

Abstract
Protein crystallography provides the structure of a protein, averaged over all elementary cells during data collection time. Thus, it has only a limited access to diffusive processes. This article demonstrates how molecular dynamics simulations can elucidate structure-function relationships in bacteriorhodopsin (bR) involving water molecules. The spatial distribution of water molecules and their corresponding hydrogen-bonded networks inside bR in its ground state (G) and late M intermediate conformations were investigated by molecular dynamics simulations. The simulations reveal a much higher average number of internal water molecules per monomer (28 in the G and 36 in the M) than observed in crystal structures (18 and 22, respectively). We found nine water molecules trapped and 19 diffusive inside the G-monomer, and 13 trapped and 23 diffusive inside the M-monomer. The exchange of a set of diffusive internal water molecules follows an exponential decay with a 1/e time in the order of 340 ps for the G state and 460 ps for the M state. The average residence time of a diffusive water molecule inside the protein is ∼95 ps for the G state and 110 ps for the M state. We have used the Grotthuss model to describe the possible proton transport through the hydrogen-bonded networks inside the protein, which is built up in the picosecond-to-nanosecond time domains. Comparing the water distribution and hydrogen-bonded networks of the two different states, we suggest possible pathways for proton hopping and water movement inside bR.
INTRODUCTION
The protein bacteriorhodopsin (bR) resides in the membrane of the archaebacterium Halobacterium salinarum and uses photonic energy for the transmembrane proton pumping. The protein incorporates a retinal chromophore bound to a lysine residue via a protonated Schiff base linkage and absorbs light at ∼568 nm. Photoexcitation triggers an isomerization of retinylidene. The photoreaction induces a vectorial transfer of a proton across the membrane, leading to the release of a proton at the extracellular side and an uptake from the cytoplasmic side. Our current knowledge of the structure and the photocycle of bR has been reviewed in detail by several authors (Haupts et al., 1999; Heberle, 2000; Lanyi, 2001, 2004). Although current opinion assumes an outward proton pumping mechanism, there has always been a discussion that bR might be not an H+ pump but an OH−. This possibility seems never to have been pursued very seriously (Stoeckenius, 1999) before the first structural evidence appeared (Luecke, 2000). Luecke (2000) proposed an inward-driven hydroxide pump model based on crystallographic evidence for rearrangement of water molecules during the photocycle. Such a model is consistent with the scenario, proposed by analogy with halide transport in halorhodopsin (Betancourt and Glaeser, 2000). Recently one more scenario has been proposed by Kouyama et al. (2004), based on the structure of the L intermediate. According to this model, the function of bacteriorhodopsin is the outward pumping of a proton and the inward pumping of a water molecule.
Since certain dynamical features of bR, important for the understanding of the proton pump, cannot be captured by crystallographic techniques, molecular dynamics simulations have been used to elucidate, among others, conformational fluctuations (Edholm et al., 1995; Xu et al., 1995; Logunov et al., 1995) and bR-water mobility (Roux et al., 1996; Baudry et al., 2001; Hayashi et al., 2002; Kandt et al., 2004). Electrostatic calculations were performed to calculate the protonational states of bR (Bashford and Gerwert, 1992; Onufriev et al., 2003; Song et al., 2003).
Although the molecular structure of bR in its ground state is now well determined, the extent of conformational changes in the late M intermediate is still controversial (Luecke et al., 1999; Sass et al., 2000; Subramaniam and Henderson, 2000). In addition, the number of buried (i.e., internal) water molecules in bR (Papadopoulos et al., 1990; Weik et al., 1998; Dencher et al., 2000; Kandori, 2000; Luecke, 2000; Zaccai, 2000; Gottschalk et al., 2001; Kandt et al., 2004), which are assumed to play a critical role in providing proton pathways and to be involved in the molecular mechanism leading to proton translocation, is still unclear.
The mean residence times of water molecules on the surface and in the interior of biomolecules and biomolecular complexes (such as ribonuclease A, lysozyme, myoglobin, trypsin, serum albumin) have been measured by oxygen-17 spin relaxation dispersion (Denisov and Halle, 1995a, b, 1996; Denisov et al., 1999). The timescale at which water penetrates or exchanges with other bulk solvent molecules (at T = 300 K, P = 1 atm) has been observed to be in the range of 10–50 ps for water residing in surface cavities (Levitt and Park, 1993), 0.1–1 ns for strongly bound water (Otting and Liepinsh, 1995; Otting et al., 1991), and nanoseconds to milliseconds for interior water (e.g., structural water molecules in proteins; see Denisov and Halle, 1995a, b).
Using NMR, Ernst et al. (1995) found that hydrophobic cavities in human interleukin-1β contain 2–4 water molecules that reside within the protein for times longer than 1 ns. These water molecules were not observed in high resolution crystal structures, since water molecules with mean-square displacement fluctuations >1 Å make negligible contributions to high resolution x-ray diffraction spectra (Yu et al., 1999). However, careful analysis of the low resolution diffraction data by Yu et al. (1999) shows that disordered water molecules observed by Ernst et al. (1995) are indeed present and it has been suggested that hydrophobic cavities commonly observed in protein structures may be filled in most cases with disordered water.
Recently two molecular dynamics studies have been performed to examine the distribution of water molecules inside membrane proteins. Kandt and co-workers (Kandt et al., 2004 and references therein) investigated the distribution of water molecules inside trimeric bacteriorhodopsin in its ground state. Water densities were shown and frequencies of H-bond contacts per residue were calculated. Olkhova et al. (2004) investigated the dynamics of water networks in cytochrome c oxidase. A much higher average number of internal water molecules than observed in crystal structures was found and the corresponding hydrogen-bonded network was investigated.
In this study we report on a molecular dynamics simulation on bR trimer, which predicts new details of the amount and the distribution of internal water molecules in bR and which describes the related hydrogen-bonded networks that constitute possible proton pathways. We have performed studies on the ground (G) and the late M states. The M intermediate has been chosen since it is a key state for understanding the mechanism of proton transfer. Comparing water distribution and hydrogen-bonded networks in two different states of bR, we suggest possible pathways for proton hopping and water movement inside bR. The main differences of the present investigations compared to the previous studies are: 1), two states of bR have been simulated; 2), precise definitions of the protein surface and the internal water molecules are introduced; and 3), probabilities of forming Grotthuss-pathways rather than single hydrogen bonds have been calculated.
We argue that crystal structures are often discussed without looking to different timescales. We show below that structures on a picosecond timescale differ considerably from time-averaged x-ray structures.
MODELS AND METHODS
Molecular model
Unfortunately there are no appropriate molecular descriptions available for archaeal lipids of purple membranes. Therefore we constructed a lipid bilayer using well-studied POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine) lipids, for which topologies and parameters have been extensively tested (Feller and MacKerell, 2000). We consider this approach reasonable given the fact that bR is fully active when reconstituted into phospholipid bilayers.
POPC membrane has a low gel-to-fluid transition temperature of Tm = −5°C. AMBER '91 parameters (Weiner et al., 1986), OPLS parameters (Jorgensen and Tirando-Rives, 1988), and our own parameters (Lin and Baumgaertner, 2000) for unsaturated hydrocarbons chains are used to model the POPC lipid. Carbon atoms with bound hydrogens are modeled as united-atoms, which reduces the total number of atoms of one lipid molecule from 134 for an all-atom model to 52 for the united-atom model. The partial charges of the headgroup are adapted from previous studies using AMBER (Essmann et al., 1995). For more details see Lin and Baumgaertner (2000). An initial equilibrated configuration for the lipid membrane in a water box consisting of 391 POPC lipids in the liquid-crystalline phase and 18,781 water molecules was generously provided by K. Gerwert (Kandt et al., 2004).
