

Abstract
We report the picosecond and nanosecond timescale rotational dynamics of a dye-labeled DNA oligonucleotide or “aptamer” designed to bind specifically to immunoglobulin E. Rotational dynamics in combination with fluorescence lifetime measurements provide information about dye-DNA interactions. Comparison of Texas Red (TR), fluorescein, and tetramethylrhodamine (TAMRA)-labeled aptamers reveals surprising differences with significant implications for biophysical studies employing such conjugates. Time-resolved anisotropy studies demonstrate that the TR- and TAMRA-aptamer anisotropy decays are dominated by the overall rotation of the aptamer, whereas the fluorescein-aptamer anisotropy decay displays a subnanosecond rotational correlation time much shorter than that expected for the overall rotation of the aptamer. Docking and molecular dynamics simulations suggest that the low mobility of TR is a result of binding in the groove of the DNA helix. Additionally, associated anisotropy analysis of the TAMRA-aptamer reveals both quenched and unquenched states that experience significant coupling to the DNA motion. Therefore, quenching of TAMRA by guanosine must depend on the configuration of the dye bound to the DNA. The strong coupling of TR to the rotational dynamics of the DNA aptamer, together with the absence of quenching of its fluorescence by DNA, makes it a good probe of DNA orientational dynamics. The understanding of the nature of dye-DNA interactions provides the basis for the development of bioconjugates optimized for specific biophysical measurements and is important for the sensitivity of anisotropy-based DNA-protein interaction studies employing such conjugates.
INTRODUCTION
Understanding the nature of dye-DNA interactions is central to the use of fluorescence methods in the study of biomolecular dynamics. The extent to which a fluorescent dye reports the mobility of the DNA molecule to which it is conjugated is a key factor in the development of fluorescence polarization experiments. Such experiments are widely applicable to the study of DNA dynamics, hybridization, and DNA-protein interactions. Dye-DNA interactions are also of crucial importance for Förster resonance energy transfer (FRET) studies that rely on orientational averaging of dyes for the unbiased determination of distance (Dietrich et al., 2002). The dynamic interaction of a dye with DNA could obscure subtle conformational dynamics of biophysical interest. Considering the widespread use of fluorescence techniques, it is crucial that such interactions are understood in detail and that, if possible, general guidelines for predicting the dye-DNA interactions be set forth.
Fluorescence polarization studies have been undertaken to understand the dynamics of dye-labeled nucleic acids (Dietrich et al., 2002; Fang et al., 2001; Ha et al., 1998, 1999; Kumke et al., 1995, 1997; McGown et al., 1995; Nazarenko et al., 2002; Reid et al., 2001; Vámosi and Clegg, 1998; Vámosi et al., 1996). Many studies have employed fluorescein-labeled single- and double-stranded oligonucleotides, in which the dye shows a high level of segmental mobility with only ∼10% of the anisotropy decay amplitude coupled to the rotation of the DNA molecule at room temperature (Fang et al., 2001; Kumke et al., 1995, 1997; Vámosi and Clegg, 1998). This coupling decreases dramatically with increases in temperature. Thus, the measured anisotropy signal corresponding to overall rotation of the aptamer comprises only 10% or less of the optically induced anisotropy. Studies on tetramethylrhodamine (TAMRA) and the sulfoindocyanine dye, Cy3, coupled to DNA demonstrated a much higher degree of coupling to DNA compared to fluorescein (Clegg et al., 1993; Norman et al., 2000). NMR studies on Cy3 showed an end-stacking arrangement with the DNA helix (Norman et al., 2000). FRET studies suggested that a different arrangement is likely observed with TAMRA where the center of the dye does not lie along the DNA helical axis (Clegg et al., 1993). Despite these recent studies, the extent and nature of the docking of dyes to DNA is not well understood, and both experimental and simulation studies are needed to provide molecular details about the nature of these interactions.
In recent years nucleic acid ligands, termed aptamers, have attracted attention as candidates for bioanalysis based on molecular recognition (McGown et al., 1995). One of the proposed methods for performing a protein bioassay is the use of fluorescence anisotropy of the fluorophore-labeled aptamer (McGown et al., 1995). This approach has a significant advantage over antibody-based methods because the aptamer is small relative to the protein and therefore experiences a large change in anisotropy upon binding. The fast kinetics and high binding affinity of DNA aptamers for protein targets allow real-time monitoring of the target molecule (Berezovski et al., 2003; Fang et al., 2001; Reid et al., 2001). The aptamer-protein interaction has been shown to be highly specific, indicating that in vivo observations of binding should be possible (Berezovski et al., 2003; Fang et al., 2001; McCauley et al., 2003).
Despite the advantages of fluorescence anisotropy as a tool to observe binding of aptamers to targets, a change in anisotropy signal can be ambiguous due to the environmental sensitivity of the fluorescence lifetime and segmental mobility of the fluorophore. The presumed cause for the change in steady-state anisotropy is the effective increase in volume of the aptamer-protein complex compared to the aptamer alone. In addition to this, changes in anisotropy could occur with changes in structure of the aptamer as segmental motions can be altered by a conformational change. Steady-state anisotropy depends not only on the rotational dynamics of the fluorophore but also on the fluorescence lifetime over which those dynamics are averaged. If binding changes the fluorescence lifetime, the steady-state anisotropy could change without any change in dynamic anisotropy of the fluorescent probe. For this reason, it is necessary to characterize the time-resolved anisotropy and fluorescence lifetime changes upon target binding.
To gain insight into the dynamic properties of labeled single-stranded DNA molecules, we have undertaken time-resolved fluorescence studies of a stem-loop dye-labeled DNA aptamer that binds to immunoglobulin E (IgE). Steady-state fluorescence anisotropy analysis of the binding interaction has been done by Gokulrangan et al. (2005). This study demonstrated linear changes in steady-state fluorescence anisotropy of the 5′ dye-labeled aptamer upon IgE binding with aptamer concentrations of 10 nM.
This study examines in detail the rotational coupling and photophysics of commonly used dyes labeled to a DNA aptamer to determine their suitability as probes of DNA rotational dynamics. In addition, we have characterized the Texas Red (TR)-DNA interaction by docking and molecular dynamics simulations. Specific issues investigated are the proportion of the anisotropy decay due to the local segmental motion of the fluorophore and the effect of temperature and target binding on this motion. The results reveal that TR and TAMRA are rotationally coupled to the DNA, in contrast to fluorescein, which rotates almost independently of the aptamer. However, TR is much less susceptible to quenching interactions with DNA, both below and above the aptamer melting temperature. Docking simulations reveal that TR interacts with the stem of the aptamer in a groove-binding conformation stabilized by van der Waals and electrostatic interactions.
