

Abstract
The relationship between the mechanical properties of cells and their molecular architecture has been the focus of extensive research for decades. The cytoskeleton, an internal polymer network, in particular determines a cell's mechanical strength and morphology. This cytoskeleton evolves during the normal differentiation of cells, is involved in many cellular functions, and is characteristically altered in many diseases, including cancer. Here we examine this hypothesized link between function and elasticity, enabling the distinction between different cells, by using a microfluidic optical stretcher, a two-beam laser trap optimized to serially deform single suspended cells by optically induced surface forces. In contrast to previous cell elasticity measurement techniques, statistically relevant numbers of single cells can be measured in rapid succession through microfluidic delivery, without any modification or contact. We find that optical deformability is sensitive enough to monitor the subtle changes during the progression of mouse fibroblasts and human breast epithelial cells from normal to cancerous and even metastatic state. The surprisingly low numbers of cells required for this distinction reflect the tight regulation of the cytoskeleton by the cell. This suggests using optical deformability as an inherent cell marker for basic cell biological investigation and diagnosis of disease.
INTRODUCTION
The cytoskeleton of cells, an intricate polymer network, is the structural framework that predominantly shapes a cell and provides its mechanical rigidity (Elson, 1988; Lodish et al., 2000). It surprises the observer by withstanding enormous external pressures, thus preserving the integrity of the cell (Janmey et al., 1991). On the other hand, the momentum carried by light and the forces it can exert upon impact on material objects are minute and not noticeable on macroscopic length scales. It is exactly these small forces induced by light, however, that prove to be ideal for the deformation of microscopic cells and the cytological detection of cytoskeleton-altering diseases such as cancer.
The cytoskeleton not only provides mechanical rigidity, but also fulfills many important cellular functions (Lodish et al., 2000). The cytoskeleton's various components—actin, microtubules, and intermediate filaments—in concert with their accessory proteins facilitate cell motility, ribosomal and vesicle transport, mitosis, and mechano-transduction (Wang et al., 1993; Wirtz and Dobbs, 1990). All of the tasks performed by cytoskeletal elements are finely tuned, regulated, and synchronized with the overall function of a specific cell. Consequently, changes to cellular function during differentiation (Olins et al., 2000) or due to disease are mirrored in the cytoskeleton: Cytoskeletal alterations cause capillary clogs in circulatory problems (Worthen et al., 1989) and various blood diseases including sickle-cell anemia, hereditary spherocytosis, or immune hemolytic anemia (Bosch et al., 1994; Williamson et al., 1985), and genetic disorders of intermediate filaments and their cytoskeletal networks lead to problems with skin, hair, liver, colon, and motor neuron diseases such as amyotrophic lateral sclerosis (Fuchs and Cleveland, 1998; Kirfel et al., 2003).
The best-known example is the malignant transformation of cells where morphological changes caused by the cytoskeleton are in fact diagnostic for cancer. During the cell's progression from a fully mature, postmitotic state to a replicating, motile, and immortal cancerous cell, the cytoskeleton devolves from a rather ordered and rigid structure to a more irregular and compliant state. The changes include a reduction in the amount of constituent polymers and accessory proteins and a restructuring of the available network (Ben-Ze'ev, 1985; Cunningham et al., 1992; Katsantonis et al., 1994; Moustakas and Stournaras, 1999; Rao and Cohen, 1991). These cytoskeletal alterations are evident because malignant cells are marked by replication and motility, both of which are inconsistent with a rigid cytoskeleton. Taken together, these changes in cytoskeletal content and structure should be reflected in the overall mechanical properties of the cell as well. Thus, measuring a cell's rigidity should provide information about its state and may be viewed as a new biological marker.
There are only a few experimental techniques capable of assessing cellular mechanical properties, but they consistently imply a correlation of cellular rigidity to cell status. Historically, the prevalent technique has been micropipette aspiration (Hochmuth, 2000). Using this technique, researchers found a 50% reduction in elasticity of malignantly transformed fibroblasts as compared to their normal counterparts (Ward et al., 1991). More recently, atomic force microscopy has been used for the determination of cellular rigidity (Mahaffy et al., 2000; Rotsch et al., 1999). Lekka et al. (1999) used atomic force microscopy to investigate normal human bladder endothelial cell lines and complimentary cancerous cell lines and found that their rigidity differed by an order of magnitude. Other researchers have employed magnetic bead rheology (Wang et al., 1993), microneedle probes (Zahalak et al., 1990), microplate manipulation (Thoumine and Ott, 1997), acoustic microscopes (Kundu et al., 2000), sorting in microfabricated sieves (Carlson et al., 1997), and the manipulation of beads attached to cells with optical tweezers (Sleep et al., 1999). In general, malignant cells responded either less elastic (softer) or less viscous (less resistant to flow) to stresses applied, depending on the measurement technique and the model employed. Metastatic cancer cells have been found to display an even lower resistance to deformation (Raz and Geiger, 1982; Ward et al., 1991). This stands to reason, because metastatic cancer cells must squeeze through the surrounding tissue matrix as they make their way into the circulatory systems where they travel to establish distant settlements (Wyckoff et al., 2000).
These findings suggest that cellular elasticity may be used as a cell marker and a diagnostic parameter for underlying disease. However, all of these techniques face major obstacles to generalized application: low cell throughput leading to poor statistics; mechanical contact of the probe leading to adhesion and active cellular response; and special preparation and nonphysiological handling leading to measurement artifacts. In this study we demonstrate that, in contrast, an optical stretcher (Guck et al., 2001) combined with microfluidic delivery can deform individual suspended cells by optically induced surface forces at rates that could eventually rival flow cytometers and thus circumvents these impediments (see Fig. 1, A and B). In addition, we show that the deformability of cells measured with a microfluidic optical stretcher is likely to be a tightly regulated inherent cell marker.
