is ascribed to Vio and/or “blue” Lut. EET from Chl b to Chl a is biphasic and characterized by time constants of ∼300 fs and 3.0 ps. These rates are ascribed to EET from Chl b spectral forms absorbing at ∼644 nm and ∼650 nm, respectively. About 25% of the excited Chls a decays very fast—within ∼15 ps. This decay is proposed to be related to the presence of the interacting Chls A5 and B5 located next to the carotenoid in the site L2 and may imply some photoprotective role for Lhca4 in the photosystem I super-complex.
INTRODUCTION
The small peripheral antenna proteins of Photosystem I (Lhca) and Photosystem II Lhcb) belong to the Lhc family (Jansson, 1999). All of them share to a large extent their amino acid sequence and bind, in a rather conservative way, 8–15 chlorophylls (Chls) and 2–4 carotenoids (Cars). Two of the sites for Cars, occupied in all family members and named L1 and L2, are located in the center of the complexes and have in most cases the highest affinity for Lut. The best characterized Lhc protein is LHCII, the most abundant antenna protein in chloroplasts and part of the photosystem II supercomplex. Its crystallographic structure with 3.4–4.9-Å resolution was published in 1994 (Kühlbrandt et al., 1994)and since then a lot of spectroscopic studies have been performed for this complex (see van Amerongen and van Grondelle, 2001for a review). Very recently, a new structure for trimeric LHCII has been published with 2.72-Å resolution (Liu et al., 2004). Fourteen Chls were identified per monomer: eight central Chls a and six Chls b at the periphery of monomers. In addition, four sites for Cars were identified including two central sites occupied by Luts in contact with six sites for Chls a, one site for Neo (Croce et al., 1999a), and one for xantophyll-cycle Cars (Ruban et al., 1999).
CP29 is another photosystem II peripheral antenna and Lhc family member, which has been studied extensively. It contains eight Chls (six Chls a and two Chls b; Bassi et al., 1999) and two Cars. In reconstituted (and likely in the native) CP29, one Car site, L1, is occupied by Lut and the other, L2, by Vio or Neo (Bassi et al., 1999; Croce et al., 2003a).
Several femtosecond transient absorption studies on LHCII and CP29 gave detailed information about EET in these complexes directly isolated from spinach (Croce et al., 2003b Gradinaru et al., 2000) or obtained from the procedure of reconstitution in vitro (Croce et al., 2001, 2003a,b). All studies agree that the Lut in L1 site transfers EE to Chl a although differences were observed for the xantophylls in the site L2: Gradinaru et al. concluded that the transfer from L2 Car occurs only to Chls a, whereas Croce et al. (2001, 2003a) suggested transfer to both Chl species. Both groups agree that EET from the central Luts to the Chls occurs with a high efficiency of ∼80% mainly from the S2 state within ∼100 fs, but the efficiency of EET from the S2 state of Vio in the L2 site in CP29 is significantly lower (below 50%; Croce et al., 2003a). The contribution of EET to Chls from the S1 Cars state is ∼20%–25% (Gradinaru et al.) or less (Croce et al., 2001, 2003a) of the total energy absorbed by the Cars. Gradinaru et al. suggest that in LHCII the EE donor from the S1 state is Lut, but this is not necessarily the case in CP29. According to Croce et al. (2003a), Lut is a very ineffective EE donor from its S1 state, and a very minor EET was observed only from the vibrationally unrelaxed S1 state. EET to Chls was also observed from the S1 state of Vio and/or Neo occupying site L2 in CP29.
EET between Chl b and Chl a in LHCII was described with up to four components ranging from subpicoseconds to a few picoseconds (Connelly et al., 1997; Croce t al., 2001; Kleima et al., 1997; Visser et al., 1996. In CP29 that contains only two Chls b, four components of Chl b→Chl a EET were resolved (150 fs, 600–800 fs, 1.2 ps, and 5–6 ps; Croce et al., 2003a,b). The 600–800-fs component was ascribed to EET from Chl b-640 nm, whereas the three others were ascribed to Chl b-652 nm. The observation that the number of components was larger than the number of Chl b binding sites was taken as evidence for the mixed character of a few Chl binding sites both in natural and reconstituted CP29 (see also Salverda et al., 2003).
In higher plants, the peripheral antenna system, LHCI, is built up from four elementary complexes, Lhca1–Lhca4. The recent crystallographic structure shows that a single set of Lhca1–Lhca4 and Lhca2 and Lhca3 dimers is located on one side of the core complex (Ben-Shem et al., 2003) confirming the earlier observation by electron microscopy (Boekema et al., 2001). Crystallographic data clearly show that there is strong homology between all Lhca complexes and LHCII but simultaneously demonstrate that there are some differences in positions, orientations, and numbers of Chls among the Lhca complexes and between them and LHCII. All Lhca complexes show emission at low temperatures more to the red (maximum at 733 nm for Lhca4) compared with Lhcb complexes (maximum at 681 nm). It has been proposed that the red-emitting forms result from excitonic interaction (Morosinotto et al., 2002). In a recent study by Morosinotto et al. (2003) it was demonstrated that a single mutation in Lhca4, replacing the asparagine that coordinates Chl A5 (notation from Kühlbrandt et al., 1994) with histidine, abolishes the red absorption and red emission. It was thus proposed that excitonic interaction in the WT occurs between two Chls a in sites A5 and B5. In the reconstituted Lhca4, there are altogether seven Chls a, three Chls b, and two Cars (1.7 Lut and 0.35 Vio). Sites for most of Chls a and Chls b were not identified although some hints can be inferred from the occupancy of particular sites in Lhca1, which is highly homologous to Lhca4, and which was studied by selective mutagenesis (Morosinotto et al., 2002).
In this study, we characterize the EE dynamics in reconstituted Lhca4 over a wide spectral range from 470/530 nm to 760 nm at room temperature and compare this system to LHCII and CP29. Application of four excitation wavelengths, 490, 514, 644, and 663 nm, allows largely selective excitation of two different spectral forms of Lut, Chl b, and Chl a. As a result we present a complete picture of EET from Cars to Chls, from Chl b to Chl a, and between different spectral forms of Chl a.
MATERIALS AND METHODS
Sample preparation
The reconstituted Lhca4 from Arabidopsis thaliana was prepared as previously described in Morosinotto et al. (2003). It contains 10 Chls (7 Chls a and 3 Chls b) and 2 Cars (1.7 Lut and 0.35 Vio). Based on analogy to LHCII, one can presume that the site L1 is occupied exclusively by Lut, whereas the site L2 shows a mixed occupancy for Lut and Vio with relative affinities of 2:1 (Croce et al., 1999b).
Femtosecond transient absorption spectroscopy
The experimental setup was described in Gradinaru et al. (2000). The duration of the excitation pulses was ∼80 fs, and the instrument response function was ∼120 fs (full width at half-maximum). For selective excitation at 490, 514, 644, and 663 nm, interference filters were used with spectral width (full width at half-maximum) of 23, 12, 16, and 8 nm, respectively. The energy of a single pulse was adjusted in such a way that independent of the excitation wavelength, the maximum photobleaching/stimulated emission signal in the Chl a Qy region was of ΔA = 0.01–0.015. It corresponds to an energy per pulse of 10, 40, 5, and 3 nJ, for excitation at 490, 514, 644, and 663 nm, respectively. The optical density of the sample at the maximum of Chl a absorption at 675.5 nm was ∼5–6 cm−1. The sample was placed in a 1-mm-thick cuvette that was shaken during the pump-probe measurement to ensure that each laser pulse illuminated a fully relaxed sample. The repetition rate of the laser was 1 kHz. Considering the small antenna size of Lhca4 (10 Chls and 2 Cars), the applied EE density was low enough to avoid any annihilation effects (<0.1 photon per Lhca4). The pump and probe light pulses were set at magic angle relative to each other. Transient absorption difference spectra were recorded at a pump-probe delay time of up to 4 ns with increasing time-delay step from 50 fs to 500 ps for the shortest and longest timescales, respectively. The total number of delay times was ∼90. Spectral resolution was 1 nm.