In both G and M states of bR, electrostatic potentials for retinylidene-Lys216 complex were calculated by the Gaussian98 (Frisch et al., 1998) program using Hartree-Fock theory with the 6-31G* basis set. Then the RESP technique (Cieplak et al., 1995) was used to calculate the partial charges. The torsional potentials for the dihedral angles of the main polyene chain of the protonated retinylidene from C5 = C6–C7 = C8 to C14–C15 = N16–Cɛ were taken from Tajkhorshid et al. (2000). Torsional potentials for the unprotonated retinylidene were set to the same values except for the dihedral angle C12 = C13–C14 = C15, for which the potential was taken from Hayashi et al. (2002).
Root mean-square (RMS) deviations between high resolution x-ray structures of bR ground state (1C3W, 1CWQ, 1QHJ), measured for the backbone atoms of residues 6–154 and 168–223, are 0.52 Å(1C3W, 1CWQ), 0.55 Å(1CWQ, 1QHJ), and 0.70 Å(1C3W, 1QHJ), respectively. Internal water molecules presented in these structures coincide quite well, except water 502 in the 1C3W, which corresponds to water 720 in the 1CWQ and has no correspondence in the 1QHJ structure, and water molecule 409 in the 1QHJ, which has no correspondence in other structures. For the late M intermediate there are two x-ray structures available. One of them is wild-type structure 1CWQ by Sass et al. (2000) and the other is D96N mutant structure 1C8S by Luecke et al. (1999). The RMS deviation between these structures is 1.22 Å. 1CWQ has four more water molecules (721, 722, 723, and 740) in the cytoplasmic part of the protein and three additional molecules (712, 714, 716) in the extracellular part in the region of Arg82 and Glu204 residues. The 1CWQ structure was chosen for the latter simulations since it is the only structure available so far for the wild-type of the late M state. We have chosen the 1CWQ M-state structure; we also took the ground (G) state structure from the 1CWQ. The crystals used for G and M states were grown and treated under the same conditions. In addition, as has been shown above, internal water molecules in the 1CWQ G-state structure coincide well with water molecules in the 1C3W structure, which has a higher resolution.
The initial structures of the bacteriorhodopsin trimer in its G and M states were constructed using crystallographic data (Sass et al., 2000; Protein Data Bank, PDB code 1CWQ). Unfortunately residues 1 and 240–248 remain unresolved in both G and M structures. They have been constructed using the XLEAP program (Pearlman et al., 1995). All crystallographically determined 77 water molecules per monomer (where only 18 and 22 molecules are actually inside the G and M states of bR, respectively) were preserved. Default protonation states were assumed for all residues apart from Asp96, Asp115, Glu204, and Schiff base, which were protonated in the G state, and Asp85, Asp96, and Asp115, which were protonated in the M state. The protein was then inserted into the membrane. The protein solvent accessible surface was constructed using the MSMS program (Sanner et al., 1996) and all water molecules overlapping with the protein surface were removed. The final system consisted of 744 amino acids, 391 lipids, and 18702 TIP3P (Jorgensen et al., 1983) water molecules.
MD simulation of 1.5 ns was performed to equilibrate the lipid-protein and water-protein interfaces. During this simulation, any motion of the protein and crystallographically determined water atoms were prohibited, in order to preserve the original protein structure. The last configuration of the whole system after this equilibration process was then used as the start configuration for the subsequent production run of 2500 ps.
Molecular dynamics simulations
The molecular dynamics (MD) simulation was performed at constant pressure, constant particle number, and constant temperature (NPT ensemble). The simulations were done using SANDER in AMBER 7.0 (Pearlman et al., 1995) installed on the CRAY T3E at the Forschungszentrum Jülich. Berendsen thermostat and barostat (Berendsen et al., 1984) were used to keep the system at the specified temperature (300 K) and at constant pressure (1 bar). All atoms were coupled independently to the thermostat with the same coupling constant of 0.2 ps, and the center-of-mass motion was removed at each picosecond, which eliminates the artifact due to the velocity rescaling scheme (Harvey et al., 1998). Anisotropic pressure scaling was used, which couples the instantaneous pressure to the reference pressure independently in each of the three directions. A coupling constant of τp = 0.1 ps for the barostat was used. The average fluctuations of the box dimensions are found to be very small, in the order of 0.5 Å. The average dimension of the periodic cell was ∼105 Å × 117 Å × 86 Å in x,y,z directions, respectively, for both G- and M-trimers. The average RMS distances between simulated and crystallographic structures, calculated for the backbone atoms of α-helices, were 0.85 Å for the G state and 1.02 Å for the M state. In particular, helix F in the M state had the largest deviation of 1.26 Å from the crystallographic structure among all the helixes. Although there is no visible tilt observed in helix F, its RMS deviation indicates that this helix is able to perform a large conformational change.
The SHAKE algorithm (Ryckaert et al., 1977) was used, which keeps the bonds of hydrogen atoms fixed. This allows the use of a larger time step of 2 fs. The Coulomb interaction was calculated using the particle-mesh Ewald method. The order of B-spline interpolation for particle-mesh Ewald algorithm was set to 4, which implies a cubic spline approximation. The direct sum tolerance was set to be 0.00001. The scale factors for 1–4 electrostatic interactions and for 1–4 van der Waals interactions were both set to 2.0 (Pearlman et al., 1995). The atomic coordinates were saved every 0.1 ps and the atomic velocities were saved every 10 ps. After 1.5 ns of equilibration, the production run of 2.5 ns followed. Trajectories of the last 1 ns were used for water statistics data gathering. MD simulations were performed on a bR trimer and the results normalized to one monomer of bR.
Hydrogen-bonded networks
Here we explain some technical details of our calculations of hydrogen-bonded chains between water molecules and amino-acid side chains. Since we are only interested in hydrogen-bonded pathways within bR, we have to define properly which water molecules, out of all water molecules in the system, are inside the protein. There have already been some studies on definitions of internal water molecules. Garcia and Hummer (2000) distinguished water molecules according to their coordination numbers. Bakowies and van Gunsteren (2002) used a triangulated surface with vertices defined by Cα atom positions to define the interior of a protein.
Here we outline our own algorithm. To each water molecule one can assign its own Cartesian coordinate system with the origin placed to the center of the oxygen atom. In this coordinate system we can define eight neighboring cubic cells of linear size a = 5 Å. Their centers are positioned at all eight possible locations out of the vectors {± a/2, ± a/2, ± a/2}, e.g., the center of one of the eight cells is given by the vector {a/2, a/2, a/2}, another one by {−a/2, a/2, a/2}, and so on. If all eight cells are occupied by at least one atom of an amino acid, then this water molecule under consideration is said to be inside the protein and termed as an internal water molecule (IWM). In this sense we can also define the surface of the protein, which separates internal from external water molecules.
Accordingly, a hydrogen-bonded chain exists when the protein surface or a residue inside the protein is connected via internal water molecules to a particular target atom of an internal amino acid. We can construct a particular hydrogen-bonded path from any internal amino-acid residue, which must consist of internal water molecules and terminates at another residue or at the first external water molecule encountered during construction of such a path toward the surface. Each walk is constructed to be self-avoiding. An example of such a network for the G state is shown in the Supplementary Material. A hydrogen bond is said to exist if the distance between H and O of two different water molecules is ≤2.2 Å and the angle of this hydrogen bond, H⋯O, and the subsequent O–H bond of the second water molecule, i.e., H⋯O–H, is >120° angle.