FIGURE 1.

Optically induced surface forces lead to trapping and stretching of cells. Cells flowing through a microfluidic channel can be serially trapped (A) and deformed (B) with two counterpropagating divergent laser beams (an animation can be found as supplementary material online). The distribution of surface forces (small arrows) induced by one laser beam with Gaussian intensity profile incident from the left (indicated with large triangles) for a cell that is (C) slightly below the laser axis and (D) on axis. An integration of these forces over the entire surface results in the net force Fnet shown as large arrows. The corresponding distributions for two identical but counterpropagating laser beams are shown for a cell on axis (E). This is a stable trapping configuration. When the cell is displaced from the axis (C and F), the symmetry of the resulting force distribution is broken, giving the net force a restoring component perpendicular to the laser axis. The inset shows the momenta of the various light rays and the resulting force at the surface.
MATERIALS AND METHODS
Cell lines
All cell lines (BALB/3T3, SV-T2, MCF-7, MCF-10, MDA-MB-231) were obtained from the American Type Culture Collection (Rockville, MD) and cultured according to the protocols provided. In addition, chemically modified MCF-7 cells (modMCF-7) were generated by treating MCF-7 cells with 100 nM 12-O-tetradecanoylphorbol-13-acetate (TPA) for 18 h. MDA-MB-231 cells were treated with all-trans retinoic acid in accordance with the procedures in reference (Wang et al., 2001) to prepare modMDA-MB-231 cells. Before measurement in the optical stretcher, all cells were trypsinized (0.1% Trypsin/EDTA) to detach them from the culture dish and washed with phosphate buffered saline (PBS). All cells assumed a spherical shape in suspension without any further manipulation.
Refractive index measurements of fibroblasts
The refractive index of the BALB3T3 and the SVT2 cell lines were measured by a phase matching technique described in (Barer and Joseph, 1954, 1955a, 1955b). A bovine serum albumin (BSA) stock solution in PBS was roughly adjusted to various refractive indices, nmedium, according to the Gladstone-Dale formula, nmedium = α c + nPBS. Here, α = 0.00187 is the specific refraction increment for BSA, c is the concentration of BSA in g/100 ml, and nPBS is the refractive index of PBS. The exact index of refraction was then determined in an Abbe-refractometer (AR6, Krüss Optronic GmbH, Hamburg, Germany). Cells are suspended in the different BSA solutions and observed on a phase contrast microscope. For all solutions, the percentages of cells that appear brighter and darker than the surrounding solution were determined separately. Typically, ∼200 cells were used for analysis for each solution. The results were then fitted by an error function
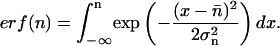 |
As an example, the inset of Fig. 3 shows the fit for the BALB/3T3 cells brighter than the surrounding medium. The inflection point of the error function (which was also the intersection point with the inverse series of measurements of cells darker than the medium; not shown) is the mean index of refraction, n̄, of the cells in the observed sample. The standard deviation, σn, can be extracted from the fit.
FIGURE 3.

Refractive index of fibroblasts. (A) Fit of an error function to the percentages of bright and dark cells compared to the surrounding medium as a function of refractive index of the medium, nmedium. Shown are the data for BALB/3T3 fibroblasts. (B) Resulting distributions of refractive indices, ncell, for BALB/3T3 (dashed line) and SV-T2 fibroblasts (solid line).
Fluorescence imaging of suspended fibroblasts
All chemicals were purchased from Sigma (St. Louis, MO) unless stated otherwise. BALB/3T3 and SV-T2 fibroblasts in suspension were allowed to reattach to a poly-L-lysine coated microscope slide for 3–5 min before washing with PBS and a 10 min fixation with 4% formaldehyde (P-6148) at 37°C. In this fashion, the cells were “frozen” in a quasi-suspended state before they had any time to start attaching. After permeabilization for 10 min with 0.1% Triton-X 100, the filamentous actin was labeled with TRITC-Phalloidin (P-1951) at a final concentration of 1 μg/ml. For microtubule staining, goat serum (G-9023) was used to block unspecific binding before the microtubules were stained with a primary β-tubulin antibody (E7, Developmental Studies Hybridoma Bank, Iowa City, IA), and a secondary AlexaFluor488 goat anti-mouse antibody (A-11001, Molecular Probes, Eugene, OR). To prevent bleaching, the cells were mounted in an appropriate medium (Prolong Antifade Kit, Molecular Probes). Images were taken on a confocal laser scanning microscope (TCS SP2, Leica, Heidelberg, Germany).
LSC measurements of the F-actin amount in fibroblasts
The staining procedure for the quantitative F-actin measurement was slightly modified from above. Both cell types were cultured separately but on the same slide so that they could be fixed and stained in an identical way. Fixation was performed on two identically prepared slides using Acetone (99.9%) for 15 min. After permeablization for 10 min with TritonX-100, cells were additionally permeabilized and washed with Tween20 for 10 min. At this point, the negative-control slide received Phalloidin (P2141) at a concentration of 1.67 mg/ml for 30 min. Both slides then were stained with Alexa-532-Phalloidin (A-22282, Molecular Probes) at a concentration of 0.18 mg/ml for 60 min, washed with PBS for 10 min, and then stained with the nuclear stain 7-AAD (A-1310, Molecular Probes) at a concentration of 25μg/ml for 15 min.