Data analysis
Global analyses based on sequential model and target analysis were applied to the recorded sequences of time-gated difference spectra as described in van Stokkum et al. (2004). The estimated spectra of the global analysis are referred to, throughout the text, as EADS. The spectra resulting from target analysis are referred to as SADS.
Fluorescence excitation
Fluorescence excitation spectra were obtained at room temperature on a Jasco (Tokyo, Japan) FP-777 fluorimeter. The samples were in 10 mM Hepes pH 7.6, 0.03% dedecyl maltoside at 0.02 optical density. The emission was recorded at 710 nm with a 3 nm bandwidth.
RESULTS
Laser pulses centered at 490, 514, 644, and 663 nm were applied to study Lhca4 with the aim to excite different pigments selectively. We estimated how much of each of the pigments was excited for the different excitation wavelengths (Tables 1 and 2). To do this, the spectrum of Lhca4 was fitted with the spectra of the pigments in a protein environment (Croce et al., 2000), and the spectral profiles of the excitation pulses were convoluted with the fitted spectra, yielding excitation probability for each of the pigments (Fig. 1). Of the two “blue” excitations, the more selective excitation was at 514 nm where only ∼20% of Chls (∼10% of Chls a and ∼10% of Chls b) and ∼80% of Cars were predicted to be excited, with the red Lut as major excited molecule (∼50%). A more uniform excitation of different Cars is predicted for the 490-nm excitation along with a strong contribution of the Soret band of Chl b (∼35%) and a negligible amount of Chl a. Overall, at 490 nm, the excitation is more evenly distributed between Chls (∼40%) and Cars (∼60%).
TABLE 1.
Percentage distribution of initial excitation over particular types of pigments at excitations in the Soret region
| Excitation wavelength | Chl a | Chl b | Vio | Lut blue | Lut red |
|---|---|---|---|---|---|
| 490 nm | 3.5 | 35 | 13.8 | 27.5 | 20 |
| 514 nm | 10.4 | 9.9 | 7.8 | 22.5 | 49.4 |
Blue and red Lut correspond to two spectral forms found in the fitting routine (Fig. 1).
TABLE 2.
Percentage distribution of initial excitation over particular types of pigments at excitations in the Chl Qy region
| Excitation wavelength | Chl a | Chl b |
|---|---|---|
| 644 nm | 40.9 | 59.1 |
| 663 nm | 85.4 | 14.6 |
FIGURE 1.
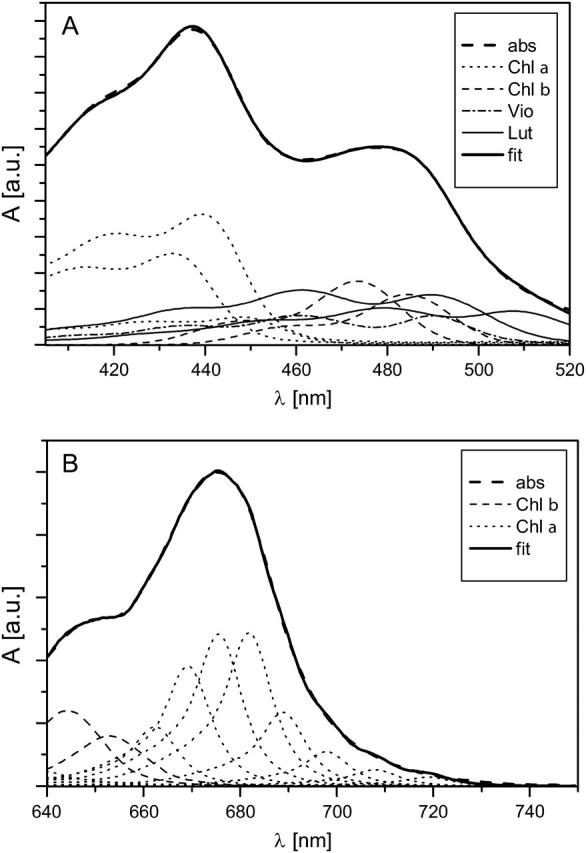
Decomposition of the Lhca4 steady-state absorption spectra in the Soret (A) and Qy (B) regions onto spectra of individual types of pigments. The shapes of pigments' spectra are taken from the measurements in a protein environment. Their amplitudes and shifts along the wavelength axis were fitted.
Excitation at 514 nm
The time-evolution in the transient absorption spectra recorded for Lhca4 after excitation at 514 nm was analyzed with six lifetime components. Fig. 2 A shows EADS (see Materials and Methods) resulting from this analysis. The initial EADS (75 fs), which represents the difference spectrum at time zero, is dominated by two negative bands: one at ∼550 nm and the other at ∼620 nm. The former band is due to SE from the S2 state (compare to Gradinaru et al., 2000), whereas the latter one originates most likely from Raman scattering in water. Small bands at 645, 682, and 667 nm are ascribed to PB/SE of Chl b and two spectral forms of Chl a, respectively, excited at low amounts at 514 nm (Table 1). The red part of this spectrum, at >∼640 nm, is lifted above zero level and is ascribed to the short-wavelength tail of the S2→SN transition band of Lut (compare to Papagiannakis et al., 2003).
FIGURE 2.
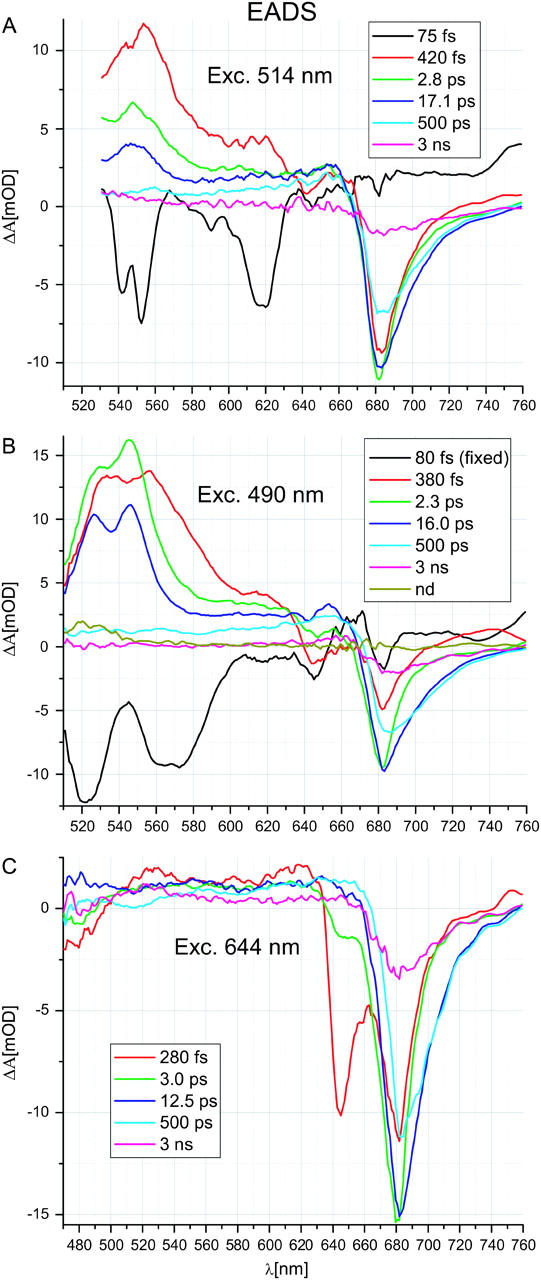
EADS for Lhca4 excited at (A) 514 nm, (B) 490 nm, and (C) 644 nm.
