

Abstract
We study the packaging of DNA into a bacteriophage capsid using computer simulation, specifically focusing on the potential impact of twist on the final packaged conformation. We perform two dynamic simulations of packaging a polymer chain into a spherical confinement: one where the chain end is rotated as it is fed, and one where the chain is fed without end rotation. The final packaged conformation exhibits distinct differences in these two cases: the packaged conformation from feeding with rotation exhibits a spool-like character that is consistent with experimental and previous theoretical work, whereas feeding without rotation results in a folded conformation inconsistent with a spool conformation. The chain segment density shows a layered structure, which is more pronounced for packaging with rotation. However, in both cases, the conformation is marked by frequent jumps of the polymer chain from layer to layer, potentially influencing the ability to disentangle during subsequent ejection. Ejection simulations with and without Brownian forces show that Brownian forces are necessary to achieve complete ejection of the polymer chain in the absence of external forces.
INTRODUCTION
The ability of bacteriophage to package, transport, and deliver its genome to a host bacterium involves the precise manipulation of DNA throughout the life cycle of the virus. Despite the apparent simplicity of most species of bacteriophage, their ability to control the conformation of DNA through physical manipulation is quite remarkable, a feat that is desirable in many different biological and technological settings. Therefore, developing a fundamental understanding of the physical mechanisms responsible for the exquisite control that bacteriophage has over its DNA is important for understanding the principles of DNA manipulation.
In this article, we focus on the processes involved in the packaging of DNA into an empty viral capsid. Specifically, we address whether the twist rigidity of the DNA molecule plays an important role in affecting its conformation during the packaging process.
Although different species of bacteriophage can vary in many ways (host species, specific structural geometry, size of virus, size of genome/number of genes, as well as many important biochemical distinctions), they also share several important similarities that are relevant to our current discussion. A bacteriophage assembles its capsid particle before the genome is packaged; then the genome is inserted into the empty capsid by an ATP-driven packaging motor. As the capsid is filled with DNA, the packaging motor works against the increasing resistance associated with compacting the long polymer chain to near-crystalline density into the relatively small cavity. Once the new bacteriophage is packaged, the now-virulent viruses lyse the host cell and escape into the surrounding medium to infect other hosts, producing further progeny. The viruses no longer have access to the energy source ATP once outside their host; therefore, any work performed during infection must be stored within the virus particles before they leave the cell that produced them. Because of these similarities, experimental observations of specific bacteriophage species can be incorporated into the general framework of DNA packaging.
The force generated by the bacteriophage φ29 packaging motor has been directly measured using optical tweezers (Smith et al., 2001), which showed a continuous buildup of the internal pressure as packaging proceeds. The measured stall force of 57 pN makes the φ29 packaging motor one of the most powerful protein motors known to date. Similarly, the pressurization of packaged phage-λ has been confirmed by using osmotic pressure as a resistive force against DNA ejection, causing the ejection to cease and leaving partially filled capsid particles (Evilevitch et al., 2003). The internal pressure within these packaged viruses is directly related to the free-energy cost of forcing the DNA into a small cavity and provides the energy for conversion to work in the injection of the DNA into the host cell.
A number of studies have addressed the DNA conformation within the capsid interior using various experimental techniques for several different viral species (North and Rich, 1961; Earnshaw and Harrison, 1977; Widom and Baldwin, 1983; Aubrey et al., 1992; Serwer et al., 1992; Lepault et al., 1987; Olson et al., 2001; Black et al., 1985; Cerritelli et al., 1997; Zhang et al., 2000). The experimental evidences have inspired potential models for DNA arrangement within the packaged capsid, including liquid crystal (Lepault et al., 1987), folded toroid (Hud, 1995), coaxial spool (Richards et al., 1973; Earnshaw and Harrison, 1977), folded coaxial spool (Serwer, 1986), and concentric spool (Hall and Schellman, 1982a,b). By electron microscopy with image reconstruction, the conformation of the packaged genome of phage T7 (Cerritelli et al., 1997), T4 (Olson et al., 2001), and P22 (Zhang et al., 2000) appears to adopt a spool-like conformation with several layers of order, consistent with the coaxial spool model, where the spools are ordered parallel to each other and perpendicular to the axis of the tail. However, flow linear dichroism studies of phage T4 suggest that the mean square value of the cosine of the angle between the tail axis and the DNA strand is inconsistent with the coaxial spool model, lending credit to the concentric spool model, where the spools are arranged at angles to each other (Hall and Schellman, 1982a,b). Recent theoretical work shows that the energetically preferred conformation of a polymer in a spherical confinement is consistent with the concentric spool model (LaMarque et al., 2004). The packaged conformation within a viral capsid remains an intriguing subject of debate.
Details of the packaging motor of φ29 suggest the potential for twist to play a role in the packaging process. From the crystal structure of the φ29 packaging motor, a mechanism for DNA translocation has been proposed whereby either the motor or the DNA itself is rotated during the process (Simpson et al., 2000). The proposed mechanism involves a 12° rotation for every 2 basepairs that enter the capsid or a rate of rotation of roughly 1/6 the periodicity of the DNA double helix. By existing experimental evidence, it is not clear whether the DNA rotates during translocation; however, some results suggest that the final packaged conformation has a nonzero linking number, either purely as writhe in a spooled conformation or by a combination of twist and writhe.