The measurement was performed with a Laser Scanning Cytometer (LSC, CompuCyte, Cambridge, MA) as previously described elsewhere (Gerstner et al., 2000) with slight modifications. Here, 7-AAD (i.e., the nuclear staining) was used as the triggering signal and F-actin was stained with Alexa-532. Both fluorochromes are excited by the Argon laser (488 nm) but their emission can be well distinguished. Instrument settings for cell triggering were optimized by visual comparison with the scan data feature of the instrument: 7-AAD intensity trigger threshold was chosen to be 1000 (instrument units), minimum trigger area 10 μm2, and for cell area 10 pixels were added to threshold area. Depending on negative-control values and staining efficiency, these instrument settings had to be changed as appropriate and were verified before each analysis. However, since both cell types were stained on one slide, the direct comparison of the relative amounts of F-actin was always possible, independent of instrument settings.
Microfluidic optical stretcher
The setup of the microfluidic optical stretcher was essentially the same as described in reference (Guck et al., 2001). Instead of the Ti-Sapphire laser system, an Ytterbium-doped fiber laser emitting at a wavelength of 1064 nm was used as light source (YLD-10-1064, IPG Photonics, Oxford, MA). The fiber laser was spliced to a 50:50 coupler to ensure equal power in the two fibers. The laser power was P = 100 mW per fiber for trapping the cells, P = 1.7 W for the stretching of the mouse fibroblasts, and P = 600 mW for the stretching of the epithelial cells, with the fiber ends spaced 130 μm apart. The laser beams, emanating from the optical fibers, were not focused as in optical tweezers but simply underwent diffraction-limited divergence; the beam diameter increased from 6.2 μm at the fiber to ∼12 μm at the location of the cell.
An aliquot of the cell suspension was placed in the microfluidic system for analysis. In this study, cells were relayed to the stretching region between the two fiber ends using a microperistaltic pump (P625, Instech Laboratories, Plymouth Meeting, PA) connected to an 80 μm (i.d.) square glass capillary (VitroCom, Mountain Lakes, NJ). The capillary tube was situated between the two fiber ends such that cells flowing within the tube passed through the center between the two laser beams, where they were trapped and stretched at the light powers mentioned above (Fig. 1, A and B, and animation provided as supplementary online material). The flow was stopped during the measurement. The entire microfluidic system was mounted on an inverted phase contrast microscope (Axiovert 25 CFL; objective: LD Achroplan, 40×/0.60 Corr. Ph2, Carl Zeiss, Thornwood, NY). Images of the cell were recorded at a rate of 10 frames/s using a digital CCD camera (A101, Basler, Exton, PA) before and during the stretching, and stored for later analysis.
Deformation analysis
The images were analyzed separately after each experiment. In general, the magnitude of the deformation is a function of time due to the viscoelastic nature of cells. Here, the major and minor axes of the cell, a and b, respectively, at trapping power (subscript o) and stretching power after some time t (subscript t) were determined using custom made video processing software. The focal depth of the imaging system was small compared to the size of the cell so that only a cross-section of the cells was imaged. Therefore, the aspect ratio (major axis divided by minor axis) was used as a measure for the deformation rather than the difference in these values directly. In this way, systematic measurement errors, due to minor differences in cell position with respect to the microscope's focal plane were avoided. The observed deformation after some time, Dt, was then
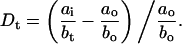 |
Smaller cells intercept less light and consequently experience a smaller stretching force. To account for variations in size of the different cells, the optically induced stress profile was calculated for each cell as described in reference (Guck et al., 2001; see also Fig. 1 E). The integrated stretching forces, F, in the direction of the major and minor axes were divided to obtain an aspect force ratio for each cell. A reference aspect force ratio (fiducial point) was then selected and used to normalize all observed deformations to yield a quantitative measure for the optical deformability, ODt,
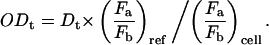 |
This quantity corresponds to the compliance in a step stress experiment (see also Results and Discussion). For the cell types that were compared directly, the same fiducial point was used. In this manner, the influence of cell size on the observed deformation was fully accounted for in the optical deformability, both within a single cell population and between different cell types.
RESULTS AND DISCUSSION
The microfluidic optical stretcher
The optical stretcher has been described previously as a two-beam laser trap that induces optical forces, which can be used to deform cells (Guck et al., 2001). In addition to deformation, these forces also provide a stable trapping situation. It is this feature that is used here to capture cells from a flow in a microfluidic system and center them automatically, thus allowing flow-cytometric single cell elasticity measurements with high throughput (see Fig. 1, A and B).
When light passes through an interface between different transparent dielectric media, e.g., the surface of a cell, it induces a force that is normal to the interface and always directed away from the optically denser medium (Ashkin and Dziedzic, 1973; Guck et al., 2000). These surface forces will cause a stretching of the object along the laser axis (Figs. 1 B, 2, A and B, and 6). Although surface forces are present in other optical traps, their specific geometry renders this effect small. The optical stretcher is optimized to maximize the deforming forces while keeping the light intensities low to ensure the viability of the cells. The magnitude of the induced forces scales linearly with the incident light power and with (n − 1), where n = ncell/nmedium is the relative refractive index. With 1W of laser power in each beam, the forces range between 200 and 500 pN even when the relative refractive indices are as low as n = 1.02–1.05, which is typical for biological materials.
FIGURE 2.

Deformation of fibroblasts in an optical stretcher. A BALB/3T3 fibroblast deforms by 6.48% ± 0.36% (measurement error) along the laser axis, as determined by image analysis, when the light power is increased from 0.1 W (A) to 1.7 W (B) in both beams. Measuring large numbers of cells (C) reveals that the optical deformability of malignantly transformed SV-T2 fibroblasts is significantly shifted to higher values compared to normal BALB/3T3 fibroblasts (ODBALB/3T3 = 8.4 ± 1.0; ODSV-T2 = 11.7 ± 1.1; mean and mean ± SE). The scale bars are 10 μm.