The spectral evolution within 75 fs (transition from the 75-fs to the 420-fs EADS) is contributed by two major processes. The first is relaxation of the Car S2 state to the hot S1 state characterized by a strong ESA band peaking at 553 nm. Strong ESA in this region is commonly observed for a range of Cars (see Polivka and Sundström, 2004 for review). Second, in the region of Chl absorption, a huge PB/SE signal appears at ∼683 nm, indicating very efficient EET from the Car S2 state to Chl a. Minor positive ΔA changes are observed in the Chl b region, around 645 nm. These changes may be the sum of several events contributing to the absorption changes in this region, like the disappearance of ESA from S2, the appearance of ESA from the S1 state and EET from the S2 state to Chl b. Because of the two former processes, it is not possible to conclude on the extent of the latest process.
After 420 fs, the 420-fs EADS in Fig. 2 A is replaced by the 2.8-ps EADS. In the Cars region these changes are ascribed to cooling of the hot S1 (compare with excitation at 490 nm) most likely accompanied by EET transfer from the hot S1 state to Chls a. Indeed, in the red part of the spectrum, a further increase of the Chl a PB/SE signal occurs, although this may largely be due to EET from Chls b, as can be seen from the disappearance of the PB “hole” in the Chl b region. Chl b to Chl a EET on this timescale is confirmed by the experiment with direct excitation of Chl b at 644 nm (see below). The relaxed S1→SN ESA spectrum of Car (2.8-ps EADS) clearly differs from that of hot S1→SN ESA, the former one peaking at ∼548 nm, 5 nm toward blue compared to the latter one (Fig. 2 A). A similar relaxation of the Car S1 state was reported for CP29 (Croce et al., 2003a) and other systems (de Weerd et al., 2002, 2003).
Further spectral evolution occurs in 2.8 ps (transition from 2.8-ps to 17.1-ps EADS) and is ascribed to EET from the S1 state of Cars to Chls a. On this timescale, a significant reduction of Car ESA occurs along with significant gain in PB/SE in the red part of the Chl a Qy region. In principle, the reduction of Car ESA could also be contributed by the natural decay of one of the Cars in Lhca4 although the observed time constant seems to be too short given natural lifetimes of ∼15 ps (Billsten et al., 2003; Frank et al., 1997) and 24 ps (Frank et al., 1994; Polivka et al., 1999) reported for Lut and Vio, respectively. On the same timescale of 2.8 ps, equilibration between bulk and red Chls a takes place, as can be seen from the decrease of the Chl a PB/SE signal amplitude at ∼682 nm accompanied by broadening of this signal toward the red. Redistribution of EE between bulk and red Chls on this timescale is also observed after direct excitation of Chls a at 663 nm (see below). No evolution is observed in the Chl b region on this timescale, indicating that the Chl b molecule(s) absorbing at ∼650 nm and transferring EE to Chl a within a few picoseconds (see below, direct excitation of Chl b at 490 and 644 nm) are not excited by the 514-nm laser pulses.
The spectral evolution on a 17.1-ps timescale (transition from the 17.1-ps to 500-ps EADS) is due to the natural decay of the Car S1 state (this time constant is close to a natural lifetime of Lut; Billsten et al., 2003; Frank et al., 1997) and the decay of a significant fraction (20%–30%) of the Chl a excited state.
After 17.1-ps no excitations are localized on Cars anymore. The two slowest lifetime components of 500 ps and 3 ns describe the decay of excitations on the remaining Chls a. Apart from a dominant PB/SE signal in the Qy region, a flat ESA is observed throughout the whole spectral region presented. The values of these two slowest lifetimes were kept the same for all the excitation wavelengths because free fits give values spreading a wide range and do not improve the quality of the fits significantly (this is due to a low amount of experimental points at a longer timescale). A more detailed description of the decay processes of Lhca proteins will be presented elsewhere (J. A. Ihalainen, R. Croce, T. Morosinotto, I. H. M. van Stokkum, R. Bassi, J. P. Dekker, and R. van Grondelle, unpublished).
Excitation at 490 nm
Excitation kinetics after 490 nm pulse remain similar to those found for 514-nm excitation (Fig. 2 B) although amplitudes and, in some cases, shapes of particular EADS show significant differences caused by the different distribution of the initial excitation over the Lhca4 pigments. Table 1 shows preferential excitation of the blue Lut/Vio (∼41%) along with strong excitation of Chl b (∼35%).
The first EADS (Fig. 2 B; 80 fs) exhibits two broad, negative bands in the blue part of the spectrum. The band peaking at ∼520 nm may correspond to the ∼550-nm double band after excitation at 514 nm and is ascribed to SE from the higher vibrational band of the Cars S2 state. Due to the fast vibrational cooling, the SE band at ∼520 nm should be significantly broadened toward the red. This is indeed observed if we assume that the two bands at ∼520 and ∼570 nm are an effect of the overlap of one broad SE band with the narrower positive ESA band peaking at ∼545 nm. The position of this band fits nicely to the S1→SN band shown in the subsequent EADS, meaning that there is at least a certain fraction of the S2→S1 IC occurring within the time resolution (see Discussion). Additionally, the ∼520-nm SE band is expected to be dominated by blue Lut/Vio (Table 1), which are 20-nm blue-shifted (Fig. 1) relative to the red Lut, which dominates the ∼550-nm SE band at 514-nm excitation. Another feature of the initial EADS is that on the top of the flat positive ΔA changes in the Chl Qy region (again ascribed to ESA from the S2 state of Cars), the negative signal in the Chl a region is of significant amplitude, similar to that in the Chl b region. This is surprising because one expects ∼35% of Chl b and only ∼3.5% of Chl a to be excited (Table 1).
Spectral evolution on the 80-fs timescale (Fig. 2 B; transition from the 80-fs to 380-fs EADS) shows that the changes in ΔA are flat between 650 and ∼700 nm and are very different compared with the 514-nm excitation, where clear formation of the negative ΔA band is observed on this timescale. Thus, the spectral evolution in this region should be mostly ascribed to the decay of the flat ESA from the S2 state of Cars, due to S2→S1 relaxation, with a minor contribution from EET to Chl a. This indicates that on the 80-fs timescale, the EET to Chl a from the blue Lut/Vio, excited preferentially at 490 nm (Table 1), is much lower than from the red Lut, excited preferentially at 514 nm. The evolution of the spectra in the Cars region is qualitatively similar to that observed at 514-nm excitation: the ESA spectrum of the hot S1 state replaces the SE signal from the Cars S2 state. Due to a better signal/noise ratio and the wider spectral window toward the blue, the double-peaked structure of the ESA bands is now visible. The shape and the position of the red-shifted peak at ∼555 nm of hot S1 ESA (also of the relaxed S1 ESA peaking at ∼545 nm) is, in contrast to the S2 spectrum, similar to that observed upon 514-nm excitation. This may be due to the fact that the S1 state is expected to be much less influenced by the environment than S2, and as a consequence it has similar spectral properties for all of the Cars. In the Chl b region, only small positive spectral changes occur on the 80-fs timescale and, similarly to what happens upon 514-nm excitation, these observations are not conclusive.
The further evolution (Fig. 2 B; transition from the 380-fs to 2.3-ps EADS) occurs with a 380-fs lifetime and is conservative in the Cars region, meaning that the loss of the oscillator strength in one spectral region (>550 nm) is compensated by the gain in another one (<550 nm). Therefore we conclude that no EET from the Car hot S1 state to Chls occurs at 490-nm excitation. Instead, pure hot S1→S1 relaxation is observed. All spectral changes occurring in the Qy Chls region are very similar to those observed after direct Chl b excitation (at 644 nm) and thus can be attributed exclusively to Chl b to Chl a EET, keeping in mind that the 490-nm pulse excites a lot of Chl b.