For example, the conformation of the genomes of phage-λ (Virrankoski-Castrodeza et al., 1982) and P22 (Casjens, 1989) exhibit supercoiling when treated with cross-linker and released from their capsids. Supercoiling of the released genome requires the packaged DNA to be writhed as a tightly spooled conformation or to have twist deformation trapped within the internalized conformation along with the writhe; further quantization of such experiments along with a prediction for the writhe of the internal conformation are required to distinguish between these two possibilities. Furthermore, bacteriophage φ29 genome with the covalently bound terminal protein gp3 exhibits supercoiling; the supercoiling is believed to be a prerequisite for efficient packaging (Grimes and Anderson, 1997).
Optical density and circular dichroism of bacteriophage T2 demonstrate anomalous features that cannot be captured if the DNA within the bacteriophage is assumed to be undeformed B-DNA (Holzwarth et al., 1974a,b); these features are better captured when the internalized DNA is assumed to have both anisotropic packaging and distorted structure. The similarities between the circular dichroism spectra of the intact phage particles (Holzwarth et al., 1974a,b) and of DNA at high salt concentrations (Tunis-Schneider and Maestre, 1970) suggest the DNA may adopt a structure that is more consistent with that of DNA at elevated salinity, which is distorted such that the helical repeat occurs at 10.2 basepairs (Baase and Johnson, 1979; Chan et al., 1979) rather than 10.4 basepairs (Wang, 1978, 1979), or its twist density is 0.0056 nm−1 relative to undeformed B-DNA.
If the DNA rotates as it enters the capsid, then either the DNA relaxes the twist deformation by axial rotation of the strand, or the conformation of the strand responds to the twist deformation in some way. Inspired by this possible mechanism, we address the problem of what qualitative features are altered in the packaged conformation if the DNA is rotated while it is fed into the capsid.
Twist plays an essential role in the biophysical behavior of DNA, particularly manifest in torsionally constrained DNA elements commonly present in plasmids of prokaryotes and transiently closed loops in gene regulation in eukaryotes. For example, binding of the lac repressor of Escherichia coli is modulated by the length of DNA between binding sites, resulting in oscillations in the repression level as the length passes through integer numbers of helical turns (Müller et al., 1996). Twist affects the conformation of DNA through a well-known topological constraint on torsionally constrained paths, which states that the linking number is a sum of the twist and the writhe of the curve (White, 1969). The underlying mathematical expressions for these topological entities dictate the allowable states of a closed chain; however, the thermodynamically preferred distribution between twist and writhe is determined by the energetics of the chain conformation. The importance of chain topology on DNA biomechanics is evidenced by the existence of topoisomerases, a class of enzymes that control the topology of DNA (Wang and Cozzarelli, 1986; Schvartzman and Stasiak, 2004).
Provided the packaging motor of a bacteriophage rotates the DNA as it enters the capsid, the conformation of the DNA within the capsid interior would be impacted by the twist deformation. Specifically, we propose that twist deformation could provide the driving force for adopting an ordered, spool-like conformation, as is observed in T7 (Cerritelli et al., 1997, T4 (Olson et al., 2001), and P22 (Zhang et al., 2000). The concept of local action influencing global behavior is a common theme in the biological function of DNA, and twist is an important mechanism for such “action at a distance” (Wang and Giaever, 1988). Previous theoretical treatments of the packaging process have neglected the role of twist on the packaged conformation (Kindt et al., 2001; Tzlil et al., 2003; Purohit et al., 2003a,b, 2004; Arsuaga et al., 2002; LaMarque et al., 2004; Marenduzzo and Micheletti, 2003; Odijk, 1998; Odijk and Slok, 2003), based on the assumption that either the motor does not introduce twist into the conformation or that this twist is relaxed by axial rotation on timescales shorter than the timescale for the packaging process (Krasilnikov et al., 1999; Thomen et al., 2002). Although we do not suggest that neglecting twist is without justification, it is not currently known whether the end of the DNA is completely free to relax. Alternatively, twist may be locked in the conformation by a number of possible mechanisms, including DNA interaction with proteins, either unattached to the DNA (capsid proteins, for example) or covalently bound to the DNA (the φ29 terminal protein gp3, for example), or anomalous twist relaxation rates owing to the crowded electrolyte environment within the capsid interior. Therefore, a reasonable question is how the conformation of DNA is altered by twist deformation if the DNA is torsionally constrained.
In this article, we address this question using computer simulation. Because the end condition of the DNA inside the capsid is unknown at present, we adopt the simplest model of torsional constraint by taking the end to be clamped and fixed on the capsid wall opposite to the portal of entry. We then perform two dynamic simulations of a polymer chain fed into a spherical confinement: one where the chain is rotated at the portal as it is fed, and one where the chain is fed without rotation. We compare the packaged conformation of the chain in these two cases, and examine the consequences for ejection from these different conformation structures. To focus on the most important aspects of the problem, we move on directly to the description of the results and relegate the details of the model and simulation method to the Appendix.