FIGURE 6.

Typical examples of the stretching of breast epithelial cells. The images in the left column are taken at an incident light power of 100 mW in each beam, which is sufficient for the trapping of the cells. At an incident light power of 600 mW (right column), the cancerous MCF-7 cells (C and D) deform more than the nonmalignant MCF-10 cells (A and B). The metastatic modMCF-7 cells (E and F) deform the most. The scale bar is 10 μm.
The simplest rheologic measurement to characterize the mechanical properties of cells is a step stress experiment that extracts the compliance of cells. From this, time- or frequency-dependent complex shear moduli or other relevant mechanical parameters can be derived (Wottawah et al., 2005). For simplicity, the optical deformability used here corresponds to the compliance after a certain time t. Given similar optical properties, the optical deformability (i.e., the degree of deformation for a specified incident light power) is then a direct measure for the strength of the cytoskeleton.
A major feature of the optical stretcher is its function as an optical trap. Integration of the induced forces over the entire illuminated surface of an object results in a net force that acts on the object as a whole (Fig. 1, C and D). The optical stretcher is based on a double-beam trap comprised of two identical coaxially aligned, counterpropagating divergent laser beams (Ashkin, 1970). Here the net forces from the two beams balance each other on the beam axis at a location equidistant from the light sources (Fig. 1 E). This is a stable trapping configuration because any displacement from the center will result in a restoring net force (Fig. 1 F). The optical stretcher has been integrated with a microfluidic system that can serially deliver individual cells into the trapping and stretching region. Using rudimentary automation of flow control, trapping, and stretching, we were able to trap individual cells from a flowing cell suspension and measure their deformability at a rate of ∼1 per min (Fig. 1, A and B, and animation provided as supplementary online material). This compares favorably with previous single cell elasticity measurement techniques that could handle only a few cells per day. Single cell measurements offer the advantage over ensemble measurements to allow the sorting of the cells of interest, which can be easily implemented with standard microfluidic techniques (Fu et al., 2002; Unger et al., 2000), and subsequent culturing or further analysis. With the microfluidic optical stretcher we now have a scaleable tool that could be incorporated in lab-on-a-chip systems to exploit optical deformability as an inherent cell marker and, ultimately, to screen cell populations for the presence of cytoskeleton-altering conditions.
Optical deformability of mouse fibroblasts
To establish the connection between cellular function and optical deformability of cells, we investigated BALB/3T3 and SV-T2 fibroblasts with the microfluidic optical stretcher (Fig. 2, A and B). BALB/3T3 is a fibroblast cell line established in 1968 from disaggregated BALB/c mouse embryos. SV-T2 cells are derived from BALB/3T3 cells by transformation with the oncogenic DNA virus SV40 (Aaronson and Todaro, 1968b). Both are well-studied cell lines and have often served as models for the study of malignant transformation in general (Aaronson and Todaro, 1968a; Thoumine and Ott, 1997). We found that the optical deformability of the SV-T2 cells was significantly increased compared to the BALB/3T3 cells (Fig. 2 C). Based on a Student's t-test, the two populations were distinguishable with 99% confidence.
Optical deformability is in general a function of time due to the viscoelastic nature of cells. Here, the cells were stretched for t = 1 s. A more detailed analysis of the rheological behavior shows that this timescale exploits both elastic and viscous contributions to deformability in this cell type (Wottawah et al., 2005). With better resolution of the deformation, the cells should be distinguishable already after fractions of seconds, which would allow higher measurement rates in general. On the other hand, this quantifiable difference in optical deformability is already evident after measuring only 30 cells each, a tiny fraction of the cells required for proteomic techniques. Thus, very high measurement rates as in fluorescence activated cell sorters (FACS) machines might not be required in the microfluidic optical stretcher for handling statistically sufficient sample sizes.
Since the optical deformability of cells, i.e., their displayed deformation for a given laser power, in general depends on both their mechanical properties as well as their optical properties, we determined the refractive index of the two cell populations. Fig. 3 shows the distribution of the refractive index, ncell, for BALB/3T3 and SV-T2 cells. They are almost identical. The numerical values are nBALB = 1.3722 ± 0.0036 (mean and SD) and nSV-T2 = 1.3711 ± 0.0039, respectively. For all practical purposes, the two cell lines cannot be distinguished based on refractive index. Thus, the difference in optical deformability can be solely attributed to their mechanical properties. To this end, optical deformability can be equated with compliance.
Cytoskeleton of suspended mouse fibroblasts
The optical stretcher operates in an intrinsic noncontact mode on cells in suspension. Up until now, however, most research on the cytoskeleton of cells has been done on cells attached to a substrate or in contact with a material probe. To verify the existence and gain insight into the structure of the cytoskeleton in suspended cells, confocal microscopy was used to image F-actin in normal and malignantly transformed fibroblasts. As can be seen in Fig. 4, cells retain an extensive polymeric network even when in suspension and not attached to a substrate. The actin cytoskeleton features a dense cortical layer underneath the plasma membrane and an isotropic network throughout the cell body (see Fig. 4, A and B). It differs from the actin cytoskeleton in adherent cells by lacking stress fibers, which is consistent with the missing focal adhesions that usually serve as anchoring points. The microtubule network resembles the one found in adherent cells, with microtubules spanning the space between nucleus and cell membrane (see Fig. 4 C), but does not significantly contribute to a cell's interphase elasticity (Janmey et al., 1991; Rotsch and Radmacher, 2000).
FIGURE 4.

Cytoskeleton in suspended fibroblasts. Fluorescence confocal images clearly show the actin (red) and the microtubule network (green) in normal BALB/3T3 (A and C) and malignantly transformed SV-T2 (B) fibroblasts. The shadows within the cells coincide with the nucleus as checked with a nuclear stain (not included in the image). The scale bars are 10 μm.