There is a relatively large fraction of the Car S1 state that decays with a short time of 2.3 ps (transition from the 2.3-ps to 16.0-ps EADS), suggesting EET to Chls. However, in the Chls Qy region it is not obvious that there is a gain of PB/SE due to EET from Cars. Actually, similarly to the 380-fs component, the spectral changes in this region could be explained exclusively by EET from Chl b to Chl a with a contribution from equilibration between bulk and red Chls a. Thus, there is a possibility that the 2.3-ps process is contributed by unusually fast, protein-induced IC decay of the S1 state. A similarly fast IC was considered before for LHCII (Croce et al., 2001).
The further spectral evolution occurs in 16.0 ps (transition from the 16.0-ps to 500-ps EADS) and is very similar to that observed at 514-nm excitation: complete loss of S1→SN ESA of Car is accompanied by a significant decay of the signal in the blue part of the Chl a Qy region. Additionally, the 16.0-ps component contains some contribution from slow equilibration between bulk and red Chls a as was observed for LHCII (Kleima et al., 1997) and mixture of LHCI dimers (Gobets et al., 2001). This is indicated by an increase of the negative amplitude of the 500-ps EADS relative to the 16.0-ps EADS above 700 nm. The ∼15-ps EE equilibration contribution is clearly seen only at 490-nm excitation. This may be due to the fact that at this wavelength a relatively highest amount of Vio (and Vio-containing Lhca4 complexes) is excited. Consequently, we speculate that the presence of Vio instead of Lut in L2 slows down the EE equilibration between bulk and red Chls in Lhca4 by introducing structural/functional changes in the vicinity of L2 and red Chls sites.
In addition to the two slow components (500 ps and 3 ns) observed at all excitation wavelengths, a nondecaying spectrum of unclear origin was resolved at 490-nm excitation showing a small nonzero amplitude only in the 520–530 nm region (Fig. 2 B).
Excitation at 644 nm
Excitation at 644 nm is distributed among Chl b (∼60%) and Chl a (∼40%; Table 2). In the Chl Qy region, the temporal evolution (Fig. 2 C) of the transient spectra resembles that after excitation at 490 nm except for the fastest process resolved only in the latter case (the 80-fs EADS in Fig. 2 B) and ascribed mainly to Car's dynamics. The EET from Chl b to Chl a, seen as a loss and gain of PB/SE signal in Chl b and Chl a Qy regions, respectively, is biphasic (280 fs and 3.3 ps) with the slower phase having a contribution from bulk to red Chl a equilibration (loss and gain of PB/SE signal in blue and red parts of Chl a Qy region, respectively). There is again a 12.4-ps decay component in the blue part of the Chl a Qy spectrum. The decay of the equilibrated excitation localized on Chls a is described by the two slowest components. The biphasic PB/SE signal decay in the Chl b Qy region is accompanied by a similar biphasic decay in the Soret region seen below 500 nm.
Excitation at 663 nm
Direct excitation of Chls a at 663 nm leads to transient absorption dynamics fitted with 5 components (∼200 fs, 1.7 ps, 15.0 ps, 500 ps, and 3 ns; not shown). The two fastest components describe two energy equilibration phases in the Qy region. The 15.0-ps component corresponds to the ∼15-ps decay of relatively blue Chls found at other excititation wavelengths. No ΔA changes in the blue part of the spectrum, down to 470 nm, were observed that could be ascribed to a response from Cars.
Fluorescence excitation studies
Based on the fluorescence excitation spectra (not shown) and the relative contribution of particular pigments in the absorption spectra (Fig. 1, Tables 1 and 2), the average efficiency of Cars to Chls energy transfer was calculated to be 58% ± 5%.
DISCUSSION
EET from the state S2 of Cars to Chls
In Lhca4, the light of two different wavelengths excites different populations of Cars: 514 nm, mainly red Lut, and 490 nm, mainly blue Lut and Vio (Table 1). Global analysis indicates clear and extensive EET from the Car S2 state to Chls a in the former case (Fig. 2 A). The results are more difficult to interpret in the latter case because of the unexpected observation of the big signal from Chl a in the Qy region at time zero (80-fs EADS; Fig. 2 B), whereas the amount of directly excited Chl a should be negligible (Table 1). At least three explanations can be given for the origin of this signal. First, there is an ultrafast Cars→Chl a EET not resolved in the experiment. Second, this signal originates from an electrochromic response of Chl a to the appearance of a highly polarized S2 state of Cars. A similar effect was proposed for CP29 by Gradinaru et al. (2000). Third, a relatively large amount of Chls a is excited by the 490-nm pulse, in clear contradiction to the estimate in Table 1 (this could happen via excitonic interaction between directly excited Chls b and Chls a). We favor the first explanation because it implies effective EET from the S2 Car state to Chls. This is in line with the situation in LHCII and CP29 where very efficient EET from the state S2 of both Luts to Chls was reported (60%–80%; Croce et al., 2001, 2003a; Gradinaru et al., 2000). Furthermore, ultrafast EET from Cars to Chls was reported earlier (Krueger et al., 2001; Zigmantas et al., 2002). The second explanation may be questioned based on the lack of similar electrochromic shift at 514-nm excitation. The third explanation excludes efficient EET because very little of EET from Cars to Chls occurs on the resolved timescales. Moreover, a hypothetical strong excitonic coupling between Chls a and b would be expected rather to affect the blue-most forms of Chl a, energetically more feasible to interact with Chl b, and not bulk Chls a. Very direct excitation of Chl a at 490 nm seems impossible (Table 1, Fig. 1).
Despite the argumentation above, the analysis of the relative amplitudes in the Car and Chls regions does demonstrate a reduced efficiency of EET from Cars to Chls upon excitation at 490 nm compared to the case of 514-nm excitation. For the 514-nm excitation, the maximal amplitude of SE from the S2 state of Cars (Fig. 2 A; 75-fs EADS at ∼550 nm), representing the population of initially excited Cars, is smaller than the Chl a PB/SE signal in the Qy region (17.1-ps EADS at ∼680 nm), representing the population of eventually excited Chls. For 490-nm excitation, this relation is inverted (Fig. 2 B): the Chl a signal (16.0-ps EADS at ∼680 nm) is smaller than the Car signal (80-fs EADS at 520 nm). If, in addition, one takes into account that at 490 nm there is at least ∼40% of direct Chls excitation whereas at 514 nm there is only ∼20% (Table 1), it follows that the ratio of Car to Chl a (excluding directly excited Chls) maximal amplitudes is ∼2 times smaller in the latter case. This rough estimation indicates that the total efficiency of EET from the S2 Car state to Chls after 490-nm excitation is ∼2 times smaller than after 514-nm excitation. This reasoning is further confirmed by the relative amplitudes associated with the relaxed S1 Car state (17.1-ps and 16.0-ps EADS in Fig. 2, A and B, respectively, at ∼545 nm): much less signal from this state appears after excitation at 514 nm than at 490 nm. It should be mentioned here that free fits of the data for 490-nm excitation had a tendency to yield the fastest component even faster than 80 fs (fixed in presented fits due to limited time resolution of our setup), thus indicating the possibility that a fast S2→S1 IC may be responsible for both unresolvable EET from Car to Chls and its decreased efficiency. This decreased efficiency may be additionally explained by the excitation of relatively more Vio (at 490 nm), which was reported to be a poor EE donor from its S2 state to Chls (Croce et al., 2003a).
In conclusion, our data demonstrate that EET to Chls from the S2 state of blue Lut is significantly different from that of red Lut. To our knowledge, this feature has not been reported before for the two central Lut in any of the Lhc proteins.
EET from the state S1 of Cars to Chls
The spectral evolution within 420 fs, 2.8 ps (Fig. 2 A), and 2.3 ps (Fig. 2 B) indicates some EET from (hot) S1 states of Cars. The quantitative analysis of the extent of this EET is presented in the Target analysis section.