MATERIALS AND METHODS
Materials
The TR12-aptamer and the TAMRA-aptamer were obtained from Integrated DNA Technologies (Coralville, IA). The TR6-aptamer was obtained from Midland Certified Reagents (Midland, TX). The aptamer (D17.4) was developed using the SELEX method by Wiegand and et al. (1996). The nucleic acid sequence of the aptamer is 5′-GGGGCACGTTTATCCG TCCCTCCTAGTGGCGTGCCCC -3′. Texas Red cadaverine (TR-cadaverine) was purchased from Molecular Probes (Eugene, OR). Both the labeled aptamers and cadaverine were mixed isomers with the tether attached at either the ortho-position or the para-position. DNA samples were procured in high-performance liquid chromatography or gel-purified form. Their purity was checked on 15% nondenaturing polyacrylamide gels. Melting of the aptamer was observed by measuring absorption at 260 nm. Aptamer solutions were melted at 70°C and renatured before each usage. Dilutions and experimentation were performed with the appropriate binding buffer based on IgE aptamer selection experiments. The binding buffer consists of 1 mM MgCl2, 2.7 mM KCl, 150 mM NaCl, and 8 mM phosphate buffer at pH 7.4 (Gokulrangan et al., 2005). IgE was obtained from Athens Research and Technology (Athens, GA). Buffers and salts were used at the highest purity available. Unless otherwise specified, for TR-labeled substances the concentration was ∼20 nM, for fluorescein studies the concentration was 200 nM, and for TAMRA studies the concentration was 50 nM.
Time-resolved fluorescence
The instrumental setup used for this experiment has been described in detail previously (Unruh et al., 2005). The normalization of collection intensity of parallel and perpendicular anisotropy decays was accomplished by collecting all decays to the same integrated intensity in a tail-matching region at long time delays such that no anisotropy effects are seen within the tail-matching region. For measurements in which the aptamer was bound to protein or for the TAMRA-aptamer, the tail-matching method was not employed because anisotropy was still present at long time delays. For these studies, a scaling factor for the perpendicular decay was determined from the steady-state anisotropy values such that the anisotropy calculated from the integrated parallel and perpendicular intensities was identical to the steady-state anisotropy value.
Data fitting
Data were analyzed with the Globals Unlimited decay analysis software (Laboratory for Fluorescence Dynamics, University of Illinois at Urbana-Champaign, Urbana, IL). The instrument response function was collected from a dilute colloidal solution of nondairy creamer. The fit to the decays was convoluted with the instrument response for every set of vertical, horizontal, and magic-angle files. The magic-angle intensity decays were fit to a sum of exponentials in the form I(t) = Σ ai exp(−t/τi), where ai is the amplitude of component i and τi is the fluorescence lifetime of component i.
The vertically and horizontally polarized decays were globally fit to equations of the form,
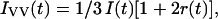 |
(1) |
and
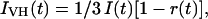 |
(2) |
respectively (Fleming, 1986). The anisotropy, r(t), is described by
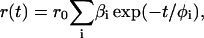 |
(3) |
where r0 is the anisotropy at time 0 for the fluorophore, βi is the amplitude of component I, and φi is the rotational correlation time of component i. In all studies except those with the fluorescein (fl)-aptamer, the r0 value was fit with the fluorescence lifetimes fixed at a value determined from the magic-angle decays. For fl-aptamer studies the r0 value was fixed to 0.37 as determined by Johansson (1990).
Time-resolved fluorescence cannot be reliably used to measure rotational correlation times that are more than an order of magnitude longer than the fluorescence lifetime (Rachofsky and Laws, 2000). The rotational correlation time expected for overall rotation of the IgE molecule is more than an order of magnitude greater than the fluorescence lifetime of any of the probes used in this study. Therefore, a rotational correlation time was added to the analysis with a fixed value of 50 ns to account for the long anisotropy decay component, allowing the amplitude of the long anisotropy decay to be determined (Kumke et al., 1995). For purposes of visualizing the trends in the time-resolved anisotropy, raw anisotropy decays were calculated from the equation (Fleming, 1986),
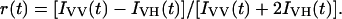 |
(4) |
Because IVV(t) and IVH(t) are convoluted with the instrument response function, this is only a visual tool and values of the anisotropy calculated in Eq. 4 are incorrect at short timescales. Therefore, all plots of raw anisotropy decays omit the initial data points. All rotational correlation times except the associated analyses for the TAMRA-aptamer were determined from Eqs. 1 and 2 by iterative reconvolution with the instrument response function. Estimates of error limits for global nonlinear least-squares fits were obtained using the support plane method (Johnson and Faunt, 1992; Lakowicz, 1999). All errors reported in this work are at 1 standard deviation.
Heterogeneous photophysical behavior of a dye-DNA conjugate may be indicative of heterogeneous interactions or the fluorophore with the DNA molecule. In this case, one might expect the anisotropy decay components to be associated with the individual fluorescent species. In this case Eqs. 1 and 2 can be rewritten as:
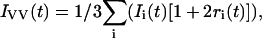 |
(5) |
and
 |
(6) |
where Ii(t) is the intensity decay function for the species i, and ri(t) is the anisotropy decay function for species i. Each ri(t) need not be single exponential, as species decaying with different lifetimes may themselves experience heterogeneous dynamics. Equations 5 and 6 were used in Globals Unlimited for analysis of the TAMRA aptamer anisotropy decays with discrete lifetimes and rotational correlation times.
Docking simulations
To gain a better understanding of the nature of the interaction of TR with the aptamer, molecular docking simulations were carried out with the program Autodock (Morris et al., 1998). The secondary structure of the aptamer was predicted to be a stem loop by Wiegand et al. (1996). This structure provided the basis for our molecular model of the aptamer. The single- and double-stranded nucleic acid builder utilities of SYBYL 6.9 (The Tripos Associates, St. Louis, MO) were used to create the loop and stem portions of the aptamer. The single-stranded region was formed into a loop structure by making small adjustments to appropriate dihedral angles within the sugar-phosphate backbone. After connection of the two regions, the aptamer structure was optimized in vacuo using SYBYL's molecular mechanics utility, the Tripos force field (Clark et al., 1989), and Gasteiger Marsili charges (Gasteiger and Marsili, 1980). This structure was then solvated in Insight II (InsightII-0907, Accelrys, San Diego, CA) with water, and sodium ions were placed at appropriate sites on the DNA backbone (Martinez et al., 2001; Young et al., 1997). This structure was equilibrated with a 20-ps molecular dynamics run using LAMMPS (Plimpton, 1995) using the complete valence force field (Dauber-Osguthorpe et al., 1988). The TR structure was created in SYBYL and minimized with molecular mechanics (Tripos force field; Gasteiger Marsili charges).
Docking of minimized dye structure to the equilibrated aptamer structure with Autodock was accomplished with the Lamarckian genetic algorithms method for structural searching, Gasteiger Marsili charges for dye-aptamer electrostatics (Gasteiger and Marsili, 1980), and all other parameters retained at their default AutoDock settings. Four unique runs were carried out with the dye placed in different starting positions relative to the aptamer: one in which the dye was stacked to the end of the helical portion, another in which the dye was placed in a groove-binding position on one side of the DNA, another in a groove-binding position on the opposing side of the helix, and a structure where the dye was positioned close to the phosphate backbone. Docked structures were chosen for molecular dynamics equilibration based on energy and distance of the chromospheres from the 5′ end of the DNA. The lowest energy structure that met the distance criterion was solvated and equilibrated with a 40-ps molecular dynamics run (20 ps linear warm-up from 100 to 300 K; 20 ps constant temperature (300 K)). Finally, a 140-ps molecular dynamics production run was carried out to assess the stability of the chosen docking configuration.