RESULTS
To simulate genome-length DNA at biologically relevant timescales, it is necessary to adopt a simplified model of the DNA/capsid system. We model the DNA using a discrete wormlike chain that includes the twist degrees of freedom (Allison et al., 1989; Chirico and Langowski, 1994); each discrete segment of chain represents ∼8 basepairs of DNA. The elastic properties of the chain are chosen to match the persistence length and torsional persistence length of DNA, with a large stretching modulus that essentially renders the chain inextensible. The chain self-interaction includes a screened electrostatic potential to approximate the influence of physiological salt concentration (∼140 mM monovalent salt) and a hard-core repulsive potential that addresses the short-range repulsive behavior of DNA (modeled using the repulsive part of a Leonnard-Jones potential). The capsid is modeled as a spherical confinement through a simple repulsive potential that scales as the radius to the fourth power outside the capsid radius (Kindt et al., 2001). With this model, we perform dynamic simulations, both with and without the effect of Brownian forces and torques, to track the temporal evolution of the chain conformation as it is fed into the spherical confinement. Further details of our model are found in the Appendix of this article.
We perform two sets of packaging simulations. The first simulation involves the simultaneous rotation of the chain segment at the portal of entry as the chain is fed into the spherical confinement; thus, the linking number increases linearly with the number of beads fed. As mentioned in the introduction, the proposed mechanism for DNA translocation by the φ29 motor involves ∼60° of rotation of the chain per helical-turn fed. Because it is not clear whether this mechanism involves rotation of the DNA or the motor, and the packaging of φ29 is marked by frequent pauses and slips of the chain (Smith et al., 2001), we choose to reduce the rate of rotation to one-third of the predicted rate from the proposed mechanism of DNA translocation (Simpson et al., 2000). The second simulation is performed without rotation of the chain as it enters; thus, the linking number remains at zero. In both sets of simulations, the feeding is performed by alternating between feed intervals and relaxation intervals until the desired volume fraction ΦP is achieved. One feed interval involves feeding 10 beads into the spherical cavity at a feed rate of 1 per time step (tB, defined in the Appendix), and one relaxation interval involves an equal amount of time (10 time steps) to relax the conformation to a local minimum energy. In the feeding simulations, we neglect the role of Brownian forces and torques to focus on the mechanical aspects of the packaging process and to develop a basis for comparison with the case when Brownian fluctuations are included.
In Fig. 1, we show snapshots from the packaging dynamics for packaged volume fraction ranging from ΦP = 0.063 to ΦP = 0.463 in even increments of 0.050. The snapshots in Fig. 1 A show the progress of packaging for coupled translocation and rotation during the packaging process, whereas the snapshots in Fig. 1 B are for packaging without rotation. The most clear distinction between the two sets of snapshots is the qualitative manner in which the packaged conformation evolves. The conformations in panel A (with rotation) appear to evolve in a spool-like manner, whereas the process of packaging without rotation in panel B occurs via a folding-type mechanism. Notably, the final conformations in panels A and B appear to have distinctly different chain orientations. The final conformation in panel A is wrapped in a spool that is on average perpendicular to the direction of entry, whereas in panel B the chain segments tend to locally align along the longitudes.
FIGURE 1.

Snapshots during the process of feeding a single polyelectrolyte into a spherical confinement. The snapshots in panel A track the progress for feeding with rotation (as described in the text) for polymer volume fraction ranging in even increments from 0.063 to 0.463. The snapshots in panel B are for feeding without rotation for the same polymer volume fractions as panel A.
The snapshots presented in Fig. 1 display the structure of the outermost layers of the packaged polymer chain; however, it is desirable to look at the polymer density in the interior of the spherical confinement. Taking the final conformation from the two simulations, we perform Brownian dynamics simulations (thus including Brownian forces and torques) to evaluate a thermally averaged density of beads as a function of position within the capsid interior (averages are performed over 100 time steps). In Fig. 2 A, we show the angle-averaged radial density ρ versus the radial distance R for the final packaged conformation for the case of feeding with rotation (solid line) and feeding without rotation (dashed line). In the case of feeding with rotation, four peaks are clearly visible in the density plot, indicative of four layers of order, with the outermost peak dominating; this layer ordering is quite reminiscent of the experimentally observed ordering in T7 (Cerritelli et al., 1997), T4 (Olson et al., 2001), and P22 (Zhang et al., 2000). For feeding without rotation, the peaks are not as easily discernible after the first two layers, indicative of the reduced degrees of layer order after the outermost layers.
FIGURE 2.

Panel A shows the density of polymer ρ versus the radial distance from the sphere center R for feeding with rotation (solid line) and without rotation (dashed line). Panel B gives the average radial distance of polymer segments versus the chain contour distance s/L for feeding with rotation. We note that s/L = 0 corresponds to the first segment inserted into the spherical confinement.
The density plot gives the positional order of the chain segments within the spherical confinement; however, the density does not resolve the sections along the chain that contribute to the layer ordering. Therefore in Fig. 2 B, we plot the average radial distance versus the rank order along the polymer chain s/L (s = 0 at the chain end attached to the capsid wall, and s = L at the chain end that is fed through the portal) for the final packaged conformation for feeding with rotation. The average for each data point in this figure is performed over time (100 time steps) for each point along the chain. The layering of the polymer within the sphere displays a preference for the beads first fed into the sphere to be located near the sphere surface, and later beads fed tend to be localized closer to the sphere center. However, the later-fed beads frequently penetrate out to the sphere surface; thus, the plot exhibits frequent spikes associated with these “out of rank” penetrations. A similar trend is displayed in the case of feeding without rotation (not shown).