Target analysis
To get a quantitative and physical model of the EET pathways in Lhca4, a target analysis was performed. In principle, one common scheme should describe all possible EET pathways independent of excitation wavelength and spectral range. However, due to the complexity of the system (three types of Cars, each one having four states; two types of Chl b; a few types of Chl a) and the relative selectivity of excitation at different wavelengths, we have worked on three simplified and partial models presented below.
The dynamics of transient absorption from 530 to 760 nm after excitation at 514 nm in Lhca4 was modeled with the compartmental scheme shown in Fig. 3. The compartment S2 is dominated by red Lut (Table 1) but also has a contribution from the two remaining Cars. Also hot S1 state (hS1) is contributed by all Cars, however, with decreased representation of red Lut due to very efficient EET to Chls from the S2 state of this Car. There are two more Car states, S1 and S1′, reflecting the biphasic decay observed in Fig. 2 A (2.8 and 17.1 ps EADS). Much lower relative amplitudes associated with these two decays at 514-nm than at 490-nm excitation (compare 2.8/2.3-ps and 17.1/16.0-ps EADS in Fig. 2, A and B) strongly indicate that they should be ascribed mostly to blue Lut/Vio rather than to red Lut, at least at 490-nm excitation. This assignment contradicts the idea that the two peaks observed in 16.0-ps EADS (Fig. 2 B) at 527 nm and 545 nm belongs to blue and red Lut (compare to Polivka et al., 2002). Most likely, the compartment S1, as indicated by its short lifetime, represents Car(s) transferring the energy to Chls (see Results); and the state S1′ belongs to Car(s) decaying naturally via IC. The shape of the S1 spectrum (Fig. 3) is different from that of the S1′ spectrum but resembles the hot S1 spectrum (this is more evident at 490-nm excitation; Fig. 4). This indicates that either S1 and S1′ represent relaxed states of different Cars (likely Vio and blue Lut) or S1 is another, slowly relaxing, hot state of one or more Cars. The latter possibility is particularly plausible because Lut is thought to be a very poor energy donor from its relaxed S1 state (Croce et al., 2001, 2003a; Polivka et al., 2002; Walla et al., 2002). On the other hand, EET from the hot S1 state of the mixture of Cars (Lut/Vio/Neo) was clearly observed in LHCII by using the fluorescence up-conversion technique (Walla et al., 2000).
FIGURE 3.
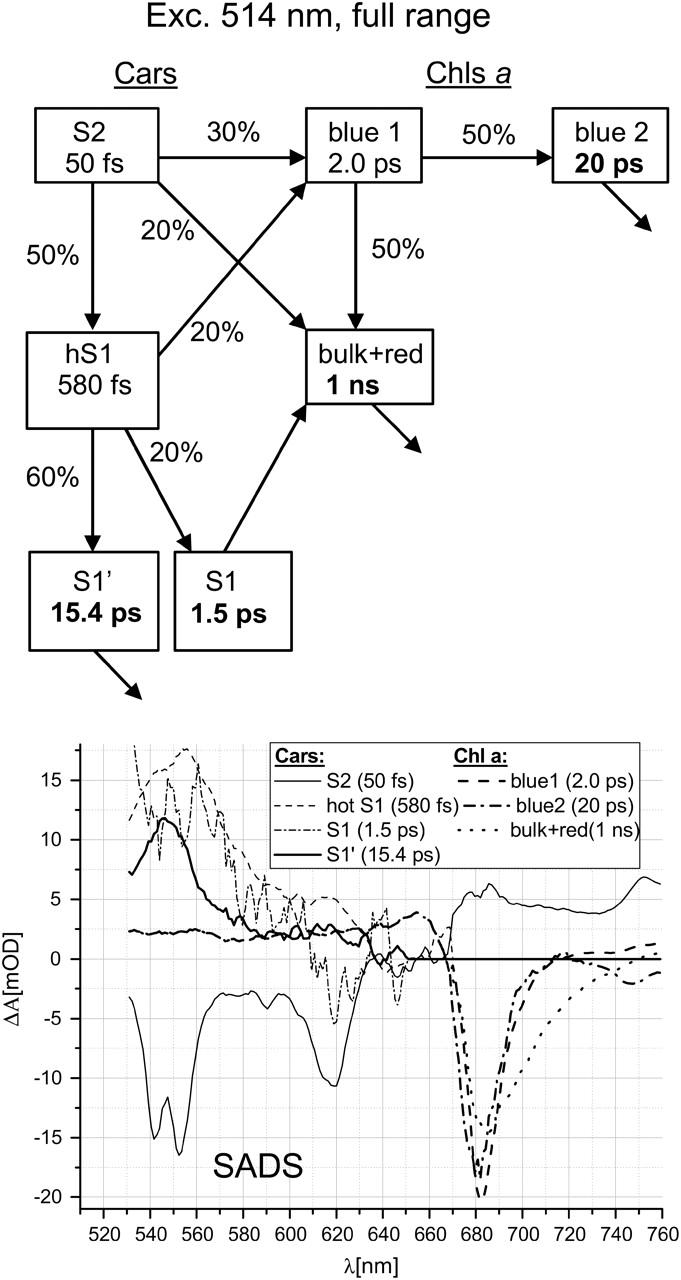
Scheme of compartments and SADS for 514-nm excitation in Lhca4. Numbers in rectangles are the intrinsic lifetimes of corresponding compartments, and numbers at arrows are efficiencies of decay routes of particular compartments estimated from the fit. Wherever there is no percentage number associated to an arrow, there is only one 100% decay channel. The scheme neglects direct excitation of Chl a (10%; Table 1) and a minor contribution of Chl b to Chl a EET due to direct excitation of Chl b (10%; Table 1) and describes EET pathways starting from the excitation of the S2 state of Cars only. To get satisfactory fits, some reasonable constraints have been put on the spectra: spectra of S1 and S1′ have been assumed to have zero amplitudes above 650 nm, amplitudes of the hot S1 spectrum were forced to be zero above 670 nm, and amplitudes of blue 1, blue 2, and bulk + red Chls a were forced to be the same in the ESA region of Chl a below 670 nm.
FIGURE 4.
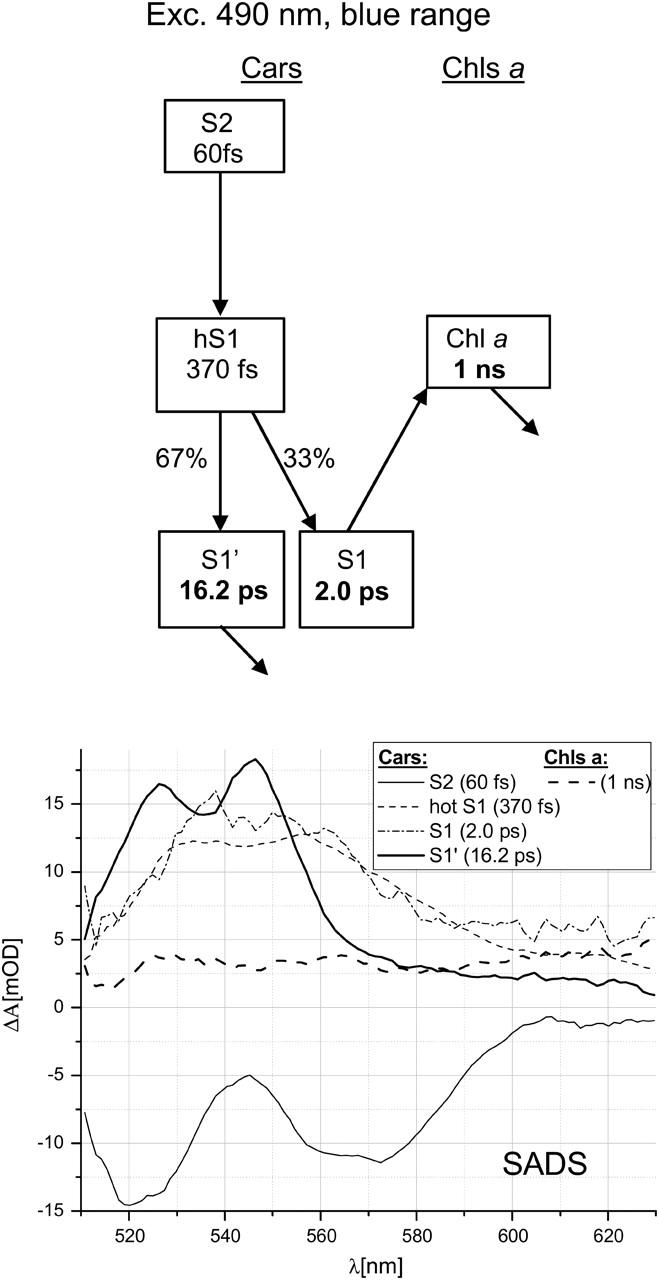
Scheme of compartments and SADS for 490-nm excitation in Lhca4 in the blue spectral range. The meaning of the numbers is as in Fig. 3.