Although a fibroblast in suspension is certainly not in a physiological environment due to lack of cell-cell contact and the sensing of a much more solid-like structure in tissue, it should be pointed out that cells attached to a rigid substrate are also not in their native environment because contact with a flat and hard surface in tissue is rare. In addition, the lack of stress fibers in suspended cells offers the advantage of allowing the measurement of a more homogeneous and isotropic structure, for which theories from polymer physics are more directly applicable.
Theory predicts a strong dependence of the shear modulus of isotropic networks of semiflexible polymers on the concentration of filaments (Gardel et al., 2003; Janmey et al., 1991; Wilhelm and Frey, 2003). Therefore, we compared the relative amounts of F-actin in BALB/3T3 and SV-T2 cells by staining the cells with Alexa-532 Phalloidin and measuring their integrated intensity distributions with an LSC. Fig. 5 shows the distribution of Alexa-532 signal measured for cells of the two cell types. Since Phalloidin binds only to F-actin, this shows that the total amount of filamentous actin in the malignantly transformed cells is reduced by ∼40%. This confirms previous ensemble measurements with standard proteomic techniques, which showed the same decrease in the F-actin content of malignantly transformed fibroblasts (Moustakas and Stournaras, 1999). Due to the reduced size of the SV-T2 cells, the concentration of F-actin (i.e., Alexa-532 intensity divided by cell area) is reduced by ∼50% (see inset of Fig. 5).
FIGURE 5.

F-actin in normal and malignant fibroblasts. The distributions of integrated Alexa-532 fluorescence intensity, which corresponds to the total amount of F-actin in BALB/3T3 (crosses) and SV-T2 fibroblasts (circles), was measured by LSC and fitted to log-normal distributions. The inset shows the distributions of total F-actin amount per cell divided by projected cell area.
The reduction in the absolute amount of F-actin is accompanied by a restructuring in the actin cytoskeleton of the malignantly transformed SV-T2 cells compared to the normal BALB/3T3 cells (see Fig. 4, A and B). Both reduced amount and different structure of the actin cytoskeleton, in combination with identical optical properties, explains the increased optical deformability of the cancerous cells. Consistent with a critically important role for F-actin, latrunculin-treated cells (BALB/3T3 and SV-T2) soften by about half.
Optical deformability of human breast epithelial cells
We also measured the optical deformability of well-characterized cell lines of human breast epithelial cells and their cancerous counterparts. MCF-cells and MDA-MB-231 cells are standard model cell lines for the study of breast cancer (Johnson et al., 1999; Soule et al., 1973; Tait et al., 1990; Wang et al., 2001). MCF-10 is a nontumorigenic epithelial cell line derived from the benign breast tissue of a 36-year-old woman with fibrocystic disease. These cells are immortal, but otherwise normal, noncancerous mammary epithelial cells. MCF-7 is a corresponding line of human breast cancer cells (adenocarcinoma), obtained from the pleural effusion of a 69-year-old Caucasian female. These cells are nonmotile, nonmetastatic epithelial cancer cells. When the phorbol ester TPA is added to MCF-7 cells (modMCF-7 cells), a dramatic increase (18-fold) in the invasiveness and the metastatic potential of these cells occurs (Johnson et al., 1999). In addition to inducing motility correlated to metastatic potential, TPA induces the release of matrix metallo-proteases in MCF-7 cells. These enzymes digest the surrounding tissue matrix, are associated with metastatic behavior in vivo, and have been strongly implicated in the progression of breast cancer (Himelstein et al., 1994). Fig. 6 shows the typical stretching behavior, whereas Fig. 7 A illustrates quantitatively the trimodal distribution of optical deformability that was observed for these cells. Confirming the result found with normal and malignant fibroblasts, the cancerous MCF-7 deformed more than the normal MCF-10 cells. Demonstrating the sensitivity of the measurement, the metastatic modMCF-7 deformed even more than the nonmetastatic MCF-7. The three populations are distinguishable with 99.9% confidence based on a Student's t-test. Considering typical biological variances, surprisingly few cells are required for this distinction (NMCF-10 = 36, NMCF-7 = 26, NmodMCF-7 = 21), indicating that, for the cell lines tested, optical deformability is a tightly regulated cell marker.
FIGURE 7.

Optical deformability of normal, cancerous, and metastatic breast epithelial cells. (A) The three populations of the MCF cell lines and (B) the two populations of the MDA-MB-231 cell lines are clearly distinguishable in the histograms of the measured optical deformability (ODMCF-10 = 10.5 ± 0.8; ODMCF-7 = 21.4 ± 1.1; ODmodMCF-7 = 30.4 ± 1.8; ODMDA-MB-231 = 33.7 ± 1.4; ODmodMDA-MB-231 = 24.4 ± 2.5; mean and mean ± SE). The values were measured at t = 60 s.
In addition, we also measured the optical deformability of MDA-MB-231 breast cancer cells. MDA-MB-231 cells are highly metastatic breast cancer cells. When treated with all-trans retinoic acid (modMDA-MB-231), they become less aggressive (Wang et al., 2001). As can be seen in Fig. 7 B, the optical deformability decreased with loss of metastatic competence. Again, based on a Student's t-test, the two populations can be distinguished with 97.5% confidence.