Topological quantities including the linking number ΔLk (solid line), twist ΔTw (open down triangles), and writhe Wr (open up triangles) for packaging with rotation are plotted in Fig. 3 against the volume fraction of packaged polymer ΦP. We identify the topological quantities ΔLk = Lk − Lk0 and ΔTw = Tw − Tw0 as the difference between their values and the undeformed values given by the natural helicity of the DNA strand (Lk0 = Tw0 = Nbp/10.4 for B-DNA; Wang, 1978, 1979). Because the undeformed writhe is zero, we can write the writhe as ΔWr = Wr − Wr0 = Wr. All three topological quantities are calculated independently. We find that the topological constraint ΔLk = ΔTw + Wr is satisfied throughout the simulation (to within machine precision), which is confirmation that the chain does not cross itself during the simulation.
FIGURE 3.

The topological quantities twist ΔTw (▿), writhe Wr (▵), and linking number ΔLk (solid line) of the packaged conformation for feeding with rotation versus the volume fraction of polymer added ΦP. The inset focuses on the early packaging behavior. Specifically, we show in the inset the twist ΔTw (▿ connected by solid lines) and writhe Wr (▵ connected by solid lines) for packaging with rotation and the twist ΔTw (□ connected by dotted lines) and writhe Wr (○ connected by dotted lines) for packaging without rotation versus the volume fraction of polymer added ΦP.
At the initial stage of the packaging process (ΦP < 0.163), the linking number is almost entirely in the form of writhe. The curved surface of the spherical confinement appears to strongly bias the chain conformation toward a writhed one. This effect is more clearly seen in the case of packaging without end rotation, where the chain writhes at the expense of incurring twist deformation. In the inset of Fig. 3 we show the twist ΔTw (open down triangles connected by solid lines) and writhe Wr (open up triangles connected by solid lines) for packaging with end rotation and the twist ΔTw (open squares connected by dotted lines) and writhe Wr (open circles connected by dotted lines) for packaging without end rotation versus the volume fraction of polymer added ΦP during the initial stage of packaging (ΦP < 0.163). For ΦP < 0.043 (first three data points in the inset of Fig. 3), the writhe increases linearly at the same rate for packaging both with and without end rotation. The writhe continues to increase at this rate for packaging with end rotation for 0.043 < ΦP < 0.163; however, the writhe dips back toward zero for packaging without end rotation for ΦP > 0.043. In the case of packaging with end rotation, the added twist facilitates the writhing by eliminating the twist produced during the spooling process. In the case of packaging without end rotation, the spooling cannot proceed in the same direction indefinitely due to the buildup of twist from the corresponding writhe.
For larger volume fractions (ΦP ≥ 0.163), twist increases essentially linearly with the added polymer; a linear fit of the twist gives a twist density of 0.0052 nm−1 relative to the undeformed twist. The twist remains much smaller than the writhe as the chain is fed into the sphere, presumably due to the propensity for the chain to adopt the spool-like conformation, which converts most of the twist from the loading into writhe. As the twist and writhe increase linearly with the volume fraction, they maintain essentially a constant ratio between them. Similarly, the process of DNA supercoiling generally involves a distribution between twist and writhe, strongly influenced by salt concentration (Bednar et al., 1994). We note that the twist and writhe remain near zero for most of the packaging process for packaging without chain rotation (maximum value of |ΔTw| = |Wr| = 2.14 and final value at fully packaged of ΔTw = −Wr = 0.39).
In Fig. 4 A, the total energy E (open circles, measured in kBT(∼4.1 pN nm = 0.6 kcal/mol)), compression energy Ecom (+), bending energy Ebend (open up triangles), twisting energy Etwist (open squares), self-interaction energy Eint (open down triangles), and capsid-interaction energy Ecapsid (×) are plotted against the packaged volume fraction ΦP for packaging with rotation (refer to the Appendix for energy definitions). The solid line is a polynomial fit (fourth order) to the total energy E, which we make use of later to determine the force opposing the packaging process. Early in the packaging process, the total energy E is dominated by the bending deformation energy Ebend; however, at approximately ΦP = 0.32, the bending energy Ebend is overcome by the self-interaction energy Eint. The capsid-interaction energy Ecapsid remains small for small volume fraction polymer (ΦP < 0.3) but rises noticeably at large volume fractions as the packing presses the polymer against the confining wall. The compression energy Ecom and twisting energy Etwist are insignificant contributions throughout the packaging process. Although packaging involves chain rotation, the twist is efficiently converted to writhe, thus the twisting energy Etwist remains quite small. Specifically, the final value of the twisting energy Etwist is 17.3 kBT for packaging with chain rotation, and if all of the linking number remained in the form of twist, the final twisting energy Etwist would be 234.8 kBT.
FIGURE 4.