RESULTS AND DISCUSSION
Dynamics of water molecules
Following the definition of internal and external water molecules, as given in the previous section, the number of internal water molecules in bR was calculated. In addition, we distinguished between water molecules which are diffusive, i.e., they have entered during the simulations and eventually have left the protein before the end of the simulation, and trapped water molecules, which were inside the protein during the simulation time of 2.5 ns. In Fig. 1, the number 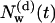 of diffusive and
of diffusive and 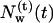 of trapped water molecules as a function of time t is shown. Since the number of trapped water molecules is constant,
of trapped water molecules as a function of time t is shown. Since the number of trapped water molecules is constant, 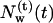 is just a straight horizontal line. Inasmuch as there are many water molecules entering the protein transiently, a precise definition for a set of diffusive water molecules is necessary. In particular, those water molecules crossing the mathematically defined protein surface and penetrating up to a few Ångstroms before exiting again have to be excluded from this set. Therefore, we determined a residence time threshold τ0 of water molecules inside the protein and defined those water molecules as diffusive water molecules, which reside in the protein longer than τ0. For this purpose we calculated from the trajectory the penetration depth of a water molecule into the protein within its residence time. The relation between the residence time τres and the penetration depth λ is shown in Fig. 2 a. The penetration depth λ is the distance from a water molecule to the protein surface in projection to the normal of the membrane. We have chosen a threshold residence time τ0 = 10 ps, which corresponds according to Fig. 2 a to λ = 2 Å and defined diffusive water molecules as water molecules with τres > τ0 and surface water molecules as water molecules with τres < τ0. The large scattering of the data in Fig. 2 a is mainly due to the definition of λ, which is the projection of the distance on the membrane normal. An important practical implication from Fig. 2 a is that all those IWMs, which have a penetration depth of λ < 2 Å (corresponding to residence times τres < 10 ps), should not be included in the comparison between the numbers of IWMs found by crystallographic methods and by simulation.
is just a straight horizontal line. Inasmuch as there are many water molecules entering the protein transiently, a precise definition for a set of diffusive water molecules is necessary. In particular, those water molecules crossing the mathematically defined protein surface and penetrating up to a few Ångstroms before exiting again have to be excluded from this set. Therefore, we determined a residence time threshold τ0 of water molecules inside the protein and defined those water molecules as diffusive water molecules, which reside in the protein longer than τ0. For this purpose we calculated from the trajectory the penetration depth of a water molecule into the protein within its residence time. The relation between the residence time τres and the penetration depth λ is shown in Fig. 2 a. The penetration depth λ is the distance from a water molecule to the protein surface in projection to the normal of the membrane. We have chosen a threshold residence time τ0 = 10 ps, which corresponds according to Fig. 2 a to λ = 2 Å and defined diffusive water molecules as water molecules with τres > τ0 and surface water molecules as water molecules with τres < τ0. The large scattering of the data in Fig. 2 a is mainly due to the definition of λ, which is the projection of the distance on the membrane normal. An important practical implication from Fig. 2 a is that all those IWMs, which have a penetration depth of λ < 2 Å (corresponding to residence times τres < 10 ps), should not be included in the comparison between the numbers of IWMs found by crystallographic methods and by simulation.
FIGURE 1.

Number of diffusive and trapped internal water molecules, 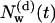 and
and 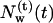 , respectively, as a function of time t, in the G and M states.
, respectively, as a function of time t, in the G and M states.
FIGURE 2.
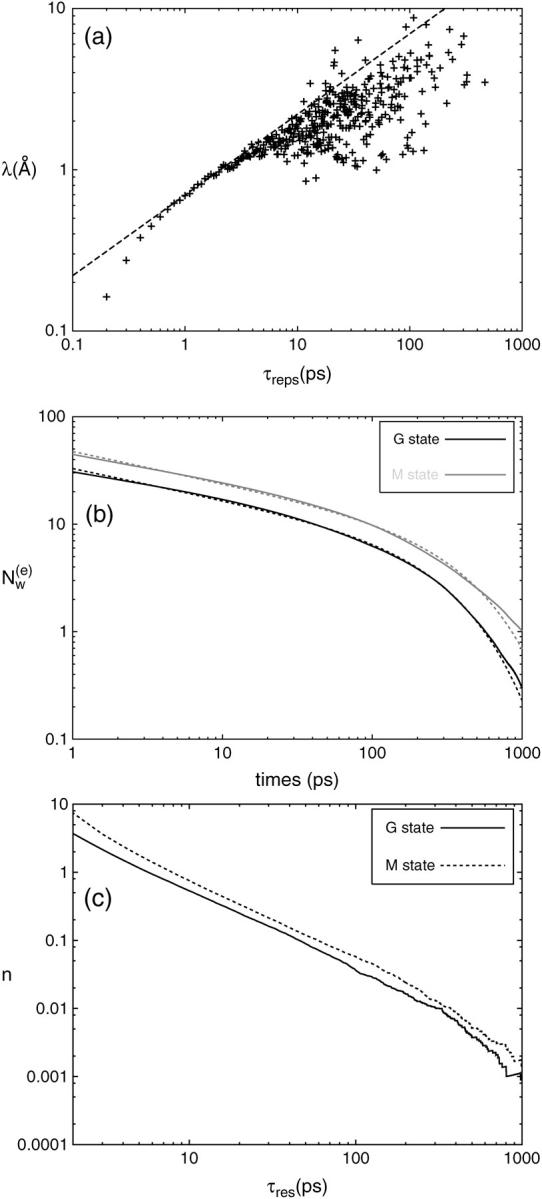
(a) Log-log plot of penetration depth λ versus residence times τres (crosses). The value λ is measured with respect to the protein surface along the membrane normal. The broken line represents the classical diffusion law 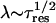 , for comparison. (b) Log-log plot of the decay of a set of diffusive internal water molecules (IWMs) as a function of time for the G and M states.
, for comparison. (b) Log-log plot of the decay of a set of diffusive internal water molecules (IWMs) as a function of time for the G and M states.  is the average number of diffusive IWMs of a set after time t. The broken lines correspond to t−α × exp(− t/τcor). (c) Probability distribution n(τres) of the number of diffusive water molecules as a function of their residence time τres.
is the average number of diffusive IWMs of a set after time t. The broken lines correspond to t−α × exp(− t/τcor). (c) Probability distribution n(τres) of the number of diffusive water molecules as a function of their residence time τres.
From Fig. 1, averaging the data for the last 1-ns, one obtains for the G state nine trapped water molecules and an average of 19 diffusive water molecules, whereas in the M state one obtains 13 trapped and an average of 23 diffusive water molecules. For comparison, the crystal structure by Sass et al. (2000) contains 18 IWMs in the G and 22 IWMs in the M states. Edholm et al. (1995) performed MD simulation of bR-trimer in its ground state and obtained nine trapped water molecules. The simulation data of Baudry et al. (2001) provided only eight trapped water molecules in the G state. Kandt et al. (2004) reported ∼11 trapped water molecules in the G state, two more than in our case. These two additional trapped molecules are located in the extracellular domain of the protein near residue Glu194. In our simulated structure this part of the protein is accessible for the bulk water and, as will be shown below, only diffusive water molecules reside here.
A significant decrease in the number of diffusive water molecules is seen during the first 1.5 ns, indicating the effect of the structural fluctuations of the protein. The initial configuration of the whole system at time t = 0 contained a fully equilibrated ensemble of lipids and water molecules, albeit with a bR structure frozen at the crystallographic coordinates. Therefore, the decrease of 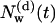 to a much smaller value than initially observed indicates that a rigid porous protein structure may incorporate on average considerably more water molecules than a soft flexible structure.
to a much smaller value than initially observed indicates that a rigid porous protein structure may incorporate on average considerably more water molecules than a soft flexible structure.
The explanation for the large difference between the numbers of IWMs found by simulation and in the crystal structure consists in the high exchange rate of diffusive water molecules. Therefore, it is of interest to characterize the time-dependent properties of the IWMs. We have estimated from the simulation data typical residence times and correlation times. This is shown in Fig. 2, b and c.