Similar to the fibroblast results, the differences in refractive index of all five cell lines were within the statistical errors and lay around 1.365 (data not shown). Thus, the difference in optical deformability seems to be correlated with a reduced cytoskeletal resistance to deformation of the cancerous cells compared to normal cells. In this case, cancerous cells with acquired metastatic competence are marked by an even greater reduction in structural strength, which is consistent with their ability to move through tissue, to intravasate into the blood and lymph systems, to circulate through these microvascular systems, and to extravasate eventually to form metastases. Although a major cytoskeletal element in epithelial cells is the intermediate filament keratin (Kirfel et al., 2003), its contribution to mechanical properties only becomes important at strains larger than those employed in this study (Janmey et al., 1991; Wang and Stamenovic, 2000). The differences in optical deformability can, most likely, be ascribed to the reduction in F-actin during malignant transformation in this cell type by ∼30% (Katsantonis et al., 1994).
CONCLUSION AND OUTLOOK
These results demonstrate that optical deformability measured with a microfluidic optical stretcher can serve as a sensitive indicator of the state of cellular development during normal differentiation and disease. Definitive changes in optical deformability are detectable in response to subtle changes in a cell's metabolism, which are manifested in cytoskeletal composition and structure. This dependence seems to fit to results from polymer physics where even minute variations in the concentration of cytoskeletal filaments are nonlinearly enhanced in the network's overall elasticity (Gardel et al., 2003; Janmey et al., 1991; Wilhelm and Frey, 2003). Since the optical deformation as defined in this study (i.e., the size-corrected deformation after a certain time t) allows already the sensitive distinction between different cell types, a detailed microrheologic characterization of cells using step-stress or sinusoidal stress experiments to extract usual viscoelastic parameters (e.g., plateau modulus, long-time viscosity, etc.) should always lead to detectable differences between different cells, if the optical properties of the cells are similar. The most discriminative mechanical parameter can then be chosen as cell marker, according to the situation at hand. If cells also differ in their optical properties, this is an additional discriminating feature that results in different induced forces and observed deformations. Thus, optical deformability in general should be a unique and useful cell marker that is sensitive to cytoskeletal changes convolved with the cell's optical properties.
Under the assumption that these findings, obtained with cultured cell lines, can be extended to primary cells, there could be immediate diagnostic relevance to these results. Many diseases, and especially cancer, are often curable when detected early but fatal otherwise. The primary basis for pathology evaluation remains morphological change in suspect tissue, but access to coherent tissue requires biopsy and can necessarily only be performed in later stages of the disease after a noticeable collection of cells has occurred. The microfluidic optical stretcher permits the investigation of samples of individual cells obtained by exfoliative cytology (Caraway et al., 1993; Epstein et al., 2002; Sherman and Kurman, 1996), enabling the quantitative screening for cytoskeletal change inherent to cancer. In a clinical situation, there are other cells present in an aspirate or other tissue sample. Besides fibroblasts and other blood cells, motile lymphocytes are present in inflammatory reactions often accompanying cancer. These cells will either have to be identified by their optical deformability, or have to be deleted using lysis, density-gradient centrifugation, magnetic bead sorting, or FACS sorting. The effect of these manipulations on mechanical parameters is currently being investigated in preclinical trials.
Cells with abnormal functioning may not only be identified with the microfluidic optical stretcher, but also isolated through microfluidic sorting. Since this is based solely on their inherent optical deformability, there is no need for preparation with fluorescent dyes or magnetic beads. Subsequently, pure samples of isolated cells can be investigated without artificial corruption using standard genomic or proteomic techniques, which are otherwise often impeded by the scarcity of the cells of interest. The aspect of noncontaminating separation of cells might be important for therapeutic uses of stem cells, which also differ in their cytoskeletal composition from differentiated cells (Olins et al., 2000) and could be identified and sorted with a microfluidic optical stretcher. This seems especially relevant because no set of molecular markers has yet been found to unambiguously define a stem cell.
In conclusion, optical deformability is an inherent cell marker that offers a sensitive cellomic alternative to current proteomic techniques and opens the door to novel investigations into all cellular processes that involve the cytoskeleton.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
We thank Chieze Ibeneche and Carole Moncman for technical help and advice, and Robin Goodman for editorial assistance. We thank Dr. Attila Tarnok for his assistance and advice with LSC measurements, which were done at the Interdisciplinary Center for Clinical Research (IZKF, Z10, Core Unit Fluorescence Technologies) at the University of Leipzig.
The research was funded by Evacyte Corporation and through the Wolfgang-Paul Prize awarded to J.K. by the Humboldt-Foundation.