Panel A shows the total energy E (○), compression energy Ecom (+), bending energy Ebend (▵), twisting energy Etwist (□), self-interaction energy Eint (▿), and capsid-interaction energy Ecapsid (×) versus the packed volume fraction ΦP for packaging with rotation. The solid line is a polynomial fit (fourth order) to the total energy E. Panel B is the difference between the total energy for packaging without rotation and packaging with rotation ΔE versus the packaged volume fraction ΦP. Note, packaging without rotation leads to a larger total energy (ΔE > 0 in our simulations).
Because the range in the figure is so large, the complimentary plot for packaging without rotation is virtually indistinguishable from the one shown. Therefore, to facilitate comparison, we plot in Fig. 4 B the difference between the total energy of the packaged conformation without rotation and the total energy for packaging with rotation ΔE against the polymer volume fraction ΦP. The energy for packaging without rotation is larger than the energy for packaging with rotation, thus ΔE > 0 (with two exceptions at ΦP = 0.053 and ΦP = 0.263), with a maximum value of 81.0 kBT. We note that, in the case of feeding with rotation, the added twist deformation represents an additional energetic contribution that presumably is not present in the case of feeding without rotation. In the case of feeding without rotation, we restrict both ends from rotating, thus any writhe produced results in an equal and opposite amount of twist to maintain the linking number. This local twist results in twist deformation energy; however, this is small in comparison to the twist present in the case of feeding with rotation. Therefore, it is quite remarkable that the energy of the conformation resulting from packaging with rotation is less than that without rotation; this is due to the reduced bending energy of the spooled conformation in the former versus the folded conformation in the latter.
The polynomial fit to the total energy E in Fig. 4 A is used to calculate the force opposing packaging F, from F = ∂LE. This assumes that the conformation within the confinement remains at equilibrium throughout the packaging process, requiring that the packaging is sufficiently slow to maintain equilibrium. In Fig. 5, we show the predicted force opposing packaging (units of pN) using a fourth-order polynomial fit to the total energy data (as in Fig. 4 A) for packaging with rotation (solid line) and without rotation (dashed line) against the packaged volume fraction ΦP. In both cases, the force opposing packaging increases with the packaged length to a final value of ∼20 pN. This value is about three times smaller than the stall force of φ29 (57 pN) (Smith et al., 2001). Because we have neglected the thermal contributions to the osmotic pressure of the chain segments by ignoring the Brownian forces and have used a crude interaction potential between the segments, a factor of three discrepancy is not unreasonable.
FIGURE 5.

The force opposing packaging (expressed in pN) versus volume fraction ΦP for packaging with rotation (solid line) and without rotation (dashed line) determined using a fourth-order polynomial fit to the total energy E (see text and Fig. 4).
From the final twist value in Fig. 3, we estimate the value of the torque τEND = 2πCΔTw/(N − 2) necessary to disallow the attached end from rotating to be 1.2 kBT. If all of the linking number remained as twist, the predicted end torque τEND would be 4.5 kBT. We note that these values of torque are within a reasonable range where typical molecular forces (for example, hydrogen bonding and van der Waals forces) could stabilize the end.
Beginning with the conformations predicted from the packaging simulations, we simulate the ejection of the chain from the spherical confinement by eliminating the feeding force/torque and allowing the chain to exit through the portal of entry. In Fig. 6, we show the results of the ejection simulations by plotting the volume fraction ΦP left within the sphere versus time (reduced by tB). For each starting conformation obtained from packaging with and without rotation, we have performed two ejection simulations, one that included the Brownian forces and one that did not. We find that Brownian forces dramatically influence the ability of the packaged sphere to eliminate the polymer within. Specifically, in the absence of Brownian forces, ejection is unable to reach full completion for both conformations obtained from packaging with (solid line) and without rotation (dashed line), indicated by the internal volume fraction tending to a nonzero value at long times. (We note that full ejection occurs at ΦP = 0.017 because of the chain end attachment to the sphere wall). When Brownian forces are included, ejection is marked by frequent fluctuations in the length of chain within the sphere. Such length fluctuations are most likely related to interior conformation fluctuations that may help to alleviate self-entanglement, thus allowing the chain to fully eject.
FIGURE 6.

The volume fraction ΦP versus time for ejection from the sphere beginning with the final conformation from packaging with chain rotation (ejection with Brownian forces (dashed-dotted line) and without Brownian forces (solid line)) and from packaging without chain rotation (ejection with Brownian forces (dotted line) and without Brownian forces (dashed line)). Beneath the figure, we show the final conformations from eject without Brownian forces beginning from packaging with chain rotation (left) and without chain rotation (right).
For ejection without Brownian forces, the ejection halts at a larger value of the volume fraction when the initial conformation is from packaging without rotation, presumably because of a higher degree of entanglement in the folded conformation from packaging without rotation than the spool-like conformation from packaging with rotation. However, when Brownian forces are included, the ejection proceeds to completion in both cases, and the conformation from packaging without rotation (dotted line) ejects faster than the conformation from packaging with rotation (dashed-dotted line). Further studies are needed to explain this latter observation.