Consider a particular set of diffusive IWMs at a certain time t0. For this set we can define the exchange (or turnover) time τcor as the time after which the initial number, 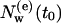 , of diffusive IWMs of this set has decreased to a fraction
, of diffusive IWMs of this set has decreased to a fraction 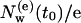 . The average decay of such a set is shown in Fig. 2 b, where
. The average decay of such a set is shown in Fig. 2 b, where 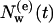 is the average number of the remaining diffusive IWMs still in internal positions after time t. From Fig. 2 b, where
is the average number of the remaining diffusive IWMs still in internal positions after time t. From Fig. 2 b, where 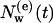 is presented in a log-log plot, two time regimes are observed. At larger times, t > 10 ps, the function follows an exponential law,
is presented in a log-log plot, two time regimes are observed. At larger times, t > 10 ps, the function follows an exponential law,
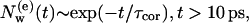 |
(1) |
where the correlation time is τcor ≈ 340 ps for the G state and τcor ≈ 460 ps for the M state, characterizing the time evolution of the exchange process of those diffusive IWMs that penetrate deeply into bR.
Makarov et al. (2000), having used MD simulations, calculated residence times for different hydration sites of myoglobin. Many sites, depending on their position, have residence time >100 ps. The longest time is 456 ps. Although myoglobin is a water-soluble protein, the residence time of some of their hydration sites are comparable with our results.
At shorter times, t < 10 ps, there are rapid exchange processes due to the surface water molecules, which do not perform a deeper penetration into bR. Hence 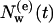 decreases in this time regime more rapidly than at larger times, and obeys an approximate power law of
decreases in this time regime more rapidly than at larger times, and obeys an approximate power law of
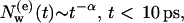 |
(2) |
where the coefficient α ≈ 0.29 for both G and M states, which clearly shows that protein and water dynamics on the short timescale t ∼10 ps are independent of the particular protein conformation. These times are in good agreement with experiments of Denisov and Halle (1995a, b, 1996) and Denisov et al. (1999).
A second quantity that corroborates this view and that characterizes the dynamics of diffusive IWMs is the distribution of residence times in a given set of diffusive IWMs. The residence time, τres, is the time that one of the diffusive IWMs spends inside the protein. Defining n(τres) as the probability distribution of IWMs with residence time τres found in a given set of diffusive IWMs, the average over many sets (taken from our simulation data) can be calculated, which is presented in Fig. 2 c. From this distribution n(τres) we have estimated the average residence time of a diffusive water molecule as
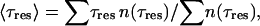 |
(3) |
which yields 〈τres〉 ≈ 95 ps for the G state, and 〈τres〉 ≈ 110 ps for the M state. Again, this result explains why it is very difficult to detect by crystallographic methods all water molecules inside bR.
Distribution of water molecules
Within the time range of 2.5 ns our simulations demonstrate that, with respect to the distribution of water molecules, bR is divided into extracellular and cytoplasmic parts separated by an impermeable structural boundary both for the G and the M states. This is demonstrated in Fig. 3 for one bR molecule of the G-trimer (Fig. 3, left) and the M-trimer (Fig. 3, right), respectively, taken from crystallographic structures (Sass et al., 2000). The volume occupied by one bR molecule is represented by the gray surface. Superimposed on the gray surface as depicted by blue and yellow triangulated nets are the surfaces of the volumes accessible to diffusive and trapped water molecules, respectively. The red balls indicate the positions of water molecules as found by crystallographic studies (Sass et al., 2000). Water-accessible volumes in the late M state are larger both for trapped and diffusive water molecules. This is explained by the structural changes in this M state, which cause a total volume increase of internal cavities. Our diffusive water distribution differs significantly from the results presented by Baudry et al. (2001). They reported that external water molecules penetrate into the protein up to residue Asp96 in the cytoplasmic part and to residue Arg82 in the extracellular part. On the contrary, in our G state simulation residues Asp96 and Arg82 are inaccessible for the diffusive (or external, in their notation) water molecules. Kandt et al. (2004) showed a similar water distribution pattern for the central part of bR.
FIGURE 3.
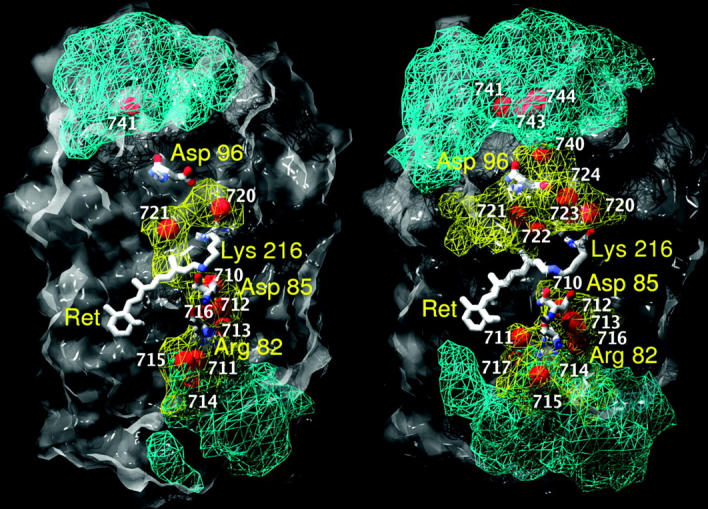
Accessible volumes for internal water molecules of the G state (left) and the M state of bR (right). The surfaces of the volumes for trapped and diffusive water molecules are represented by yellow and blue triangulated nets, respectively. The red balls represent the positions of internal water molecules as identified by crystallographic studies (Sass et al., 2000).
The equilibrium distribution of internal water molecules (IWMs) in bR is shown in Fig. 4 for the G (a) and M (b) states, respectively. We have calculated the number density nw(z) with respect to the z axis (membrane normal), i.e., the average number of IWMs within a slab of thickness δz = 1 Å. The origin z = 0 is placed at the center of mass of the protein, which is very close to the Schiff base nitrogen at z = +2.5 Å. As indicated in Fig. 4, we have discriminated between diffusive and trapped water molecules. The corresponding analysis of these sets in the G and M states yields 19 and 23 diffusive IWMs and 9 and 13 trapped IWMs, respectively.
FIGURE 4.