References
- Aaronson, S. A., and G. J. Todaro. 1968a. Basis for the acquisition of malignant potential by mouse cells cultivated in vitro. Science. 162:1024–1026. [DOI] [PubMed] [Google Scholar]
- Aaronson, S. A., and G. J. Todaro. 1968b. Development of 3T3-like lines from Balb-c mouse embryo cultures: transformation susceptibility to SV40. J. Cell. Physiol. 72:141–148. [DOI] [PubMed] [Google Scholar]
- Ashkin, A. 1970. Acceleration and trapping of particles by radiation pressure. Phys. Rev. Lett. 24:156–159. [Google Scholar]
- Ashkin, A., and J. M. Dziedzic. 1973. Radiation pressure on a free liquid surface. Phys. Rev. Lett. 30:139–142. [Google Scholar]
- Barer, R., and S. Joseph. 1954. Refractometry of living cells, Part I. Basic principles. Q. J. Microsc. Sci. 95:399–423. [Google Scholar]
- Barer, R., and S. Joseph. 1955a. Refractometry of living cells, Part II. The immersion medium. Q. J. Microsc. Sci. 96:1–26. [Google Scholar]
- Barer, R., and S. Joseph. 1955b. Refractometry of living cells, Part III. Technical and optical methods. Q. J. Microsc. Sci. 96:423–447. [Google Scholar]
- Ben-Ze'ev, A. 1985. The cytoskeleton in cancer cells. Biochim. Biophys. Acta. 780:197–212. [DOI] [PubMed] [Google Scholar]
- Bosch, F. H., J. M. Werre, L. Schipper, B. Roerdinkholder-Stoelwinder, T. Huls, F. L. Willekens, G. Wichers, and M. R. Halie. 1994. Determinants of red blood cell deformability in relation to cell age. Eur. J. Haematol. 52:35–41. [DOI] [PubMed] [Google Scholar]
- Caraway, N. P., C. V. Fanning, E. M. Wojcik, G. A. Staerkel, R. S. Benjamin, and N. G. Ordonez. 1993. Cytology of malignant melanoma of soft parts: fine-needle aspirates and exfoliative specimens. Diagn. Cytopathol. 9:632–638. [DOI] [PubMed] [Google Scholar]
- Carlson, R. H. G., C. V. Gabel, S. S. Chan, R. H. Austin, J. P. Brody, and J. W. Winkelman. 1997. Self-sorting of white blood cells in a lattice. Phys. Rev. Lett. 79:2149–2152. [Google Scholar]
- Cunningham, C. C., J. B. Gorlin, D. J. Kwiatkowski, J. H. Hartwig, P. A. Janmey, H. R. Byers, and T. P. Stossel. 1992. Actin-binding protein requirement for cortical stability and efficient locomotion. Science. 255:325–327. [DOI] [PubMed] [Google Scholar]
- Elson, E. L. 1988. Cellular mechanics as an indicator of cytoskeletal structure and function. Annu. Rev. Biophys. Biophys. Chem. 17:397–430. [DOI] [PubMed] [Google Scholar]
- Epstein, J. B., L. Zhang, and M. Rosin. 2002. Advances in the diagnosis of oral premalignant and malignant lesions. J. Can. Dent. Assoc. 68:617–621. [PubMed] [Google Scholar]
- Fu, A. Y., H. P. Chou, C. Spence, F. H. Arnold, and S. R. Quake. 2002. An integrated microfabricated cell sorter. Anal. Chem. 74:2451–2457. [DOI] [PubMed] [Google Scholar]
- Fuchs, E., and D. W. Cleveland. 1998. A structural scaffolding of intermediate filaments in health and disease. Science. 279:514–519. [DOI] [PubMed] [Google Scholar]
- Gardel, M. L., M. T. Valentine, J. C. Crocker, A. R. Bausch, and D. A. Weitz. 2003. Microrheology of entangled F-actin solutions. Phys. Rev. Lett. 91:158302. [DOI] [PubMed] [Google Scholar]
- Gerstner, A., W. Laffers, F. Bootz, and A. Tarnok. 2000. Immunophenotyping of peripheral blood leukocytes by laser scanning cytometry. J. Immunol. Methods. 246:175–185. [DOI] [PubMed] [Google Scholar]
- Guck, J., R. Ananthakrishnan, H. Mahmood, T. J. Moon, C. C. Cunningham, and J. Käs. 2001. The optical stretcher: a novel laser tool to micromanipulate cells. Biophys. J. 81:767–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guck, J., R. Ananthakrishnan, T. J. Moon, C. C. Cunningham, and J. Käs. 2000. Optical deformability of soft biological dielectrics. Phys. Rev. Lett. 84:5451–5454. [DOI] [PubMed] [Google Scholar]
- Himelstein, B. P., R. Canete-Soler, E. J. Bernhard, D. W. Dilks, and R. J. Muschel. 1994. Metalloproteinases in tumor progression: the contribution of MMP-9. Invasion Metastasis. 14:246–258. [PubMed] [Google Scholar]
- Hochmuth, R. M. 2000. Micropipette aspiration of living cells. J. Biomech. 33:15–22. [DOI] [PubMed] [Google Scholar]
- Janmey, P. A., U. Euteneuer, P. Traub, and M. Schliwa. 1991. Viscoelastic properties of vimentin compared with other filamentous biopolymer networks. J. Cell Biol. 113:155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, M. D., J. A. Torri, M. E. Lippman, and R. B. Dickson. 1999. Regulation of motility and protease expression in PKC-mediated induction of MCF-7 breast cancer cell invasiveness. Exp. Cell Res. 247:105–113. [DOI] [PubMed] [Google Scholar]
- Katsantonis, J., A. Tosca, S. B. Koukouritaki, P. A. Theodoropoulos, A. Gravanis, and C. Stournaras. 1994. Differences in the G/total actin ratio and microfilament stability between normal and malignant human keratinocytes. Cell Biochem. Funct. 12:267–274. [DOI] [PubMed] [Google Scholar]
- Kirfel, J., T. M. Magin, and J. Reichelt. 2003. Keratins: a structural scaffold with emerging functions. Cell. Mol. Life Sci. 60:56–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu, T., J. Bereiter-Hahn, and I. Karl. 2000. Cell property determination from the acoustic microscope generated voltage versus frequency curves. Biophys. J. 78:2270–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lekka, M., P. Laidler, D. Gil, J. Lekki, Z. Stachura, and A. Z. Hrynkiewicz. 1999. Elasticity of normal and cancerous human bladder cells studied by scanning force microscopy. Eur. Biophys. J. 28:312–316. [DOI] [PubMed] [Google Scholar]
- Lodish, H. B., A. Berk, S. L. Zipursky, P. Matsudaira, D. Baltimore, and J. E. Darnell. 2000. Molecular Cell Biology. W.H. Freeman and Company, New York.