DISCUSSION
This preliminary study on the packaging of DNA within a bacteriophage capsid focuses on the potential impact of twist on the packaged conformation, specifically if the packaging motor rotates the DNA as it feeds. Toward this end, we perform two computer simulations of a single polyelectrolyte actively inserted into a spherical confinement: one where the polymer chain is rotated as it is fed, and one where the polymer chain is not rotated during the feeding process. We choose to fix the nonfeeding end to the capsid wall directly across from the portal. This does not necessarily imply a proposed biological phenomenon; rather, it is a convenient way to elucidate the effects of twist by imposing the topological constraint.
Our results show distinct differences between the conformation of the packaged polymer in packaging with rotation and without rotation. Most notably, the final conformation in packaging with rotation exhibits a spool-like character, perhaps more consistent with a concentric spool (Hall and Schellman, 1982a,b) than a coaxial spool (Richards et al., 1973; Earnshaw and Harrison, 1977), whereas the final conformation for packaging without rotation is folded with chain segments generally oriented parallel to the axis of feeding. We note that the preference for the concentric spool is consistent with energy-minimized conformations found in LaMarque et al. (2004) for feeding without rotation and with a free polymer end in the sphere interior. However, our approach is not to seek global energy minima as in LaMarque et al. (2004), but rather to track the process of packaging as it proceeds along a dynamically accessible trajectory.
Rotating the polymer chain while feeding acts to break chiral symmetry of the packaged conformation, whereas feeding without rotation favors neither chirality. Packaging a torsionally constrained polymer without rotation begins by coiling the chain in one direction at random; this writhing gives rise to twist that eventually forces the chain to coil in the opposite direction to alleviate the developing twist. This scenario is reminiscent of coiling a garden hose without rotation: coiling may proceed in one direction for several wraps but must eventually coil in the opposite direction or suffer other twist-reducing instabilities (formation of plectonemes, for example). On the other hand, orderly coiling of a garden hose is accomplished by twisting the hose as it lies down (Mahadevan and Keller, 1996). The energetically preferred conformations found in LaMarque et al. (2004) are invariant to reflection because they have no twist deformation, which is assumed to be alleviated by axial rotation of the free end. Thus, presumably an average over many packaged conformations predicted by a model that neglects twist deformation would not exhibit chiral preference. Packaging with rotation would provide the driving force for spool-like packaging with a definitive chiral preference.
However, the adopted spool-like conformation from packaging with rotation is not a perfect, idealized spool; rather it contains many defects of chain segments jumping from interior layers to exterior layers. Although the density plot (Fig. 2 A) exhibits layer ordering (notably more pronounced for packaging with rotation than without rotation), the rank ordering plot (Fig. 2 B) shows that the conformation does not proceed by filling the outmost layer first before filling the interior shells but by placing the chain in available spaces, preferring the outmost shell throughout the packaging process. We note that the layer jumping exhibited in our simulations is consistent with experimental studies of phage-λ by Widom and Baldwin (1983). After the phage is lysed with EDTA and spread in a cytochrome c film, electron micrographs show the DNA contacts the capsid at random locations along its entire length.
The physical argument of the curvature stress offsetting the self-repulsion is certainly at work, as argued by continuum modeling of the packaging process (Odijk, 1998); however, the curvature stress also drives inner loops toward the outer layers when transient gaps or defects present themselves near the sphere surface. Continuum models of the packaging process assume sequential spooling (Odijk, 1998; Purohit et al., 2003b) without these layer-jumping instances, because the continuum treatment does not account for local inhomogeneities. However, it is worth noting that such a continuum treatment (Purohit et al., 2003b) achieves quantitative agreement with the experimentally measured force opposing packaging in φ29 (Smith et al., 2001). Nonetheless, the instances of defects may lead to entanglements as the polymer chain is ejected from the confinement. Our ejection results (presented in Fig. 6) demonstrate that Brownian forces are necessary to achieve complete ejection of the chain from the spherical cavity. We propose that this is due to the entanglements arising from interior defects, which are not predicted from continuum treatments of the packaging process.
Packaging with chain rotation suggests that the increasing linking number of the strand is introduced in the form of twist as the straight section of chain entering the spherical confinement rotates, and conversion from twist to writhe occurs once the entering bead is released into the capsid interior. During the initial stage of packaging (ΦP < 0.163 in Fig. 3), our simulation shows that all of the twist is converted into writhe, and further packaging results in most of the twist being converted into writhe, as they both increase linearly but with different slopes. This suggests that during the initial stage, the driving force for coiling is dominated by nontwist related forces (self-interaction and bending, mainly), whereas further ordered packaging in coils occurs due to self-interaction, bending, and stresses arising from twist deformation energy.
Notably, the manner in which the polymer chain alleviates the twist deformation is to form coils or solenoid-like structures, which is completely dissimilar from the more prevalent plectonemic structures formed in a twisted polymer chain in the absence of confinement. The preference for the plectonemic form in free space is due to the fact that plectonemes more efficiently convert twist into writhe than solenoid structures (Marko and Siggia, 1994). Although not shown in the previous section, we find that performing packaging simulations with a larger rotation rate (larger linking number) results in plectonemic structures in the sphere confinement rather than coils or solenoids.