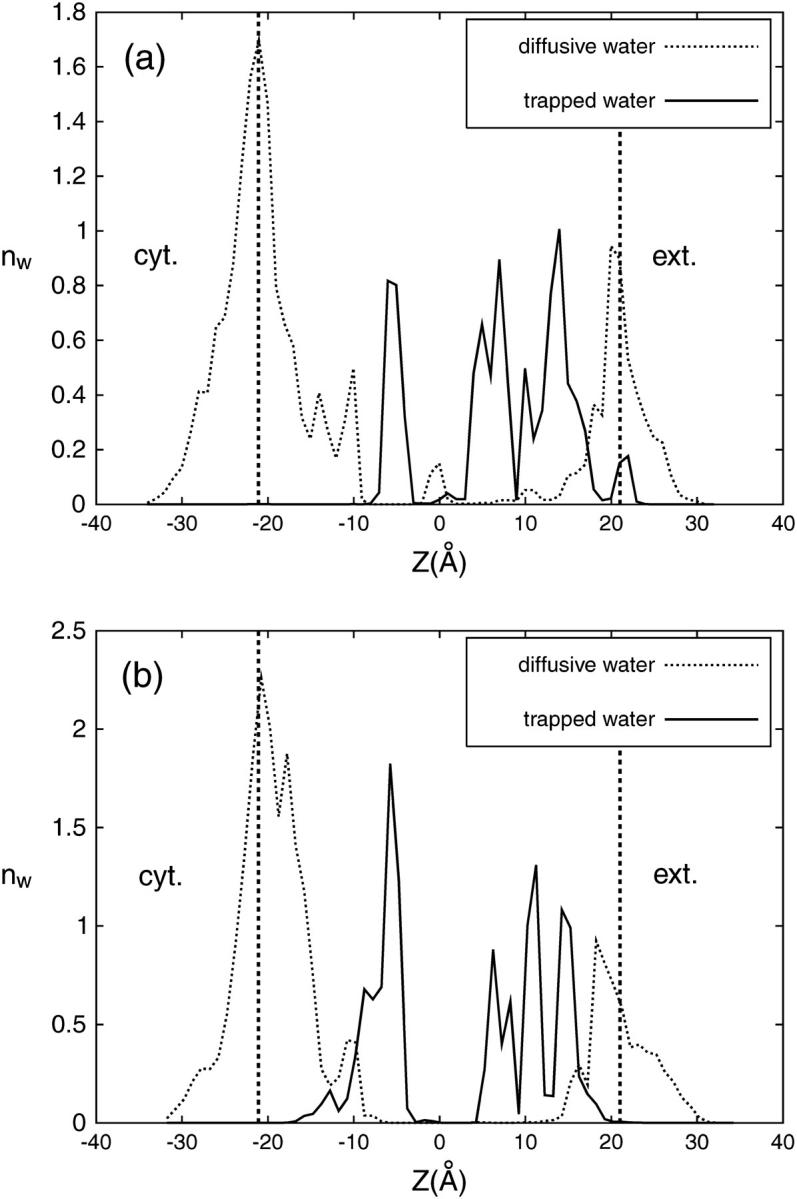
One-dimensional number density nw(z) of water molecules in projection to the membrane normal in the G state (a) and M state (b) as found by simulation. The full and the dotted lines denote diffusive and trapped water molecules, respectively. The two broken vertical lines indicate the average positions of the two membrane surfaces defined by the average locations of the nitrogen atoms of the lipid headgroups. Zero point on the z axis corresponds to the center of mass of the protein. The cytoplasmic part of the protein is on the left and extracellular part on the right. The nitrogen atom of the Schiff base, as a reference point, has a z coordinate of +2.5.
The first remarkable feature of the distribution nw(z) is the gap in the distribution of diffusive water molecules. This suggests that migration of water does not occur between the extracellular and cytoplasmic parts of bR during our simulation and that this gap reflects a kind of water barrier in the protein. However, there might exist a tiny water population in this region, which is not distinguishable in the distribution nw(z). To confirm the existence of this water barrier in the protein we also analyzed all internal water trajectories and did not find any migration of water molecules between extracellular and cytoplasmic parts of bR. This observation is in agreement with the data provided by Baudry et al. (2001) and Kandt et al. (2004), where no external water molecules move to the retinal binding site. The existence of this impenetrable structural interface between the cytoplasmic and extracellular parts of bR was already concluded from the crystallographic data, and is a necessary feature of bR prohibiting a leakage of water across the membrane.
Comparing positions of trapped water molecules with those determined from crystallographic studies (Fig. 3), we found more differences in the M state than in the G state, which is related to the higher water mobility in the M state. To estimate water mobility we calculated for every trapped water molecule the radial probability distribution nr(d) as a function of distance d between the positions from crystallographic and simulation data. This is shown in Fig. 5 for the G state (the a plot) and the M state (the b and c plots). The peak of the distribution corresponds to a most probable deviation from the crystallographic position. Water molecules with distribution peaks at d < 1 Å may be considered as immobile. As can be seen, all nine trapped water molecules in the G state are immobile, whereas in the M state they are only 5 out of 13. MD simulation of the G state performed by Kandt et al. (2004) revealed a high mobility of water molecules 710, 712, 716 (in their notation 402BL, 406BL, 401BL, respectively) with fluctuations of >4 Å and a standard deviation of 0.6 ± 0.4 Å. Our data of the G state show less fluctuations in the position of the trapped water molecules with the most probable deviations from the initial structure of <0.7 Å. We also fitted radial probability distribution functions in Fig. 5 with Gaussians and obtained standard deviations in the G state for the water molecules 710, 712, and 716 of 0.30 ± 0.10 Å, 0.42 ± 0.12 Å, and 0.40 ± 0.13 Å, respectively. These values are smaller than reported by Kandt et al. (2004). In general, our trapped water molecules match better with the x-ray model. For instance, water molecule 720, which has left its initial position in the simulations by Kandt et al. (2004), is, in our case, distant from the starting position by 0.3 ± 0.05 Å with a standard deviation of 0.22 ± 0.15 Å. The highest deviation from its crystallographically determined position has water molecule 710. This is apparently due to the highly polarizable Asp85-Asp212-SB interior, which cannot be sufficiently well described by a classical model (Hayashi and Ohmine, 2000). In Supplementary Material we represent trapped water molecules inside bR in a different saturation of red color. The more the color is saturated, the more the water molecule is stable.
FIGURE 5.
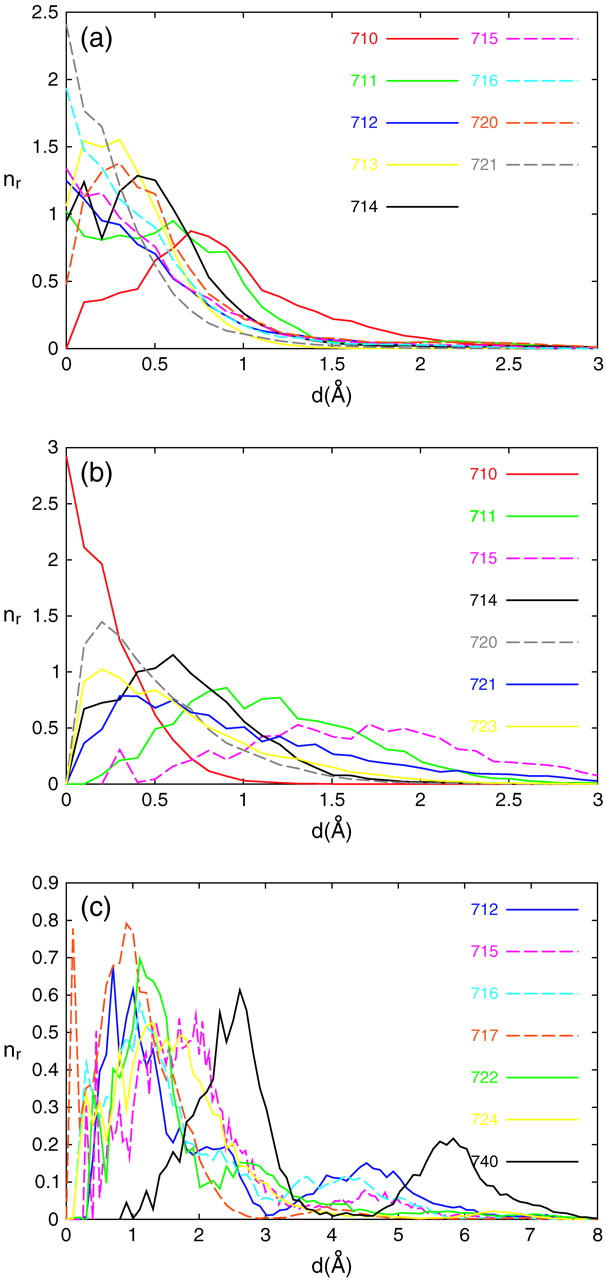
Radial probability distribution nr versus distance d between simulated and crystallographically defined positions of water molecules. In a, data for the G state is presented, with data for the M state split between b and c for clarity. For the numbering of water molecules, see Fig. 3.