Chls a are divided into three different compartments (Fig. 3). Compartments “blue 1” and “bulk + red” are inferred directly from the observation of EET from Cars to two different spectral pools of Chls a (Fig. 2 A). Actually, the “bulk + red” compartment should be split even further since a biphasic decay of the fully equilibrated EE is observed (Fig. 2), but for the sake of clarity they were lumped together. The third compartment “blue 2” has to be introduced to account for the fast, ∼15-ps, decay of excitation of short-wavelength Chls a (Fig. 2).
The numbers shown in Fig. 3 demonstrate that ∼50% of energy absorbed by Cars is transferred to Chl a from the S2 state and ∼20% from the S1 state and ∼30% is converted into heat via IC. The low amount of EE transferred from the Car S1 state to Chls is in line with earlier reports for LHCII and CP29 (Croce et al., 2001, 2003a; Gradinaru et al., 2000). Considering that blue Lut/Vio are poorer EE donors than red Lut, it may be estimated, based on numbers in Table 1, that the EET efficiency from the S2 state of red Lut could amount to up to 60%–70%, a value similar to that for the EET efficiency from the S2 state of Lut in LHCII and CP29 (Croce et al., 2001, 2003a; Gradinaru et al., 2000).
Target analysis for the Cars' region of spectrum (510–630 nm) after 490-nm excitation was based on the model presented in Fig. 4. The compartment S2 represents mainly blue Lut and Vio, and contribution from Car S2→Chl EET was ignored as it is apparently minor (see Results). The hypothetical ultrafast EET from the state S2 to Chls was also ignored in this modeling as it was not resolved in transient spectra. Numbers in Fig. 4 show that the total Cars to Chls EET efficiency is 33% and occurs from the S1 state. However, if one takes into account that unresolved EET from the state S2 to Chls affects ∼25% (half of the efficiency of EET from the S2 state of Cars to Chls at 514-nm excitation—see above) of initially excited Cars, the Cars to Chls EET efficiency via S1 state drops to ∼25%. Even this number may be overestimated because, as it was noticed in Results, a part of the S1 state may also decay via IC within ∼2 ps without transferring the EE to Chls. The remaining part of the excited Cars decays via IC from the S1′ state on an ∼15-ps timescale.
Transient absorption dynamics after 644-nm excitation in the Chls' Qy region, 630–760 nm, was modeled based on the compartmental scheme in Fig. 5. We distributed initial excitation over two Chl b compartments (60%), in line with observation of two different Chl b→Chl a EET phases (Results) and two Chl a compartments (40%; Table 2). Two spectral forms of Chl b are distinguished, one centered at ∼644 nm, and the other one at ∼650 nm. These wavelengths fit quite well the positions of the maximum absorption of the two spectral forms of Chl b found from the steady-state absorption spectrum decomposition (Fig. 1 B). The lifetimes associated with these spectra, 250 fs and 3.3 ps, respectively, are in agreement with those found from global analysis (Fig. 2 C). It was noted in Results that after excitation at 514 nm, only the fast EET from Chl b to Chl a was observed, meaning that the slow Chl b was not excited. This implies that the redder Soret band (at ∼485 nm; Fig. 1 A) belongs to the fast Chl b showing the bluer Qy band, and the bluer Soret band (at ∼472 nm) belongs to the slow Chl b showing the redder Qy band. The previous study on Chl b to Chl a EET in reconstituted Lhca4 containing a similar ratio of Chl a/b, however at 77 K, also revealed two spectral forms of Chl b, absorbing at 640 nm and 650 nm, respectively, but resolved only one lifetime of EET to Chl a of 500–600 fs (Melkozernov et al., 2000). Biexponential EET from Chl b to Chl a with lifetimes of 0.5 and 2–3 ps, however without resolving spectral differences between Chls b, was reported for Lhca1 (Melkozernov et al., 2002) and a mixture of LHCI dimers (Gobets et al., 2001). The spectra and lifetimes of the three pools of Chl a are similar to those observed after excitation at 514 nm (Fig. 3), thus making the results internally consistent. The same compartmental scheme was applied for target analysis of the data collected in the same red spectral range at 490-nm excitation yielding almost identical SADS (not shown).
FIGURE 5.
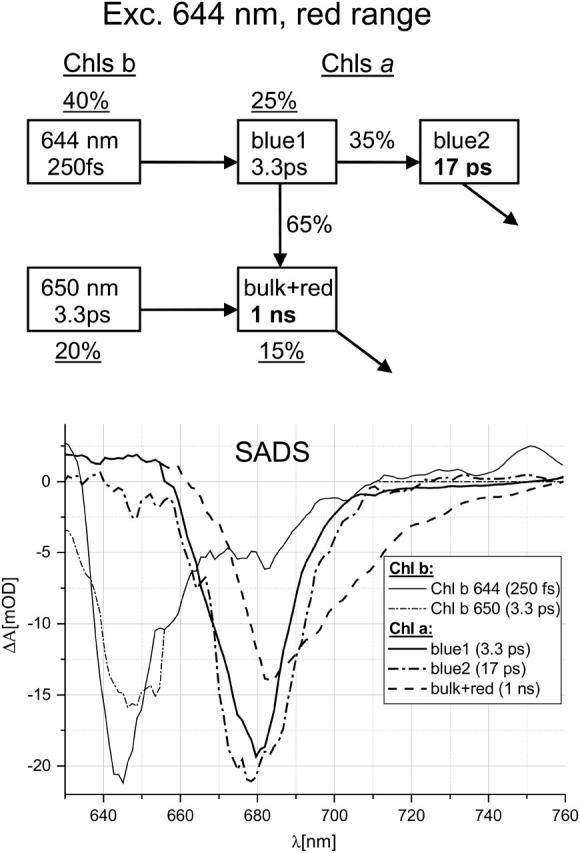
Scheme of compartments and SADS for 644-nm excitation in Lhca4 in the red spectral range. The meaning of the numbers is as in Fig. 3. Underlined numbers denote distribution of initial excitation over particular compartments. Spectral shapes of Chl b-644 and Chl b-650 were forced to be identical between 655 and 710 nm and, additionally, spectrum of Chl b-650 was forced to have zero amplitude above 710 nm. Also spectra of Chl a “blue 1” and “bulk + red” were forced to be identical below 655 nm. These reasonable constraints were necessary to get a good fit.