RESULTS
To understand the dynamics of the fluorophores attached to the DNA aptamer we carried out time-resolved fluorescence anisotropy decay experiments. The anisotropy parameters for all dye-labeled aptamers are shown in Table 1. Structures of these conjugates are given in Unruh et al. (2005). The r0 value was 0.37 ± 0.03 for all TR samples and 0.36 ± 0.01 for all TAMRA samples. The r0 value for the fl samples was fixed to a value of 0.37 (see Materials and Methods).
TABLE 1.
Fluorescence anisotropy parameters at 25°C
| Sample | φ1 (ns) | β1 | φ2 (ns) | β2 | χ2 |
|---|---|---|---|---|---|
| 20 nM TR-cadaverine*† | 0.23 ± 0.02 | 1.0 | – | – | 1.38 |
| 10 nM TR12-aptamer*‡ | 0.7 ± 0.2 | 0.27 ± 0.05 | 5.1 ± 0.4 | 0.73 ± 0.05 | 1.68 |
| 20 nM TR12-aptamer*‡ | 0.5 ± 0.2 | 0.28 ± 0.04 | 5.0 ± 0.4 | 0.72 ± 0.04 | 1.91 |
| 50 nM TR12-aptamer*‡ | 0.6 ± 0.2 | 0.27 ± 0.05 | 4.9 ± 0.4 | 0.73 ± 0.05 | 1.13 |
| 20 nM TR6-aptamer*‡ | 0.5 ± 0.2 | 0.26 ± 0.04 | 5.1 ± 0.4 | 0.74 ± 0.04 | 1.56 |
| 200 nM fl-aptamer§† | 0.49 ± 0.03 | 1.0 | – | – | 2.70 |
| 50 nM TAMRA-aptamer (lifetime-associated anisotropy)¶‖ | 1.41 | ||||
| 0.14-ns component | 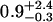 |
1.0 | – | – | |
| 0.63-ns component | 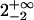 |
1.0 | – | – | |
| 2.6-ns component | 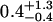 |
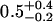 |
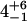 |
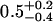 |
Samples excited at 590 nm and emission collected at 590 nm.
Anisotropy fitting function is: r(t) = r0 exp(−t/φ).
Anisotropy fitting function is: r(t) = r0 [β1 exp(−t/φ1) + β2 exp(−t / φ2)].
Samples excited at 440 nm and emission collected at 612 nm.
Samples excited at 563 nm and emission collected at 590 nm.
Lifetime-associated anisotropy fitting was accomplished as described in Materials and Methods.
To understand the complex anisotropy decays of the dye bound to DNA, we measured the anisotropy of TR cadaverine (Texas Red with the tether attached). For this sample, we observed a rotational correlation time of 230 ± 20 ps at room temperature. In contrast, the anisotropy decay of the TR12-aptamer was biexponential. At room temperature, the rotational correlation time of the fast component was ∼600 ps whereas the rotational correlation time of the slow component was ∼5 ns. This fast rotational correlation time is on the same order of magnitude as expected for local motion of the fluorophore. However, it is longer than the reorientational time of TR cadaverine (which has a linker of the same length), probably as a result of coupling to the DNA molecule. It is also possible that the linker arm itself interacts with the DNA, reducing the mobility of the dye. The slow rotational correlation time is on the same order of magnitude as predicted for a hydrated DNA molecule of 37 basepairs (Gokulrangan et al., 2005). Therefore, it can be inferred that the reorientational dynamics include motions of both the tethered dye molecule and the aptamer as a whole. The relative amplitude of the TR12-aptamer fast motion as a function of temperature is shown in Fig. 1. The anisotropy decay was dominated by the slow rotational motion at low temperatures, indicating that the dye motion was strongly coupled to the aptamer, even though it is attached by a 12-carbon linker. As temperature increases, the fast rotational motion became more prominent and dominates the anisotropy at 65°C. This coincides with the melting temperature of the aptamer tertiary structure at ∼65°C (see Supplementary Material).
FIGURE 1.

Comparison of the (A) rotational amplitude of the fast component, (B) fast rotational correlation times, and (C) slow rotational correlation times of the TR12-aptamer (□) and TR6-aptamer (▪) as a function of temperature. The rotational amplitude of the slow rotational component is given by 1 − β where β is the fast rotational amplitude plotted in panel A.
We also examined a six-carbon linker TR label (TR6-aptamer). This aptamer showed almost identical anisotropy and fluorescence characteristics to the 12-carbon linker. A comparison of the rotational amplitudes and rotational correlation times is shown in Fig. 1 B. Thus, the length of the linker arm has minimal effect on the anisotropy properties of the dye-labeled aptamer for TR. In contrast, a dependence on the length of the linker has been shown for fluorescein, where the value of the fast rotational correlation time decreases with increasing linker length (Kumke et al., 1997).
The anisotropy decay of the fl-aptamer was also analyzed. The rotational correlation time at room temperature was 590 ± 30 ps. We observed no evidence for a biexponential anisotropy decay at any temperature between 15 and 75°C. The lack of a long rotational component indicates that the dye motion was, to a large extent, uncoupled from the global aptamer rotation in this system. The differences between the anisotropy decays of the TR6- and fl-aptamers are visualized by observing the actual anisotropy decays in Fig. 2 A. Previous studies have shown an ∼5% long rotational component at room temperature for fluorescein attached to a double-stranded oligonucleotide (Kumke et al., 1997).
FIGURE 2.

(A) Comparison of the raw anisotropy decays of fl-aptamer and TR6-aptamer at 25°C. The anisotropy decays were calculated from parallel and perpendicular polarized fluorescence decays. (B) Plot of the raw anisotropy data for the TAMRA-aptamer at 25°C (solid line) along with theoretical plots of the anisotropy decay for 0, 50, and 100% amplitudes of the mobile state for the long-lifetime distribution according to Eqs. 4–6 (dashed lines). For the long-lifetime distribution, the immobile state (coupled to the DNA) has a rotational correlation time of 5 ns and the mobile state has a rotational correlation time of 0.5 ns. The short-lifetime distribution is assumed to be coupled to the DNA with a rotational correlation time of 5 ns. Data at early times are omitted due to irregularities caused by the instrument response function (see Materials and Methods).