In Fig. 5 distributions with more than one peak correspond to water molecules, which have different positions in different monomers. In the case of the M state, several water molecules escaped from the cytoplasmic interior of the protein during the simulation. We observed three such events. Water molecule 740, about which x-ray density was doubtful (Sass et al., 2000), escaped from two monomers of the trimeric protein during the simulation, whereas this water molecule in the third monomer shifted its position significantly toward the retinylidene residue. We also observed one more escape (water molecule 722 in one out of three monomers) and one penetration of a bulk water molecule toward the cytoplasmic side of the protein (up to water molecules 720, 723, and 724). For the picture of the escape and penetration pathways one should look to the Supplementary Material.
Hydrogen-bonded network
During the photocycle, protons are vectorially transported from the cytoplasmic side to the extracellular environment. During this process a proton is released by the Schiff base and another is captured. This implies that hydrogen-bonded pathways must exist between the cytoplasmic surface and the Schiff base via the side-chain Asp96 and between the Schiff base and the extracellular surface of the protein via several side chains (Asp85, Glu204, Glu194) as shown by infrared spectroscopy (Gerwert et al., 1989; Heberle et al., 2000; Dioumaev et al., 1998).
With regard to the previous section, which is concerned with the water population in bR, we now raise the question of the quantitative contribution of water molecules in generating hydrogen-bonded pathways between the surface and the core of bR.
To construct chains of hydrogen bonds (hydrogen-bonded pathways) we have used the Grotthuss relay model, or the structural-proton diffusion model, for a proton transport. Since there is a vast amount of literature on this subject, we simply cite a few of the more recent articles (Knapp et al., 1980; Tuckerman et al., 1995; Agmon, 1995; Pomes and Roux, 1998; Vuillemier and Borgis, 1998; Mei et al., 1998; Schmitt and Voth, 1999). In their studies ordered chains of water molecules are considered, where one path consists of an alternating sequence of hydrogen bonds between water molecules, H⋯O, separated by O–H bonds of water molecules. The protons are assumed to hop along such a path, which results in a reorientation of the participating water molecules. Many refinements of this model have been proposed. The most accurate approach is based on a density functional calculation (Marx et al., 1999). Despite the simplicity of the Grotthuss model, its application, in particular to biological systems, has led to valuable insights of proton transport governed by the concerted actions of spontaneously forming hydrogen-bonded networks and the structural fluctuations of the embedding proteins (Sagnella et al., 1996; Brewer et al., 2001; Pomes and Roux, 2002). In the present study, however, the Grotthuss-path model is used as a static geometrical construct rather than a dynamical one. A dynamical picture would include certain time-dependent correlations between proton and water displacements, which is out of the scope of the present work. Using the static picture, the present approach can be considered as a description of the capability of bR to form spontaneously Grotthuss-pathways, which are relevant for the proton transport. There might be different mechanisms for proton translocation inside the protein. Here we only consider the Grotthuss model with a continuous chain of water-water hydrogen bonds, which connects proton donor and acceptor sites of two different residues. We assume that a proton moves fast between the residues and can be stored only at specific sites of polar amino acids. Reorientation of the internal water molecules then might occur, which will make it possible for the proton to move to the next residue. Such a Grotthuss mechanism with a continuous chain of water-water hydrogen bonds is essentially barrierless and the most favorable for fast proton transport (Pomes and Roux, 1998). Hydrogen bonded pathways between different nearby residues inside the protein consist only of a few water molecules. Hence, we do not consider water molecule reorientations and a break of the hydrogen bonded network during a virtual proton move from one residue to another.
Based on the analysis of MD trajectories, we have calculated all possible Grotthuss-pathways inside the protein. Typical snapshots for the G and M states are depicted in Supplementary Material. From these trajectories we observed that several key residues are involved in the pathway. We have also calculated probabilities of forming different Grotthuss-pathways between polar residues and the surface of the protein for the G and M states. The probability map is presented in Fig. 6. We find these pathways to be quite different for the G and M states. For instance, we see a connection between residue Glu204 and the protein surface only in the M state (see also Supplementary Material). There are possible connections between the Schiff base and residues Asp85, Asp212, and Arg82 only in the G state. We did not observe any connections between Asp96 and cytoplasmic surface of the protein in either the G or the M states. We only found a connection in the M state between Asp96 and Lys216. It is interesting to note that in the M state Asp85 and Asp212 are always linked by a hydrogen-bonded network, which can help a proton to hop from one residue to the other. It is difficult to draw any precise conclusions about the pathway from Asp96 to the Schiff base. In the M state we found a possible connection from Asp96 carboxyl group to the backbone oxygen of Lys216. We should address this question to a structure of the N intermediate, since hopping processes are assumed to take place between the late M and N states.
FIGURE 6.

All possible Grotthuss-pathways in the protein are shown with arrows for the G state (left) and for the late M state (right). The numbers at the arrows denote probabilities of the corresponding connections.
In both G and M states, we found a pathway from Asp85 to the possible proton release groups Glu194 and Glu204. This pathway includes internal water molecules along with the residues Asp212 and Arg82. Although residue Glu194 is assumed to assist the proton movement inside bR (Brown et al., 1995; Balashov et al., 1997; Dioumaev et al., 1998), we did not find a noticeable connection from this residue to Glu204 and bulk.
Despite the fact that several pathways can be constructed, the possible existence of such pathways is not sufficient for an actual proton translocation to take place. The stabilization of a proton in a hydrogen-bonded complex AH⋯B depends on the local environment of the complex. The electric field, induced by the nearby dipoles, can dramatically change the shape of the double-well potential in the complex and force the proton to move from one well into the other (Staib et al., 1994). Since highly polarizable medium cannot be well described by the presented classical model (Hayashi and Ohmine, 2000), the effect of the polarizability on the proton translocation in bR will be the subject of our future study.
SUMMARY AND CONCLUSIONS
Using molecular dynamics simulation we have studied the spatial distribution of water molecules and their corresponding hydrogen-bonded networks inside bacteriorhodopsin in its G and M state conformations.
We found a much higher number of internal water molecules (trapped + diffusive) in the simulations, 28 and 36 in G and M states, respectively, as compared to 18 and 22 (Sass et al., 2000) from crystallographic data.
We described the mobility of trapped water molecules and calculated the radial distribution function for each of them. The typical residence time of a diffusive water molecule inside the protein is 95 ps for the G state and 110 ps for the M state. We calculated the probabilities of forming Grotthuss hydrogen-bonded networks between different residues inside bR. This network is different in the G and M states. Using this information we suggest possible pathways for proton hopping and water penetration inside bR.
It is of interest to extend and compare the present study to other intermediates of bacteriorhodopsin, particularly later intermediates. This should provide new insights to the proton transport across the cytoplasmic and extracellular domains.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
We are grateful to C. Kandt and K. Gerwert for providing the equilibrated structure of POPC membrane and for useful discussions. We also thank J. Heberle for numerous discussions and suggestions.
This study was supported by the Alexander-von-Humboldt Foundation.