Structural assignments
Comparative studies of Lhca4 WT and a mutant missing the red-most absorption indicate that red Lut, preferentially excited at 514 nm, is located in the site L2 (K. Gibasiewicz, R. Croce, T. Morosinotto, J. A. Ihalainen, I. H. M. van Stokkum, J. P. Dekker, R. Bassi, and R. van Grondelle, unpublished). As L1 is known to have very specific affinity for Lut (Croce et al., 1999b), it is most likely occupied by the blue Lut only, whereas the L2 site has mixed occupancy and accommodates both red Lut and Vio. This assignment is in line with the relative absorbance of these three Cars found from the decomposition of the Lhca4 spectra (Fig. 1, A and B). Clearly, the most abundant Car is blue Lut. Taking additionally into account that the contribution of Lut and Vio in Lhca4 is 1.7 and 0.35, respectively, one can estimate that the occupancy ratio of the site L2 is red Lut/Vio ≈ 2:1 and that there is ∼3 times more blue Lut than Vio per Lhca4. In other words, in our preparation, ∼66% of particles contain two Luts and ∼34% of particles contain one Lut and one Vio.
EE quenching
Application of four different excitation wavelengths consistently show that in Lhca4 there is a fast, ∼15-ps, decay of excitation localized on relatively blue Chls a. The amount of energy quenched in this way is estimated to be ∼25% of total energy reaching Chl a (calculated from numbers in schemes in Figs. 3 and 5 and easily seen from loss of the relative area related to the ∼15 ps process in Fig. 2, A–C). Although the loss of excitation is seen on the blue side of the Qy spectrum—we propose that the candidate for this quencher is the A5-B5 pair of Chls, as in the mutant for which the red absorption was shown to be abolished (Morosinotto et al., 2003)—the ∼15-ps phase is virtually absent (data not shown). It was recently speculated that a pair of closely located Chls a in LHCII may serve as EE quencher, enhancing the quenching effect of xantophyll-cycle Cars (Beddard et al., 1976; Liu et al., 2004). Since in a fraction of Lhca4 (34%; see above) the Chl A5-B5 pair is proposed here to be located in direct contact with Vio (in L2 site), which is a precursor of xantophyll-cycle Cars, we speculate that the pigments in this region may play some kind of photoprotective role.
CONCLUSIONS
We propose that the site L1 in reconstituted Lhca4 is occupied by the blue Lut, whereas the site L2 is occupied by red Lut and Vio with the ratio 2:1. We have shown that the interactions between blue Lut and Chls a differ significantly from those between red Lut and Chls a. EET efficiency from the state S2 of the red Lut is 60%–70%, whereas that of the blue Lut is estimated to be about half of this, most likely due to a fast IC competing with EET. EET from the Car state S1 with the efficiency of up to 20%–25% was inferred mainly from the fast decay of Cars S1→SN ESA signal although this number may be overestimated if fast, ∼2-ps, IC is induced in the Cars by the protein environment. It is likely that all this EET occurs from the vibrationally hot S1 state. Red Lut and either both or one of the blue Lut/Vio contribute to EET from the (hot) S1 state to Chls. Average Car→Chls EET efficiency of 58%, found from the fluorescence excitation experiment, agrees with the average value obtained from the EET efficiencies found at 514-nm and 490-nm excitations. No EET from Cars to Chl b was clearly detected, however, possibly due to compensating effects of ESA in the Chl b Qy region. Two distinct spectral forms of Chl b transfer the EE to Chl a with two lifetimes: fast transfer of ∼300 fs occurs from Chl b-644 nm, and an order of magnitude slower transfer of ∼3 ps occurs from Chl b-650 nm. The presence of an excitonically coupled pair of Chls in the vicinity of the L2 site is proposed to induce fast, ∼15-ps, and effective quenching of excitation. This may imply some kind of photoprotective role of Lhca4.
Acknowledgments
We thank Mikas Vengris for his help with the pump-probe setup.
This work was supported by the Access to Research Infrastructures activity in the Sixth Framework Programme of the European Union (contract RII3-CT-2003-506350, Laserlab Europe), the PSICO Research Training Network of the European Union, grant HPRN-CT-2002-00248, and “MIUR” Progetti FIRB No. RBAU01E3CX and FIRB No. RBNE01LACT. K.G. gratefully acknowledges the Marie-Curie Individual Fellowship, contract HPMF-CT-2002-01812, and Netherlands Organisation for Scientific Research grant (B 81-734).
Abbreviations used: EET, excitation energy transfer, EE; excitation energy; PB, photobleaching; SE, stimulated emission; ESA, excited state absorption; IC, internal conversion; WT, wild-type; EADS, evolution associated difference spectra; SADS, species associated difference spectra; Lut, lutein, Vio, violaxanthin; Neo, neoxanthin.
References
- Bassi, R., R. Croce, D. Cugini, and D. Sandona. 1999. Mutational analysis of a higher plant antenna protein provides identification of chromophores bound into multiple sites. Proc. Natl. Acad. Sci. USA. 96:10056–10061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beddard, G. S., S. E. Carlin, and G. Porter. 1976. Concentration quenching of chlorophyll fluorescence in bilayer lipid vesicles and liposomes. Chem. Phys. Lett. 43:27–32. [Google Scholar]
- Ben-Shem, A., F. Frolow, and N. Nelson. 2003. Crystal structure of plant photosystem I. Nature. 426:630–635. [DOI] [PubMed] [Google Scholar]
- Billsten, H. H., P. Bhosale, A. Yemelyanov, P. S. Bernstein, and T. Polivka. 2003. Photochem. Photobiol. 78:138–145. [DOI] [PubMed] [Google Scholar]
- Boekema, E. J., P. E. Jensen, E. Schlodder, J. F. L. van Breemen, H. van Roon, H. V. Scheller, and J. P. Dekker. 2001. Green plant photosystem I binds light-harvesting complex I on one side of the complex. Biochemistry. 40:1029–1036. [DOI] [PubMed] [Google Scholar]
- Connelly, J. P., M. G. Müller, M. Hucke, G. Gatzen, C. W. Mullineaux, A. V. Ruban, P. Horton, and A. R. Holzwarth. 1997. Ultrafast spectroscopy of trimeric light harvesting complex II from higher plants. J. Phys. Chem. B. 101:1902–1909. [Google Scholar]
- Croce, R., G. Cinque, A. R. Holzwarth, and R. Bassi. 2000. The Soret absorption of carotenoids and chlorophylls in antenna complexes of higher plants. Photosynth. Res. 64:221–231. [DOI] [PubMed] [Google Scholar]
- Croce, R., M. G. Müller, R. Bassi, and A. R. Holzwarth. 2001. Carotenoid-to-chlorophyll energy transfer in recombinant major light-harvesting complex (LHCII) of higher plants. I. Femtosecond transient absorption measurements. Biophys. J. 80:901–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce, R., M. G. Müller, R. Bassi, and A. R. Holzwarth. 2003b. Chlorophyll b to chlorophyll a energy transfer kinetics in the CP29 antenna complex. A comparative femtosecond absorption study between native and reconstituted proteins. Biophys. J. 84:2508–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce, R., M. G. Müller, S. Caffarri, R. Bassi, and A. R. Holzwarth. 2003a. Energy transfer pathways in the minor antenna complex CP29 of Photosystem II: a femtosecond study of carotenoid to chlorophyll transfer on mutant and WT complexes. Biophys. J. 84:2517–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce, R., R. Remelli, C. Varotto, J. Breton, and R. Bassi. 1999a. The neoxanthin binding site of the major light harvesting complex (LHCII) from higher plants. FEBS Lett. 456:1–6. [DOI] [PubMed] [Google Scholar]