The anisotropy decay of the TAMRA-aptamer is quite complex due to the fact that the lifetime of TAMRA is heterogeneous, with a distribution of decay rates (Unruh et al., 2005). One might expect the anisotropy to exhibit an associated anisotropy decay because the states represented by the two distinct lifetime distributions could have different mobilities. This is suggested by the observations of Vámosi and co-workers who assigned the short lifetime to a state where the dye is strongly coupled to the DNA molecule (Vámosi et al., 1996). However, the analysis of associated anisotropy decay for a system with a distribution of fluorescence lifetimes is beyond the scope of this work. Therefore, we chose to analyze our data through an associated anisotropy model with three discrete fluorescence lifetimes to describe the fluorescence decay. The associated anisotropy results are summarized in Table 1. The resulting lifetimes were 0.14, 0.63, and 2.6 ns. The first two lifetimes belong to a distribution of quenched lifetimes, whereas the long lifetime describes a long-lifetime distribution (Unruh et al., 2005). The anisotropies of the short lifetimes were well described by single exponentials. The rotational correlation times are 0.9 ns for the 0.14-ns lifetime and 2 ns for the 0.63-ns lifetime. The upper error limit on the 0.9-ns rotational correlation time was 3.3 ns and the upper error limit on the 2-ns rotational correlation time was not defined. The lack of sensitivity in determining these rotational times was a result of the short lifetime with which they are associated. Therefore, it was not possible to accurately assess the mobility of the state associated with the short lifetime. In contrast, the long lifetime state had a well-resolved anisotropy decay that could not be described by a single rotational component. The rotational correlation times were 0.4 and 4.1 ns with amplitudes for both components of 50%. Therefore, the long lifetime state demonstrates a significant coupling to the DNA. This coupling can also be visualized by the presence of significant anisotropy at times longer than three nanoseconds where the contribution from the short lifetime state was negligible (Fig. 2 B). Fig. 2 B demonstrates the dependence of the predicted anisotropy decay on the mobility of the long-lifetime state for that TAMRA aptamer at room temperature. Comparison with the measured anisotropy decay provides clear evidence for the rotational coupling of the long-lifetime state to the DNA.
Concentration dependence
One of the major concerns in using DNA ligands as analytical tools is the possibility of self-association at higher DNA concentrations. We checked the concentration dependence of the anisotropy of TR12-aptamer, and Table 1 shows the effect of concentration on the rotational correlation times and rotational amplitudes at 10, 20, and 50 nM. The values of the rotational parameters are identical within experimental error at these concentrations. Therefore, it is evident that self-association is not an issue at the concentrations used in this study. Steady-state data led to the same conclusion up to 100 nM with both the fl-aptamer and the TR12-aptamer (Gokulrangan et al., 2005). At concentrations up to 200 nM the rotational correlation time of the fl-aptamer also does not show any evidence of self-association.
Docking simulations
To better understand the nature of the interaction between TR and the aptamer, we carried out docking simulations on the system with different starting orientations of the dye relative to the stem portion of the aptamer (Fig. 3, A–D). The four different starting configurations all gave low-energy structures in approximately the same conformation. This conformation is one in which the multiple-ring portion of the dye is in a minor-groove-binding configuration in which most of the contact is with the 3′ helix strand. The single-ring portion of the dye is in contact with the other strand. In all of the four docking simulations, a helix-stacked conformation was not observed, even when the dye was biased toward this structure in the starting structure (Fig. 3, B and C). In addition, no structures were observed in which the dye did not make contact with the minor groove of the DNA molecule. The dye also never traversed to the opposite side of the helix (major groove), and when it was positioned there initially (Fig. 3 A), all resulting structures were in the minor groove.
FIGURE 3.

Comparison of lowest-energy docked structures (blue) from the four Autodock simulations with the starting structure for the respective simulations (red). Guanosine bases are shown in green. The dye structures and guanosine bases are shown in bold. All other aptamer atoms are colored by atom type. Hydrogens have been removed for clarity. The aptamer and dye structures were created as described in Materials and Methods.
One of the low-energy structures from the docking simulation was chosen based on its distance from the attachment point at the 5′ end of the aptamer. A linker was created and this structure was solvated and used in a 140-ps molecular dynamics study to assess the stability of the docked structure. The dye did not move significantly from its docked position, indicating that this is a stable configuration for the dye-DNA interaction. The trajectory for the distance between the ring system of the dye and a nearby aptamer base is shown in Fig. 4 B. The histogram of distances was fit to a Gaussian distribution with a standard deviation of 0.7 Å (Fig. 4 B, inset). The final equilibrated structure is shown in Fig. 4 A.
FIGURE 4.
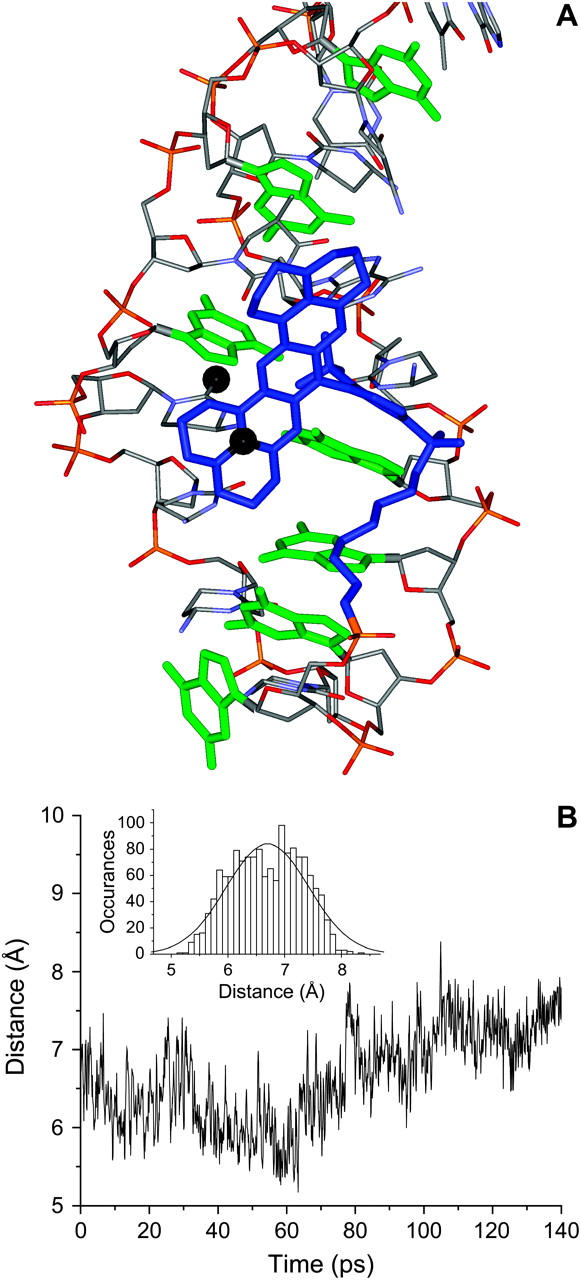
(A) Final molecular dynamics structure of the Texas Red-aptamer complex. Texas Red is blue and aptamer is colored by atom. Guanosine bases are shown in green. The dye and guanosine bases are shown in bold. Hydrogen atoms have been removed for clarity. A nitrogen on the dye and an oxygen on the nearby DNA base are shown as black balls. (B) The distance between these atoms as a function of time for the molecular dynamics run. (Inset) Histogram of the distances shown in panel B and a Gaussian fit of that histogram (curve).