References
- Agmon, N. 1995. The Grotthuss mechanism. Chem. Phys. Lett. 244:456–462. [Google Scholar]
- Baudry, J., E. Tajkhorshid, F. Molnar, J. Phillips, and K. Schulten. 2001. Molecular dynamics study of bacteriorhodopsin and the purple membrane. J. Phys. Chem. B. 105:905–918. [Google Scholar]
- Bakowies, D., and W. F. van Gunsteren. 2002. Water in protein cavities: a procedure to identify internal water and exchange pathways and application to fatty acid-binding protein. Proteins. 47:534–545. [DOI] [PubMed] [Google Scholar]
- Balashov, S. P., E. S. Imasheva, T. G. Ebrey, N. Chen, D. R. Menick, and R. K. Crouch. 1997. Glutamate-194 to cysteine mutation inhibits fast light-induced proton release in bacteriorhodopsin. Biochemistry. 36:8671–8676. [DOI] [PubMed] [Google Scholar]
- Bashford, D., and K. Gerwert. 1992. Electrostatic calculations of the pKa values of ionizable groups in bacteriorhodopsin. J. Mol. Biol. 224:473–486. [DOI] [PubMed] [Google Scholar]
- Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- Betancourt, F. M. H., and R. M. Glaeser. 2000. Chemical and physical evidence for multiple functional steps comprising the M state of the bacteriorhodopsin photocycle. Biochim. Biophys. Acta. 1460:106–118. [DOI] [PubMed] [Google Scholar]
- Brewer, M. L., U. W. Schmitt, and G. A. Voth. 2001. The formation and dynamics of proton wires in channel environments. Biophys. J. 80:1691–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, L. S., J. Sasaki, H. Kandori, A. Maeda, R. Needleman, and J. K. Lanyi. 1995. Glutamic acid 204 is the terminal proton release group at the extracellular surface of bacteriorhodopsin. J. Biol. Chem. 270:27122–27126. [DOI] [PubMed] [Google Scholar]
- Cieplak, P., W. D. Cornell, C. Bayly, and P. A. Kollman. 1995. Application to the multimolecule and multiconformational RESP methodology to biopolymers: charge derivation for DNA, RNA and proteins. J. Comput. Chem. 16:1357–1377. [Google Scholar]
- Dencher, N. A., H. J. Sass, and G. Büldt. 2000. Water and bacteriorhodopsin:structure, dynamics, and function. Biochim. Biophys. Acta. 1460:192–203. [DOI] [PubMed] [Google Scholar]
- Denisov, V. P., and B. Halle. 1995a. Hydrogen exchange and protein hydration: the deuteron spin relaxation dispersions of bovine pancreatic trypsin inhibitor and ubiquitin. J. Mol. Biol. 245:698–709. [DOI] [PubMed] [Google Scholar]
- Denisov, V. P., and B. Halle. 1995b. Protein hydration dynamics in aqueous solution: a comparison of bovine pancreatic trypsin inhibitor and ubiquitin by oxygen-17 spin relaxation dispersion. J. Mol. Biol. 245:682–697. [DOI] [PubMed] [Google Scholar]
- Denisov, V. P., and B. Halle. 1996. Protein hydration dynamics in aqueous solution. Faraday Discuss. 103:227–244. [DOI] [PubMed] [Google Scholar]
- Denisov, V. P., B. H. Johnson, and B. Halle. 1999. Hydration of denatured and molten globule proteins. Nat. Struct. Biol. 6:253–260. [DOI] [PubMed] [Google Scholar]
- Dioumaev, A. K., H. T. Richter, L. S. Brown, M. Tanio, S. Tuzi, H. Saitá, Y. Kimura, R. Needleman, and J. K. Lanyi. 1998. Existence of a proton transfer chain in bacteriorhodopsin: participation of Glu-194 in the release of protons to the extracellular surface. Biochemistry. 37:2496–2506. [DOI] [PubMed] [Google Scholar]
- Edholm, O., O. Berger, and F. Jähnig. 1995. Structure and fluctuations of bacteriorhodopsin in the purple membrane: a molecular dynamics study. J. Mol. Biol. 250:94–111. [DOI] [PubMed] [Google Scholar]
- Ernst, J. A., R. T. Clubb, H. X. Zhou, A. M. Gronenborn, and G. M. Clore. 1995. Demonstration of positionally disordered water within a protein hydrophobic cavity by NMR. Science. 267:1813–1817. [DOI] [PubMed] [Google Scholar]
- Essmann, U., L. Perera, and M. L. Berkowitz. 1995. The origin of the hydration interaction of lipid bilayers from MD simulation of dipalmitoylphosphatidylcholine membranes in gel and liquid crystalline phases. Langmuir. 11:4519–4531. [Google Scholar]
- Feller, S., and A. D. MacKerell, Jr. 2000. An improved empirical potential energy function for molecular simulations of phospholipids. J. Phys. Chem. B. 104:7510–7515. [Google Scholar]
- Frisch, M. G., G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, et al. 1998. Gaussian 98, Rev. A.6. Gaussian, Pittsburgh, PA.
- Garcia, A. E., and G. Hummer. 2000. Water penetration and escape in proteins. Proteins. 38:261–272. [PubMed] [Google Scholar]
- Gerwert, K., B. Hess, J. Soppa, and D. Oesterhelt. 1989. Role of Aspartate-96 in proton translocation by bacteriorhodopsin. Proc. Natl. Acad. Sci. USA. 86:4943–4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk, M., N. A. Dencher, and B. Halle. 2001. Microsecond exchange of internal water molecules in bacteriorhodopsin. J. Mol. Biol. 311:605–621. [DOI] [PubMed] [Google Scholar]
- Harvey, S. C., R. K.-Z. Tan, and T. E. Cheatham III. 1998. The flying ice cube: velocity rescaling in molecular dynamics leads to violation of energy equipartition. J. Comput. Chem. 19:726–740. [Google Scholar]
- Haupts, U., J. Tittor, and D. Oesterhelt. 1999. Closing in on bacteriorhodopsin: progress in understanding the molecule. Annu. Rev. Biophys. Biomol. Struct. 28:367–399. [DOI] [PubMed] [Google Scholar]
- Hayashi, S., and I. Ohmine. 2000. Proton transfer in bacteriorhodopsin: structure, excitation, IR spectra, and potential energy surface analyses by an ab initio QM/MM method. J. Phys. Chem. B. 104:10678–10691. [Google Scholar]
- Hayashi, S., E. Tajkhorshid, and K. Schulten. 2002. Structural changes during the formation of early intermediates in the bacteriorhodopsin photocycle. Biophys. J. 83:1281–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heberle, J. 2000. Proton transfer reactions across bacteriorhodopsin and along the membrane. Biochim. Biophys. Acta. 1458:135–147. [DOI] [PubMed] [Google Scholar]
- Heberle, J., J. Fitter, H. J. Sass, and G. Büldt. 2000. Bacteriorhodopsin: the functional details of a molecular machine are being resolved. Biophys. Chem. 85:229–248. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Compassion of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- Jorgensen, W. L., and J. Tirando-Rives. 1988. The OPLS potential functions for proteins. Energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 110:1657–1666. [DOI] [PubMed] [Google Scholar]
- Kandori, H. 2000. Role of internal water molecules in bacteriorhodopsin. Biochim. Biophys. Acta. 1460:177–191. [DOI] [PubMed] [Google Scholar]
- Kandt, C., J. Schlitter, and K. Gerwert. 2004. Dynamics of water molecules in the bacteriorhodopsin trimer in explicit lipid/water environment. Biophys. J. 86:705–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp, E. W., K. Schulten, and Z. Schulten. 1980. Proton conduction in linear hydrogen-bonded systems. Chem. Phys. 46:215–229. [Google Scholar]
- Kouyama, T., T. Nishikawa, T. Tokuhisa, and H. Okumura. 2004. Crystal structure of the L intermediate of bacteriorhodopsin: evidence for vertical translocation of a water molecule during the proton pumping cycle. J. Mol. Biol. 335:531–546. [DOI] [PubMed] [Google Scholar]
- Lanyi, J. K. 2001. X-ray crystallography of bacteriorhodopsin and its photointermediates: insights into the mechanism of proton transport. Biochemistry. 66:1192–1196. [DOI] [PubMed] [Google Scholar]