- Croce, R., S. Weiss, and R. Bassi. 1999b. Carotenoid-binding sites of the major Light-Harvesting Complex II of higher plants. J. Biol. Chem. 274:29613–29623. [DOI] [PubMed] [Google Scholar]
- de Weerd, F. L., J. P. Dekker, and R. van Grondelle. 2003. Dynamics of β-carotene-to-chlorophyll singlet energy transfer in the core of photosystem II. J. Phys. Chem. B. 107:6214–6220. [Google Scholar]
- de Weerd, F. L., I. H. M. van Stokkum, and R. van Grondelle. 2002. Subpicosecond dynamics in the excited state absorption of all-trans-β-carotene. Chem. Phys. Lett. 354:38–43. [Google Scholar]
- Frank, H. A., V. Chynwat, R. Z. B. Desamero, R. Farhoosh, J. Erickson, and J. A. Bautista. 1997. On the photophysics and photochemical properties of carotenoids and their role as light-harvesting pigments in photosynthesis. Pure Appl. Chem. 69:2117–2124. [Google Scholar]
- Frank, H. A., A. Cua, V. Chynwat, A. Young, D. Gosztola, and M. R. Wasielewski. 1994. Photophysics of the carotenoids associated with the xanthophyll cycle in photosynthesis. Photosynth. Res. 41:389–395. [DOI] [PubMed] [Google Scholar]
- Gobets, B., J. T. M. Kennis, J. A. Ihalainen, M. Brazzoli, R. Croce, I. H. M. van Stokkum, R. Bassi, J. P. Dekker, H. van Amerongen, G. R. Fleming, and R. van Grondelle. 2001. Excitation energy transfer in dimeric Light Harvesting Complex I: a combined streak-camera/fluorescence upconversion study. J. Phys. Chem. B. 105:10132–10139. [Google Scholar]
- Gradinaru, C. C., I. H. M. van Stokkum, A. A. Pascal, R. van Grondelle, and H. van Amerongen. 2000. Identifying the pathways of energy transfer between carotenoids and chlorophylls in LHCII and CP29. A multicolor, femtosecond pump-probe study. J. Phys. Chem. B. 104:9330–9342. [Google Scholar]
- Jansson, S. 1999. A guide to the Lhc genes and their relatives in Arabidopsis. Trends Plant Sci. 4:236–240. [DOI] [PubMed] [Google Scholar]
- Kleima, F. J., C. C. Gradinaru, F. Calkoen, I. H. M. van Stokkum, R. van Grondelle, and H. van Amerongen. 1997. Energy transfer in LHCII monomers at 77 K studied by sub-picosecond transient absorption spectroscopy. Biochemistry. 36:15262–15268. [DOI] [PubMed] [Google Scholar]
- Krueger, B. P., S. S. Lampoura, I. H. M. van Stokkum, E. Papagiannakis, J. M. Salverda, C. C. Gradinaru, D. Rutkauskas, R. G. Hiller, and R. van Grondelle. 2001. Energy transfer in the peridinin chlorophyll-a protein of Amphidinium carterae studied by polarized transient absorption and target analysis. Biophys. J. 80:2843–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühlbrandt, W., D. N. Wang, and Y. Fujiyoshi. 1994. Atomic model of plant light-harvesting complex by electron crystallography. Nature. 367:614–621. [DOI] [PubMed] [Google Scholar]
- Liu, Z., H. Yan, K. Wang, T. Kuang, J. Zhang, L. Gui, X. An, and W. Chang. 2004. Crystal structure of spinach major light-harvesting complex at 2.72 resolution. Nature. 428:287–292. [DOI] [PubMed] [Google Scholar]
- Melkozernov, A. N., S. Lin, V. H. R. Schmid, H. Paulsen, G. W. Schmidt, and R. Blankenship. 2000. Ultrafast excitation dynamics of low energy pigments in reconstituted peripheral light-harvesting complexes of photosystem I. FEBS Lett. 471:89–92. [DOI] [PubMed] [Google Scholar]
- Melkozernov, A. N., V. H. R. Schmid, S. Lin, H. Paulsen, and R. E. Blankenship. 2002. Excitation energy transfer in the Lhca1 subunit of LHC I-730 peripheral antenna of Photosystem I. J. Phys. Chem. B. 106:4313–4317. [Google Scholar]
- Morosinotto, T., J. Breton, R. Bassi, and R. Croce. 2003. The nature of a chlorophyll ligand in Lhca proteins determines the far red fluorescence emission typical of Photosystem I. J. Biol. Chem. 278:49223–49229. [DOI] [PubMed] [Google Scholar]
- Morosinotto, T., S. Castelletti, J. Breton, R. Bassi, and R. Croce. 2002. Mutation analysis of Lhca1 antenna complex. J. Biol. Chem. 277:36253–36261. [DOI] [PubMed] [Google Scholar]
- Papagiannakis, E., I. H. M. van Stokkum, R. van Grondelle, R. A. Niederman, D. Zigmantas, V. Sundström, and T. Polivka. 2003. A near-infrared transient absorption study of the excited-state dynamics of the carotenoid spirilloxanthin in solution and in the LH1 complex of Rhodospirillum rubrum. J. Phys. Chem. B. 107:11216–11223. [Google Scholar]
- Polivka, T., J. L. Herek, D. Zigmantas, H.-E. Åkerlund, and V. Sundström. 1999. Direct observation of the (forbidden) S1 state in carotenoids. Proc. Natl. Acad. Sci. USA. 96:4914–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polivka, T., and V. Sundström. 2004. Ultrafast dynamics of carotenoid excited states—from solution to natural and artificial systems. Chem. Rev. 104:2021–2071. [DOI] [PubMed] [Google Scholar]
- Polivka, T., D. Zigmantas, V. Sundström, E. Formaggio, G. Cinque, and R. Bassi. 2002. Carotenoid S1 state in a recombinant Light-Harvesting Complex of Photosystem II. Biochemistry. 41:439–450. [DOI] [PubMed] [Google Scholar]
- Ruban, A. V., P. J. Lee, M. Wentworth, A. J. Young, and P. Horton. 1999. Determination of the stoichiometry and strength of binding of xanthophylls to the Photosystem II Light Harvesting Complexes. J. Biol. Chem. 274:10458–10465. [DOI] [PubMed] [Google Scholar]
- Salverda, J. M., M. Vengris, B. P. Krueger, G. D. Scholes, A. R. Czarnoleski, V. Novoderezhkin, H. van Amerongen, and R. van Grondelle. 2003. Energy transfer in Light-Harvesting Complexes LHCII and CP29 of spinach studied with three pulse echo peak shift and transient grating. Biophys. J. 84:450–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Amerongen, H., and R. van Grondelle. 2001. Understanding the energy transfer function of LHCII, the major light-harvesting complex of green plants. J. Phys. Chem. B. 105:604–617. [Google Scholar]
- van Stokkum, I. H. M., D. S. Larsen, and R. van Grondelle. 2004. Global and target analysis of time-resolved spectra. Biochim. Biophys. Acta. 1657:82–104. [DOI] [PubMed] [Google Scholar]
- Visser, H. M., F. J. Kleima, I. H. M. van Stokkum, R. van Grondelle, and H. van Amerongen. 1996. Probing the many energy-transfer processes in the photosynthetic light-harvesting complex II at 77 K using energy-selective sub-picosecond transient absorption spectroscopy. Chem. Phys. 210:297–312. [Google Scholar]
- Walla, P. J., P. A. Linden, K. Ohta, and G. R. Fleming. 2002. Excited-state kinetics of the carotenoid S1 state in LHC II and two-photon excitation spectra of lutein and β-carotene in solution: efficient Car S1→Chl electronic energy transfer via hot S1 states? J. Phys. Chem. A. 106:1909–1916. [Google Scholar]
- Walla, P. J., J. Yom, B. P. Krueger, and G. Fleming. 2000. Two-photon excitation spectrum of Light-Harvesting Complex II and fluorescence upconversion after one- and two-photon excitation of the carotenoids. J. Phys. Chem. B. 104:4799–4806. [Google Scholar]
- Zigmantas, D., R. G. Hiller, V. Sundström, and T. Polivka. 2002. Carotenoid to chlorophyll energy transfer in the peridinin-chlorophyll-a–protein complex involves an intramolecular charge transfer state. Proc. Natl. Acad. Sci. USA. 99:16760–16765. [DOI] [PMC free article] [PubMed] [Google Scholar]