Binding to IgE
Table 2 shows the fluorescence anisotropy parameters of TR12-aptamer with varying concentrations of IgE. Fig. 5 shows the anisotropies calculated from the parallel and perpendicular fluorescence decays. We found that there are two rotational contributions in addition to the long rotational contribution that corresponds to the overall rotation of the aptamer-IgE complex. This was the case even in the limit where all of the aptamer was bound to IgE as shown by steady-state experiments (Gokulrangan et al., 2005). The intermediate rotational correlation time (φ2) thus indicates some segmental motion other than the local motion of the dye in the bound state. The fraction of long rotational population (β3) increased linearly with increasing IgE concentration (Fig. 5, inset) to a value of ∼50%. In addition, the fraction of the intermediate rotational correlation time (β2) decreased linearly in the same fashion to a final value of ∼30%. This behavior reflects the linear change in populations of bound and free aptamer, respectively, with IgE concentration as discussed in a recent manuscript describing steady-state fluorescence polarization measurements (Gokulrangan et al., 2005). The fraction of segmental motion due to local dye motion (β1) was similar to that observed in the free aptamer at all points in the titration.
TABLE 2.
Fluorescence anisotropy parameters for 10 nM TR12-aptamer-IgE binding (25°C)
| [IgE]*†‡ | φ1 (ns) | β1 | φ2 (ns) | β2 | φ3 (ns) | β3 | χ2 |
|---|---|---|---|---|---|---|---|
| 0 nM | 0.5 ± 0.2 | 0.28 ± 0.04 | 5.0 ± 0.4 | 0.72 ± 0.04 | – | – | 1.91 |
| 5 nM | 0.5 ± 0.2 | 0.26 ± 0.06 | 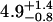 |
0.61 ± 0.05 | 50§ | 0.14 ± 0.07 | 1.47 |
| 10 nM | 0.7 ± 0.3 | 0.26 ± 0.11 | 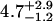 |
0.50 ± 0.08 | 50§ | 0.24 ± 0.13 | 1.91 |
| 15 nM | 1.0 ± 0.5 | 0.25 ± 0.16 | 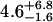 |
0.39 ± 0.13 | 50§ | 0.36 ± 0.19 | 1.43 |
| 26 nM | 0.5 ± 0.3 | 0.17 ± 0.12 | 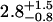 |
0.28 ± 0.07 | 50§ | 0.54 ± 0.14 | 1.74 |
| 150 nM | 0.7 ± 0.3 | 0.23 ± 0.07 | 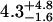 |
0.28 ± 0.05 | 50§ | 0.49 ± 0.09 | 1.55 |
All solutions are in binding buffer.
Samples are excited at 590 nm and emission is collected at 612 nm.
Anisotropy fitting function is: r(t) = r0 Σ βi exp(−t/φi).
This rotational correlation time is fixed to account for overall protein rotation.
FIGURE 5.

Raw anisotropy decays for the titration of 10 nM TR12-aptamer with IgE. (Inset) Amplitudes of the short rotational component (▪), intermediate rotational component (□), and long rotational component (○). The initial part of the decay is not shown as in Fig. 2.
DISCUSSION
Biophysical fluorescence studies of DNA dynamics require a precise understanding of the interaction between the fluorescent dye label and the fluorophore. Fluorescence anisotropy is an important tool for understanding DNA-protein interactions (Hill and Royer, 1997; LeTilley and Royer, 1993). This type of measurement is especially sensitive to changes in the dye-DNA interaction. Gokulrangan and co-workers have shown that the steady-state fluorescence anisotropy of TR12-aptamer is four times more sensitive to IgE binding than the anisotropy of the fl-aptamer (Gokulrangan et al., 2005). Therefore, it is important to study the properties of the interaction between fluorescent dyes and DNA to discover the reasons for these vastly different dynamics.
The fluorescence photophysics of dye-labeled nucleic acids are also important for the development of biophysical methods. We have recently completed a thorough study of the photophysics of the fl-, TR-, and TAMRA-aptamers (Unruh et al., 2005). The fl-aptamer exhibits a multiexponential fluorescence decay at physiological pH due to the presence of a monoanionic species with a shorter fluorescence lifetime. The pKa for this transition is environmentally dependent (Sjöback et al., 1998). Fluorescein also undergoes charge transfer with DNA, though this decay pathway does not become important until the aptamer structure melts. The TAMRA-aptamer exhibits multiexponential behavior with one highly quenched lifetime distribution that displays a temperature dependence characteristic of charge transfer and a second, broadly distributed lifetime with a temperature dependence comparable to that of free TAMRA.
In contrast, the TR-aptamer displayed little quenching of TR fluorescence and the photophysics of the TR-aptamer conjugates was not affected by the melting of the aptamer. An analysis based on the published electrochemical properties of rhodamine dyes showed that the reason that TR is not susceptible to electron-transfer quenching by guanosine is its low excitation energy rather than a significantly different ground-state reduction potential or structural perturbations (Unruh et al., 2005). The absence of highly efficient nonradiative decay pathways in the TR6-aptamer resulted in a high quantum yield of 0.54 compared to a quantum yield of 0.10 for the TAMRA-aptamer. Consequently, TR is a good choice for a fluorescence probe of DNA rotational dynamics.
Segmental motion
The steady-state anisotropy of the fl-aptamer is rather low and undergoes only small changes upon binding to target (Gokulrangan et al., 2005). Our time-resolved results show that this is due to a large amplitude of free reorientation of the dye relative to the aptamer molecule. Our data indicate that at all temperatures for the fl-aptamer, the dye rotates almost independently of the aptamer as evidenced by the absence of a long rotational correlation time corresponding to the global rotation of the system (Fig. 2 A). This factor is a severe impediment in the development of a sensitive and reproducible binding assay. Kumke et al. (1997) have shown that this can be remedied somewhat by decreasing the dye linker length. Nevertheless, the large amplitude of segmental motion in the fl-aptamer in addition to the complicated environmental dependence of fluorescein's photophysics (Unruh et al., 2005) makes quantitative assessment of IgE binding difficult with the fl-aptamer, especially in heterogeneous environments.
These results show that fluorescent dye labels other than fluorescein can exhibit significantly increased coupling of the dye to reorientational motions of the aptamer. The TAMRA conjugate demonstrates rotational coupling to the DNA, but this is complicated by the presence of two emitting species with different rotational mobilities. One species is indicative of the dye in close contact with guanosine and undergoing a photoinduced charge-transfer reaction (Unruh et al., 2005). The other species is not quenched by guanosine. Changes in anisotropy will be sensitive to changes in the populations of these species and their respective decay rates. The steady-state anisotropies of the TR-aptamers are much higher than that of fl-aptamer and show a greater change upon binding (Gokulrangan et al., 2005). Time-resolved data (Figs. 1 and 2) demonstrate that in this case the dye is strongly coupled to the rotational mobility of the DNA with limited free reorientation of the dye. The fact that this behavior is not affected by a change in linker length suggests a close interaction of the dye with the DNA. We have also shown that the TR-aptamer is not subject to the heterogeneous photophysical behavior seen with the TAMRA-aptamer (Unruh et al., 2005). Therefore, TR conjugates will likely provide sensitive information about DNA-protein interactions independent of the local environment—a significant advantage for biophysical applications.