The linear increase of the twist during the packaging process results in a constant twist density (twist per unit length) of 0.0052 nm−1 relative to the undeformed value, which very closely matches the change in twist of DNA in a high-salt environment given by 0.0056 nm−1 relative to DNA in physiological salt concentration (Wang, 1978, 1979; Baase and Johnson, 1979; Chan et al., 1979). Circular dichroism spectra of bacteriophage T2 (Holzwarth et al., 1974a,b) closely resembles that of DNA in a high-salt environment (Tunis-Schneider and Maestre, 1970), that is the packaged DNA in T2 is over-twisted by 0.0056 nm−1. Our simulations suggest that this twist distortion can be accounted for when the chain is packaged with end rotation.
However, we note that the over-twisting could also be caused by the electrolyte conditions on the interior of the capsid. Because our simple model of the DNA strand lacks molecular detail, we cannot predict the effect of electrolyte conditions on the helical pitch of the packaged DNA. One way to distinguish between these possibilities would be to measure the degree of over-twisting versus the length packaged. The case of active twisting by the motor, as in our simulations, gives a linear increase in the total twist, thus a constant twist density. If over-twisting is due to the electrolyte environment, we would see an increase in the twist density during the packaging process due to the changing charge conditions on the capsid interior, assuming that the electrolyte condition is primarily due to the DNA segments within the capsid.
The energy of the packaged conformation shows a steep increase as packaging proceeds, indicative of the escalating resistance opposing packaging, as is evident in φ29 (Smith et al., 2001). In the early stage of packaging (ΦP < 0.32 in Fig. 4 A), the bending energy dominates the total energy, and the conformation is driven to the outside wall. As packaging proceeds, the self-interaction energy overcomes the bending energy, and the conformation tends to explore more of the interior of the sphere (noting the tendency for layer jumping previously discussed). All other energetic contributions are rather small in comparison to the bending and self-interaction energies throughout the packaging process. This does not imply their influence is insignificant, as demonstrated by the qualitative difference between the final conformation from packaging with rotation and without rotation displayed in Fig. 1.
Throughout this article, we have deliberately avoided an explicit definition of the Brownian timescale in actual units. This is because of the uncertainty in the drag coefficient for DNA in a crowded, highly charged environment. It is tempting to assume that the drag is the same as in free solution (Meiners and Quake, 2000; Krasilnikov et al., 1999; Thomen et al., 2002) or alternatively to consider a more detailed model that includes hydrodynamic interactions (Jendrejack et al., 2003) (both polymer/polymer and polymer/capsid wall). However, it is very likely that the drag is not easily interpreted once we reach packaging densities where the chains are separated by only a couple of hydration layers, as is the case here. Beyond the equilibrium hydration forces that occur (Leikin et al., 1991), confined water exhibits slow relaxation behaviors that may dramatically alter the effective drag coefficient on the DNA strand (Zhu and Granick, 2001).
We neglect Brownian forces in the packaging simulations in this article, and certainly several features are not captured without their effect. For example, thermally induced reorganization may act to disentangle the polymer chain as packaging proceeds, thus eliminating some of the defects prevalent in our simulation results. However, we note that our thermal averaging of the final conformation does not exhibit large-scale reorganization in the timescale of the average (100 time steps) due to the extremely sluggish processes associated with disentanglement at this volume fraction (ΦP = 0.463). Although we do not explore this in our current work, the disentanglement process for a single chain in a confinement has several similarities to and nontrivial distinctions from the relaxation processes prevalent in entanglement of a polymer chain in a melt (de Gennes, 1971; Doi and Edwards, 1986). Brownian forces also acts as a source of transient inhomogeneities in the local ordering, thus the instances of layer jumping described earlier may be enhanced under the influence of thermal fluctuations. Thermal fluctuations influence the tendency to order; for example, molecular crowding in semiflexible polymer solutions results in ordered liquid-crystal phases. One distinct effect of semiflexibility on the liquid-crystal phase is the presence of hairpin defects or instances of sharp reversal of the chain orientation to form a loop (de Gennes, 1982; Spakowitz and Wang, 2003). These effects may play a role in the formation of ordered conformations within a packaged bacteriophage capsid, which we plan to explore further in future studies. Overall, the influence of thermal fluctuations on the packaged conformation remains an important issue that requires further investigation.
This study acts as preliminary evidence that twist may play a role in the packaged DNA conformation in a bacteriophage capsid. Further work is needed to elucidate the connection between the packaged conformation and the end goal of the viral packaging, the ability for the virus to eject its genome from the capsid into the host cell. Future work will focus more on the ejection process to make the connection between the manner of packaging and the resulting ejection behavior.
Acknowledgments
The authors thank Rob Phillips, Bill Gelbart, Sherwood Casjens, Prashant Purohit, Paul Grayson, and Paul Wiggins for helpful discussions.
This work was supported in part by the National Science Foundation (DMR-9970589) and the W. M. Keck Foundation.