- Mahaffy, R. E., C. K. Shih, F. C. MacKintosh, and J. Käs. 2000. Scanning probe-based frequency-dependent microrheology of polymer gels and biological cells. Phys. Rev. Lett. 85:880–883. [DOI] [PubMed] [Google Scholar]
- Moustakas, A., and C. Stournaras. 1999. Regulation of actin organisation by TGF-beta in H-ras-transformed fibroblasts. J. Cell Sci. 112:1169–1179. [DOI] [PubMed] [Google Scholar]
- Olins, A. L., H. Herrmann, P. Lichter, and D. E. Olins. 2000. Retinoic acid differentiation of HL-60 cells promotes cytoskeletal polarization. Exp. Cell Res. 254:130–142. [DOI] [PubMed] [Google Scholar]
- Rao, K. M., and H. J. Cohen. 1991. Actin cytoskeletal network in aging and cancer. Mutat. Res. 256:139–148. [DOI] [PubMed] [Google Scholar]
- Raz, A., and B. Geiger. 1982. Altered organization of cell-substrate contacts and membrane-associated cytoskeleton in tumor cell variants exhibiting different metastatic capabilities. Cancer Res. 42:5183–5190. [PubMed] [Google Scholar]
- Rotsch, C., K. Jacobson, and M. Radmacher. 1999. Dimensional and mechanical dynamics of active and stable edges in motile fibroblasts investigated by using atomic force microscopy. Proc. Natl. Acad. Sci. USA. 96:921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotsch, C., and M. Radmacher. 2000. Drug-induced changes of cytoskeletal structure and mechanics in fibroblasts: an atomic force microscopy study. Biophys. J. 78:520–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman, M. E., and R. J. Kurman. 1996. The role of exfoliative cytology and histopathology in screening and triage. Obstet. Gynecol. Clin. North Am. 23:641–655. [PubMed] [Google Scholar]
- Sleep, J., D. Wilson, R. Simmons, and W. Gratzer. 1999. Elasticity of the red cell membrane and its relation to hemolytic disorders: an optical tweezers study. Biophys. J. 77:3085–3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soule, H. D., J. Vazguez, A. Long, S. Albert, and M. Brennan. 1973. A human cell line from a pleural effusion derived from a breast carcinoma. J. Natl. Cancer Inst. 51:1409–1416. [DOI] [PubMed] [Google Scholar]
- Tait, L., H. D. Soule, and J. Russo. 1990. Ultrastructural and immunocytochemical characterization of an immortalized human breast epithelial cell line, MCF-10. Cancer Res. 50:6087–6094. [PubMed] [Google Scholar]
- Thoumine, O., and A. Ott. 1997. Comparison of the mechanical properties of normal and transformed fibroblasts. Biorheology. 34:309–326. [DOI] [PubMed] [Google Scholar]
- Unger, M. A., H. P. Chou, T. Thorsen, A. Scherer, and S. R. Quake. 2000. Monolithic microfabricated valves and pumps by multilayer soft lithography. Science. 288:113–116. [DOI] [PubMed] [Google Scholar]
- Wang, N., J. P. Butler, and D. E. Ingber. 1993. Mechanotransduction across the cell surface and through the cytoskeleton. Science. 260:1124–1127. [DOI] [PubMed] [Google Scholar]
- Wang, N., and D. Stamenovic. 2000. Contribution of intermediate filaments to cell stiffness, stiffening, and growth. Am. J. Physiol. Cell Physiol. 279:C188–C194. [DOI] [PubMed] [Google Scholar]
- Wang, Q., D. Lee, V. Sysounthone, R. A. S. Chandraratna, S. Christakos, R. Korah, and R. Wieder. 2001. 1,25-dihydroxyvitamin D3 and retonic acid analogues induce differentiation in breast cancer cells with function- and cell-specific additive effects. Breast Cancer Res. Treat. 67:157–168. [DOI] [PubMed] [Google Scholar]
- Ward, K. A., W. I. Li, S. Zimmer, and T. Davis. 1991. Viscoelastic properties of transformed cells: role in tumor cell progression and metastasis formation. Biorheology. 28:301–313. [DOI] [PubMed] [Google Scholar]
- Wilhelm, J., and E. Frey. 2003. Elasticity of stiff polymer networks. Phys. Rev. Lett. 91:108103. [DOI] [PubMed] [Google Scholar]
- Williamson, J. R., R. A. Gardner, C. W. Boylan, G. L. Carroll, K. Chang, J. S. Marvel, B. Gonen, C. Kilo, R. Tran-Son-Tay, and S. P. Sutera. 1985. Microrheologic investigation of erythrocyte deformability in diabetes mellitus. Blood. 65:283–288. [PubMed] [Google Scholar]
- Wirtz, H. R., and L. G. Dobbs. 1990. Calcium mobilization and exocytosis after one mechanical stretch of lung epithelial cells. Science. 250:1266–1269. [DOI] [PubMed] [Google Scholar]
- Worthen, G. S., B. Schwab 3rd, E. L. Elson, and G. P. Downey. 1989. Mechanics of stimulated neutrophils: cell stiffening induces retention in capillaries. Science. 245:183–186. [DOI] [PubMed] [Google Scholar]
- Wottawah, F., S. Schinkinger, B. Lincoln, R. Ananthakrishnan, M. Romeyke, J. Guck, and J. Käs. 2005. Optical rheology of biological cells. Phys. Rev. Lett. 94:098103. [DOI] [PubMed] [Google Scholar]
- Wyckoff, J. B., J. G. Jones, J. S. Condeelis, and J. E. Segall. 2000. A critical step in metastasis: in vivo analysis of intravasation at the primary tumor. Cancer Res. 60:2504–2511. [PubMed] [Google Scholar]
- Zahalak, G. I., W. B. McConnaughey, and E. L. Elson. 1990. Determination of cellular mechanical properties by cell poking, with an application to leukocytes. J. Biomech. Eng. 112:283–294. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.