Conformational states of the TAMRA-aptamer
The heterogeneous nature of the TAMRA-aptamer fluorescence decay allows for characterization of the relationship between the different mobility states (coupled and uncoupled to the DNA) and the different lifetime states through associated anisotropy analysis. The highly quenched nature of the short lifetime prevents a quantitative assessment of its rotational mobility, but the rotational correlation times associated with the short fluorescence decay components are longer than the rotational diffusion times of the free dye, suggesting coupling of the fluorophore to the reorientational dynamics of the aptamer. The long fluorescence lifetime lends itself to a more detailed analysis and demonstrates significant coupling to the aptamer motion. The fluorescence anisotropy decay associated with the long lifetime state is biexponential with a long rotational time amplitude of 50% (Table 2). There are, therefore, at least three distinct states in this system—one rotationally coupled to the aptamer with a short fluorescence lifetime distribution, a second rotationally coupled to the aptamer with a long fluorescence lifetime, and a third in which the dye is rotationally mobile with a long lifetime. Seidel and co-workers also observed three conformational states for TAMRA-labeled double-stranded DNA (Eggeling et al., 1998). They were able to observe three lifetime states via single-molecule burst integrated fluorescence lifetime analysis. They assigned two of the states to a structure coupled to the DNA, one quenched by a single guanosine residue and one coupled to the DNA but not quenched to the same extent. They also observed a long lifetime state that was attributed to dye uncoupled from the DNA. Our system is different from the one studied by Eggeling et al. (1998) in that we do not see a distinct intermediate lifetime state, but rather a heterogeneous anisotropy decay for the unquenched lifetime state. Nevertheless, we also observe two states in which the dye is coupled to the DNA motion and one state in which the dye moves freely.
The long-lifetime, low-mobility conformation of TAMRA-aptamer demonstrates that the efficiency of charge transfer depends not only on the presence of a donor moiety but also on the configuration in which the dye molecule interacts with the DNA. This is in contrast to TR, which is not susceptible to charge transfer even when the distance from guanosine is small (Torimura et al., 2001; Unruh et al., 2005). The observation of a TAMRA-aptamer state with a long fluorescence lifetime and a low rotational mobility may seem puzzling considering the prevalence of guanosine at the 5′ end of the DNA. If the dye is coupled to the DNA, as is demonstrated by the reduced rotational mobility, quenching might be expected to be highly efficient. This is consistent with the observation of a quenched short-lifetime state of TAMRA attached to the aptamer. Heinlein and co-workers suggested that the presence of multiple guanosines in a DNA strand increases the electron donating properties of the DNA significantly (Heinlein et al., 2003).
In the aptamer system, the last four out of 37 bases on the 5′ end are guanosine. The dye would have to interact with the DNA at a distance of >14 Å away from its attachment point to avoid contact with a guanine residue (Kelley and Barton, 1999). This is unlikely considering that the length of the dye with an extended linker is ∼15 Å (data not shown). Alternatively, the dye could interact with the opposing strand of the DNA. Our docked structures for TR demonstrate interactions with both DNA strands (Fig. 3). One might expect TAMRA to interact with the aptamer in a similar manner to TR. Prediction of charge-transfer efficiency in DNA systems is difficult due to the various configurations of electron donors and acceptors with respect to the DNA. Stacked donors and acceptors can exhibit significant charge transfer at distances as long as 14 Å (Kelley and Barton, 1999). However, in systems where charge transfer occurs between strands the efficiency is negligible as close as two bases away (Kelley and Barton, 1999). Murphy and co-workers demonstrated that groove-binding electron acceptors undergo charge transfer much less efficiently than intercalated or stacked acceptors (Murphy et al., 1994). Therefore, it is reasonable that charge transfer for a groove-binding structure could occur with low efficiency when the dye interacts with the DNA strand opposite a guanosine residue. In addition, contact between the dye ring system and the negatively charged phosphate backbone could reduce the excited-state electron affinity of the TAMRA.
Docking of TR with DNA
The nature of the interaction between dye and DNA in the TR-aptamers is not completely clear from the anisotropy studies, but segmental motion becomes predominant at a temperature close to the melting temperature for the aptamer stem structure (Fig. 1). Therefore, the interaction is probably dependent on the presence of the stem structure of the aptamer. Upon melting the stem structure disappears and the interaction is no longer favorable. A recent NMR study by Norman and co-workers suggested that Cy3 participates in a stacking interaction with terminal DNA basepair (Norman et al., 2000). FRET measurements as a function of DNA length with Cy3 as the acceptor and fluorescein as the donor fluorophore show no length-dependent modulation in the energy transfer efficiency (Norman et al., 2000). On the other hand, previous studies with TAMRA as the acceptor show significant modulation of the energy-transfer efficiency as a function of DNA length (Clegg et al., 1993). These studies show that TAMRA is coupled to the DNA in an asymmetrical manner with respect to the helical axis, whereas Cy3 is attached in a highly symmetrical manner in its end basepair stacking configuration. This behavior of TAMRA is consistent with a groove-binding configuration as opposed to the stacked configuration of Cy3. TR and TAMRA are both zwitterionic at neutral pH whereas fl is mostly dianionic at neutral pH. The nucleic acid backbone is highly electronegative. This suggests that fluorescein is electrostatically repelled from the DNA surface and therefore does not participate in a stacking interaction. Cy3, TR, and TAMRA can participate in such an interaction due to their regions of positive charge.
To confirm the possibility of groove-binding interactions between TR and the DNA helix, docking and molecular dynamics simulations were carried out on a model aptamer structure and the TR fluorophore. Docking simulations have, as a limitation, the inability to simulate large-scale rearrangements in the receptor (DNA) structure. Therefore, this approach does not rule out the presence of intercalated structures. However, Alba and co-workers have shown that intercalation prevents chemical excitation of dyes by peroxyoxalate (Alba and Daban, 1999), and TR demonstrates strong chemiluminescence, indicating that intercalation does not occur in this system (Alba and Daban, 2001).
The docking procedure was carried out with the dye in specific starting positions to bias the simulation toward different possible configurations. Four representative starting positions are shown in red in Fig. 3. The corresponding low-energy structures resulting from these simulations are shown in blue. Though the structures resulting from the docking simulations were not identical, they all shared the same binding characteristics. Most of the interactions were van der Waals interactions between the aromatic ring system and the DNA bases in a groove-binding configuration. In addition, there are also interactions between the positively charged tertiary amine groups of the TR ring system and the negatively charged phosphate backbone. Despite the fact that two of the starting structures in Fig. 3 were in an end-stacked configuration, no final end-stacked configurations were predicted throughout the simulation. This is in agreement with the energy-transfer results of Clegg et al. (1993) suggesting that the dye is positioned asymmetrically with respect to the DNA helix. We also carried out molecular dynamics calculations on this system for the groove-binding structure. The position of the dye relative to the aptamer did not change significantly throughout the 140-ps simulation as indicated by the trajectory of the distance between the ring system of the dye and the DNA bases (Fig. 4 B). The final structure resulting from the simulation is seen in Fig. 4 A.