- Lanyi, J. K. 2004. Bacteriorhodopsin. Annu. Rev. Physiol. 66:665–688. [DOI] [PubMed] [Google Scholar]
- Levitt, M., and B. H. Park. 1993. Water: now you see it, now you don't. Structure. 1:223–226. [DOI] [PubMed] [Google Scholar]
- Lin, J. H., and A. Baumgaertner. 2000. Stability of a melittin pore in lipid bilayer: a molecular dynamics study. Biophys. J. 78:1714–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logunov, I., W. Humphrey, K. Schulten, and M. Sheves. 1995. Molecular dynamics study of the 13-cis form (bR548) of bacteriorhodopsin and its photocycle. Biophys. J. 68:1270–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luecke, H., B. Schobert, H.-T. Richter, J. P. Cartailler, and J. K. Lanyi. 1999a. Structure of bacteriorhodopsin at 1.55 Å resolution. J. Mol. Biol. 291:899–911. [DOI] [PubMed] [Google Scholar]
- Luecke, H., B. Schobert, H.-T. Richter, J.-P. Cartailler, and J. Lanyi. 1999b. Structural changes in bacteriorhodopsin during ion transport at 2 Ångstrom resolution. Science. 286:255–260. [DOI] [PubMed] [Google Scholar]
- Luecke, H. 2000. Atomic resolution structures of bacteriorhodopsin photocycle intermediates: the role of discrete water molecules in the function of this light-driven ion pump. Biochim. Biophys. Acta. 1460:133–156. [DOI] [PubMed] [Google Scholar]
- Makarov, V. A., B. K. Andrews, P. E. Smith, and B. M. Pettitt. 2000. Residence times of water molecules in the hydration sites of myoglobin. Biophys. J. 79:2966–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx, D., M. E. Tuckerman, J. Hutter, and M. Parinello. 1999. The nature of the hydrated excess proton in water. Nature. 397:601–604. [Google Scholar]
- Mei, H. S., M. E. Tuckerman, D. E. Sagnella, and M. E. Klein. 1998. Quantum nuclear ab initio molecular dynamics study of water wires. J. Phys. Chem. B. 102:10446–10458. [Google Scholar]
- Olkhova, E., M. C. Hutter, M. A. Lill, V. Helms, and H. Michel. 2004. Dynamic water networks in cytochrome-c oxidase from Paracoccus denitrificans investigated by molecular dynamics simulations. Biophys. J. 86:1873–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onufriev, A., A. Smondyrev, and D. Bashford. 2003. Proton affinity changes driving unidirectional proton transport in the bacteriorhodopsin photocycle. J. Mol. Biol. 332:1183–1193. [DOI] [PubMed] [Google Scholar]
- Otting, G., E. Liepinsh, and K. Wüthrich. 1991. Protein hydration in aqueous solution. Science. 254:974–980. [DOI] [PubMed] [Google Scholar]
- Otting, G., and E. Liepinsh. 1995. Protein hydration viewed by high-resolution NMR spectroscopy: implications for magnetic resonance image contrast. Acc. Chem. Res. 28:171–177. [Google Scholar]
- Papadopoulos, G., N. A. Dencher, G. Zaccai, and G. Büldt. 1990. Water of bacteriorhodopsin localized by neutron diffraction. J. Mol. Biol. 214:15–19. [DOI] [PubMed] [Google Scholar]
- Pearlman, D. A., D. A. Case, J. W. Caldwell, W. R. Ross, T. E. Cheatham III, S. DeBolt, D. Ferguson, G. Seibel, and P. Kollman. 1995. AMBER, a computer program for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to elucidate the structures and energies of molecules. Comput. Phys. Comm. 91:1–41. [Google Scholar]
- Pomes, R., and B. Roux. 1998. Free energy profiles for H+ conduction along hydrogen-bonded chains of water molecules. Biophys. J. 75:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomes, R., and B. Roux. 2002. Molecular mechanism of H+ conduction in the single-file water chain of the gramicidin channel. Biophys. J. 82:2304–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, B., M. Nina, R. Pomes, and J. C. Smith. 1996. Thermodynamic stability of water molecules in the bacteriorhodopsin proton channel: a molecular dynamics free energy perturbation study. Biophys. J. 71:670–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert, J. P., G. Ciccotti, and H. J. C. Berendsen. 1977. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23:327–341. [Google Scholar]
- Sagnella, D. E., K. Laasonen, and M. L. Klein. 1996. Ab initio molecular dynamics study of proton transfer in a polyglycine analog of the ion channel gramicidin A. Biophys. J. 71:1172–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanner, M. F., J.-C. Spehner, and A. J. Olson. 1996. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers. 38:305–320. [DOI] [PubMed] [Google Scholar]
- Sass, H. J., G. Büldt, R. Gessenich, D. Hehn, D. Neff, R. Schlesinger, J. Berendzen, and P. Ormos. 2000. Structural alterations for proton translocation in the M state of wild-type bacteriorhodopsin. Nature. 406:649–653. [DOI] [PubMed] [Google Scholar]
- Schmitt, U. W., and G. A. Voth. 1999. The computer simulation of proton transport in water. J. Chem. Phys. 111:9361–9381. [Google Scholar]
- Song, Y., J. Mao, and M. R. Gunner. 2003. Calculation of proton transfers in bacteriorhodopsin bR and M intermediates. Biochemistry. 42:9875–9888. [DOI] [PubMed] [Google Scholar]
- Staib, A., D. Borgis, and J. T. Hynes. 1994. Proton transfer in hydrogen-bonded acid-base complexes in polar solvents. J. Chem. Phys. 102:2487–2505. [Google Scholar]
- Stoeckenius, W. 1999. Bacterial rhodopsins: evolution of a mechanistic model for the ion pumps. Protein Sci. 8:447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam, S., and R. Henderson. 2000. Molecular mechanism of vectorial proton translocation by bacteriorhodopsin. Nature. 406:653–657. [DOI] [PubMed] [Google Scholar]
- Tajkhorshid, E., J. Baudry, K. Schulten, and S. Suhai. 2000. Molecular dynamics study of the nature and origin of retinal's twisted structure in bacteriorhodopsin. Biophys. J. 78:683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
Tuckerman, M., K. Laasonen, M. Sprik, and M. Parrinello. 1995. Ab initio molecular dynamics simulation of the solvation and transport of
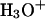 and OH− ions in water. J. Phys. Chem. 99:5749–5752. [Google Scholar]
and OH− ions in water. J. Phys. Chem. 99:5749–5752. [Google Scholar] - Vuillemier, R., and D. Borgis. 1998. Quantum dynamics of an excess proton in water using an empirical valence-bond Hamiltonian. J. Phys. Chem. 102:4261–4264. [Google Scholar]
- Weik, M., G. Zaccai, N. A. Dencher, D. Oesterhelt, and T. Hauss. 1998. Structure and hydration of the M-state of the bacteriorhodopsin mutant D96N studied by neutron diffraction. J. Mol. Biol. 275:625–634. [DOI] [PubMed] [Google Scholar]
- Weiner, S. J., P. A. Kollman, D. T. Nguyen, and D. A. Case. 1986. An all-atom force field for simulations of proteins and nucleic acids. J. Comput. Chem. 7:230–252. [DOI] [PubMed] [Google Scholar]
- Xu, D., M. Sheves, and K. Schulten. 1995. Molecular dynamics study of the M412 intermediate of bacteriorhodopsin. Biophys. J. 69:2745–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, B., M. Blaber, A. M. Gronenborn, G. M. Clore, and D. L. D. Caspar. 1999. Disordered water within a hydrophobic protein cavity visualized by x-ray crystallography. Proc. Natl. Acad. Sci. USA. 96:103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccai, G. 2000. Moist and soft, dry and stiff: a review of neutron experiments on hydration-dynamics activity relations in the purple membrane of Halobacterium salinarum. Biophys. Chem. 86:249–257. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.