Rotational dynamics
One interesting piece of dynamic information is the presence of a 500-ps component in the anisotropy decay of TR bound to the aptamer. This is significantly longer than the rotational correlation time of TR cadaverine. The fl-aptamer and TR-aptamer had similar values of the short reorientational times, yet, as described above, rotational dynamics showed that only the TR-aptamer associates with the aptamer. Therefore, the lengthening of the short reorientational time relative to that of the free dye is likely due to linker arm constraints rather than interaction with the DNA, because this interaction was not observed with fluorescein. These constraints may result from interaction of the linker arm with the DNA or simply from the energetics of bond reorientation in the linker.
IgE binding studies
To optimize signal and understand the reliability of DNA-protein interaction studies, it is important to understand the molecular details behind the changes in reorientational mobility upon binding. Changes in protein or aptamer structure could lead to anomalous changes in steady-state anisotropy due to changes in the reorientational freedom of the dye label and segmental motions of the aptamer. If the dye-aptamer interaction were affected by protein binding, the increase in anisotropy relative to the free aptamer might not be as great as expected. In addition, the extent to which the aptamer is rotationally coupled to the IgE would affect the sensitivity of the anisotropy measurement to IgE binding. It is evident from Fig. 5 and the data in Table 2 that the anisotropy components mirror the fractions of bound and unbound aptamer in solution throughout the titration. This indicates a two-state bound-unbound equilibrium system. In addition, no changes were seen in the fraction of the fast local dye motion as a function of IgE concentration. This indicates that the dye-DNA interaction is insensitive to IgE binding. Nevertheless, there was some segmental mobility other than local dye motion even at saturating concentrations of IgE. This segmental motion occurred on the same timescale as the overall rotation of the aptamer. The aptamer-IgE dissociation constant is ∼10 nM (Wiegand et al., 1996). The presence of segmental motion in the bound complex suggests either flexibility of the bound aptamer or of the IgE binding site. However, the high affinity of the aptamer for IgE suggests a tight binding configuration in which the IgE binding site would have limited flexibility.
CONCLUSION
We have found significant differences in the orientational dynamics of three commonly used fluorescent dyes attached to a DNA aptamer. The rotational dynamics significantly affect the sensitivity of anisotropy-based detection of DNA binding. In particular, the interaction between TR and DNA molecules provides a significant increase in the sensitivity of anisotropy measurements to target binding. Docking simulations showed that this interaction is not characterized by end stacking but rather by a groove-binding interaction characterized by van der Waals and electrostatic interactions. Associated anisotropy analysis also allowed three conformational states of the TAMRA-aptamer to be detected. Considering the prevalence of guanosine residues near the labeling site, the presence of both quenched and unquenched low-mobility states indicated that the dye can interact with DNA near guanosine residues and still exhibit low charge-transfer efficiencies, probably due to association of the dye with the opposing strand of DNA. However, the rotational freedom of the fl-aptamer and the lifetime-associated anisotropy of the TAMRA-aptamer indicate that these probes are poor choices for anisotropy studies of DNA dynamics. TR, on the other hand, is docked to the DNA structure without significant changes to its photophysical behavior (Unruh et al., 2005), making it ideal for such studies. TR exhibits environmentally insensitive fluorescence but is not rotationally mobile. We suggest that the segmental rotational mobility is dependent, in part, on electrostatic interactions of the fluorescent probe with DNA. Therefore, TR and dyes with similar electrostatic and electrochemical properties are ideal for biophysical fluorescence studies of DNA structure and dynamics.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
We thank Russ Middaugh and Chris Wiethoff for assistance with the spectrophotometric melting study and for helpful discussions.
J.R.U. acknowledges support from the Dynamic Aspects of Chemical Biology Training Grant (National Institutes of Health 5 T32 GM08545-09). Support from the Petroleum Research Fund, administered by the American Chemical Society, is gratefully acknowledged.
References
- Alba, F. J., and J.-R. Daban. 1999. Inhibition of peroxyoxalate chemiluminescence by intercalation of fluorescent acceptors between DNA bases. Photochem. Photobiol. 69:405–409. [PubMed] [Google Scholar]
- Alba, F. J., and J.-R. Daban. 2001. Detection of Texas red-labelled double-stranded DNA by non-enzymatic peroxyoxylate chemiluminescence. Luminescence. 16:247–249. [DOI] [PubMed] [Google Scholar]
- Berezovski, M., R. Natiu, Y. Li, and S. N. Krylov. 2003. Affinity analysis of a protein-aptamer complex using nonequilibrium capillary electrophoresis of equilibrium mixtures. Anal. Chem. 75:1382–1386. [DOI] [PubMed] [Google Scholar]
- Clark, M., R. D. Cramer, III, and N. van Opdenbosch. 1989. Validation of the general purpose Tripos 5.2 force field. J. Comput. Chem. 10:982–1012. [Google Scholar]
- Clegg, R. M., A. I. H. Murchie, A. Zechel, and D. M. J. Lilley. 1993. Observing the helical geometry of double-stranded DNA in solution by fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA. 90:2994–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauber-Osguthorpe, P., V. A. Roberts, D. J. Osguthorpe, M. Genest, and A. T. Hagler. 1988. Structure and energetics of ligand binding to proteins: Escherichia coli dihydrofolate reductase-trimethoprim, a drug-receptor system. Proteins. 4:31–47. [DOI] [PubMed] [Google Scholar]
- Dietrich, A., V. Buschmann, C. Müller, and M. Sauer. 2002. Fluorescence resonance energy transfer (FRET) and competing processes in donor-acceptor substituted DNA strands: a comparative study of ensemble and single-molecule data. Rev. Mol. Biotechnol. 82:211–231. [DOI] [PubMed] [Google Scholar]
- Eggeling, C., J. R. Fries, L. Brand, R. Günther, and C. A. M. Seidel. 1998. Monitoring conformational dynamics of a single molecule by selective fluorescence spectroscopy. Proc. Natl. Acad. Sci. USA. 95:1556–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, X., Z. Cao, T. Beck, and W. Tan. 2001. Molecular aptamer for real-time oncoprotein platelet-derived growth factor monitoring by fluorescence anisotropy. Anal. Chem. 73:5752–5757. [DOI] [PubMed] [Google Scholar]
- Fleming, G. R. 1986. Chemical Applications of Ultrafast Spectroscopy. Oxford University Press, New York.
- Gasteiger, J., and M. Marsili. 1980. Iterative partial equalization of orbital electronegativity: a rapid access to atomic charges. Tetrahedron. 36:3219–3228. [Google Scholar]
- Gokulrangan, G., J. R. Unruh, D. F. Holub, B. Ingram, C. K. Johnson, and G. S. Wilson. 2005. DNA aptamer based bioanalysis of IgE using fluorescence anisotropy. Anal. Chem. In press. [DOI] [PubMed]
- Ha, T., J. Glass, T. Enderle, D. S. Chemla, and S. Weiss. 1998. Hindered rotational diffusion and rotational jumps of single molecules. Phys. Rev. Lett. 90:2093–2096. [Google Scholar]
- Ha, T., T. A. Laurence, D. S. Chemla, and S. Weiss. 1999. Polarization spectroscopy of single fluorescent molecules. J. Phys. Chem. B. 103:6839–6850. [Google Scholar]
- Heinlein, T., J.-P. Knemeyer, O. Piestert, and M. Sauer. 2003. Photoinduced electron transfer between fluorescent dyes and guanosine residues in DNA-hairpins. J. Phys. Chem. B. 107:7957–7964. [Google Scholar]
- Hill, J. J., and C. A. Royer. 1997. Fluorescence approaches to study of protein-nucleic acid complexation. Methods Enzymol. 278:390–416. [DOI] [PubMed] [Google Scholar]
- Johansson, L. B.-Å. 1990. Limiting fluorescence anisotropies of perylene and xanthene derivatives. J. Chem. Soc. Faraday Trans. 86:2103–2107. [Google Scholar]
- Johnson, M. L., and L. M. Faunt. 1992. Parameter estimation by least-squares methods. Methods Enzymol. 210:1–37. [DOI] [PubMed] [Google Scholar]
- Kelley, S. O., and J. K. Barton. 1999. Electron transfer between bases in double helical DNA. Science. 283:375–381. [DOI] [PubMed] [Google Scholar]
- Kumke, M. U., G. Li, L. B. McGown, G. T. Walker, and C. P. Linn. 1995. Hybridization of fluorescein-labeled DNA oligomers detected by fluorescence anisotropy with protein binding enhancement. Anal. Chem. 67:3945–3951. [DOI] [PubMed] [Google Scholar]
- Kumke, M. U., L. Shu, L. B. McGown, G. T. Walker, J. B. Pitner, and C. P. Linn. 1997. Temperature and quenching studies of fluorescence polarization detection of DNA hybridization. Anal. Chem. 69:500–506. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J. R. 1999. Principles of Fluorescence Spectroscopy. Kluwer Academic/Plenum, New York.
- LeTilley, V., and C. A. Royer. 1993. Fluorescence anisotropy assays implicate protein-protein interactions in regulating trp repressor DNA binding. Biochemistry. 32:7753–7758. [DOI] [PubMed] [Google Scholar]
- Martinez, J. M., S. K. C. Elmroth, and L. Kloo. 2001. Influence of sodium ions on the dynamics and structure of single-stranded DNA oligomers: a molecular dynamics study. J. Am. Chem. Soc. 123:12279–12289. [DOI] [PubMed] [Google Scholar]
- McCauley, T. G., N. Hamaguchi, and M. Stanton. 2003. Aptamer-based biosensor arrays for detection and quantification of biological macromolecules. Anal. Biochem. 319:244–250. [DOI] [PubMed] [Google Scholar]
- McGown, L. B., M. J. Joseph, J. B. Pitner, G. P. Vonk, and C. P. Linn. 1995. The nucleic acid ligand: a new tool for molecular recognition. Anal. Chem. 67:A663–A668. [DOI] [PubMed] [Google Scholar]
- Morris, G. M., D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew, and A. J. Olson. 1998. Automated docking using a Lamarckinan genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 19:1639–1662. [Google Scholar]
- Murphy, C. J., M. R. Arkin, N. D. Ghatlia, S. Bossmann, N. J. Turro, and J. K. Barton. 1994. Fast photoinduced electron transfer through DNA intercalation. Proc. Natl. Acad. Sci. USA. 91:5315–5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarenko, I., R. Pires, B. Lowe, M. Obaidy, and A. Rashtchian. 2002. Effect of primary and secondary structure of oligodeoxyribonucleotides on the fluorescent properties of conjugated dyes. Nucleic Acids Res. 30:2089–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman, G. G., R. J. Grainger, D. Uhrin, and D. M. J. Lilley. 2000. Location of cyanine-3 on double-stranded DNA: importance for fluorescence resonance energy transfer studies. Biochemistry. 39:6317–6324. [DOI] [PubMed] [Google Scholar]
- Plimpton, S. J. 1995. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 117:1–19. [Google Scholar]
- Rachofsky, E. L., and W. R. Laws. 2000. Kinetic models and data analysis methods for fluorescence anisotropy decay. Methods Enzymol. 321:216–238. [DOI] [PubMed] [Google Scholar]
- Reid, S. L., D. Parry, H.-H. Liu, and B. A. Connolly. 2001. Binding and recognition of GATATC target sequences by the EcoRV restriction endonuclease: a study using fluorescent oligonucleotides and fluorescence polarization. Biochemistry. 40:2484–2494. [DOI] [PubMed] [Google Scholar]
- Sjöback, R., J. Nygren, and M. Kubista. 1998. Characterization of fluorescein-oligonucleotide conjugates and measurement of local electrostatic potential. Biopolymers. 46:445–453. [DOI] [PubMed] [Google Scholar]
- Torimura, M., S. Kurata, K. Yamada, T. Yokomaku, Y. Kamagata, T. Kanagawa, and R. Kurane. 2001. Fluorescence-quenching phenomenon by photoinduced electron transfer between a fluorescent dye and a nucleotide base. Anal. Sci. 17:155–160. [DOI] [PubMed] [Google Scholar]
- Unruh, J. R., G. Gokulrangan, G. S. Wilson, and C. K. Johnson. 2005. Fluorescence properties of fluorescein, tetramethylrhodamine, and Texas Red linked to a DNA aptamer. Photochem. Photobiol. In press. [DOI] [PubMed]
- Vámosi, G., and R. M. Clegg. 1998. The helix-coil transition of DNA duplexes and hairpins observed by multiple fluorescence parameters. Biochemistry. 37:14300–14316. [DOI] [PubMed] [Google Scholar]
- Vámosi, G., C. Gohlke, and R. M. Clegg. 1996. Fluorescence characteristics of 5-carboxytetramethylrhodamine linked covalently to the 5′ end of oligonucleotides: multiple conformers of single-stranded and double-stranded dye-DNA complexes. Biophys. J. 71:972–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand, T. W., P. B. Williams, S. C. Dreskin, M. Jouvin, J. Kinet, and D. Tasset. 1996. High-affinity oligonucleotide ligands to human IgE inhibit binding to Fcɛ receptor I. J. Immunol. 157:221–230. [PubMed] [Google Scholar]
- Young, M. A., B. Jayaram, and D. L. Beveridge. 1997. Intrusion of counterions into the spine of hydration in the minor groove of B-DNA: fractional occupancy of electronegative pockets. J. Am. Chem. Soc. 119:59–69. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.