APPENDIX: MODEL OF DNA/CAPSID
Modeling the process of DNA packaging within a viral capsid is challenging because of the large number of degrees of freedom of the DNA strand and the long timescale for the packaging process. Therefore, it is necessary to adopt a simplified view of the DNA/capsid system. Because our interest is in the behavior of the overall DNA conformation in the capsid, we neglect the individual atomic coordinates and reduce the description to a coarse-grained model. A suitable model for our purposes is the worm-like chain model that includes the twist degrees of freedom (Kratky and Porod, 1949; Yamakawa, 1997), which is used to study a wide range of DNA-related phenomena. The worm-like chain model is approximated as a beaded chain to make it amenable to computer simulation (Allison et al., 1989; Chirico and Langowski, 1994).
The DNA conformation is described as a string of beads with positions 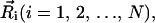 thus there are N − 1 bond vectors
thus there are N − 1 bond vectors  where
where  To account for the material twisting, two normal vectors
To account for the material twisting, two normal vectors  and
and 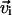 are attached to each bond vector
are attached to each bond vector 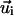 such that
such that  and
and  This leaves the polymer chain with a total of 4N − 1 degrees of freedom (Chirico and Langowski, 1994).
This leaves the polymer chain with a total of 4N − 1 degrees of freedom (Chirico and Langowski, 1994).
The behavior of the polymer chain is dictated by the total chain energy, which includes elastic deformation energy, self-interaction energy, and capsid-interaction energy, all expressible in terms of our degrees of freedom. The chain deformation energy takes the simplest form possible: the compression, bending, and twisting deformation energies are quadratic in the displacement away from a straight, untwisted conformation (Love, 1944). These energy terms are given by
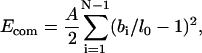 |
(1) |
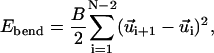 |
(2) |
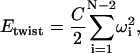 |
(3) |
where A, B, and C are, respectively, the compression, bending, and twisting moduli (units of energy), l0 is the equilibrium bond length, and ωi is the twist angle, which satisfies 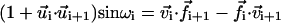 (Chirico and Langowski, 1994). The self-interaction energy takes the form
(Chirico and Langowski, 1994). The self-interaction energy takes the form
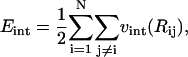 |
(4) |
where  and vint(R) is the two-body interaction potential. In our model, vint(R) is given by
and vint(R) is the two-body interaction potential. In our model, vint(R) is given by
 |
(5) |
which incorporates both a screened electrostatic potential (Debye-Hückel form) at all values of interbead distance R and a hard-core potential (repulsive Leonnard-Jones form) for interbead distance R less than the hydrodynamic radius of DNA σ (∼2.5 nm). The electrostatic interaction strength ve in Eq. 5 is given by q2kBTlB, where q is the charge per bead (in units of the electron charge), kBT is the thermal energy, and lB is the Bjerrum length (∼0.7 nm for water at room temperature); the Debye length lD scales the length of electrostatic screening. Although typical bacteriophage have icosahedral capsids, we model the confining capsid as a sphere. The interaction between a chain segment and the capsid wall is modeled as
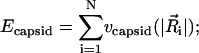 |
(6) |
as in Kindt et al. (2001), we adopt a simple form for the capsid-interaction potential vcapsid(R), given by
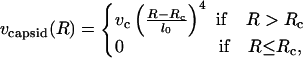 |
(7) |
where vc is the capsid-interaction strength, and Rc is the radius of the capsid.
The forces and torques are found from the total energy E = Ecom + Ebend + Etwist + Eint + Ecapsid using the method of variations. Specifically, the force 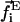 on the ith bead and the torque
on the ith bead and the torque 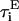 on the ith bond vector are found using the expression
on the ith bond vector are found using the expression 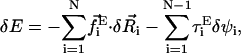 where ψi is the Euler angle of pure rotation (Rose, 1957), which is orthogonal to the drift motion of the beads
where ψi is the Euler angle of pure rotation (Rose, 1957), which is orthogonal to the drift motion of the beads 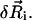 The dynamic equations of motion of the chain are found by performing a force balance on the beads and a torque balance on the bonds, which incorporates the forces and torques from the total energy E as well as terms accounting for inertia, hydrodynamic drag, and Brownian fluctuations. As we consider highly viscous dynamics, we neglect inertial forces and torques. The hydrodynamic drag for this preliminary study takes the simplest possible form: the drag force and torque is local and linear in the velocity of the bead and the angular velocity of the bond, respectively. Thus, the equations of motion are given by
The dynamic equations of motion of the chain are found by performing a force balance on the beads and a torque balance on the bonds, which incorporates the forces and torques from the total energy E as well as terms accounting for inertia, hydrodynamic drag, and Brownian fluctuations. As we consider highly viscous dynamics, we neglect inertial forces and torques. The hydrodynamic drag for this preliminary study takes the simplest possible form: the drag force and torque is local and linear in the velocity of the bead and the angular velocity of the bond, respectively. Thus, the equations of motion are given by
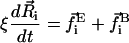 |
(8) |
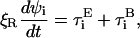 |
(9) |
where ξ is the drag coefficient for drift, and ξR is the drag coefficient for rotation. The Brownian force  and torque
and torque 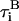 satisfy the fluctuation dissipation theorem such that
satisfy the fluctuation dissipation theorem such that  and
and  (Doi and Edwards, 1986).
(Doi and Edwards, 1986).
In our simulations, we nondimensionalize energy by kBT, length by l0, and time by  ). Choosing the equilibrium bond length to be l0 = 2.5 nm, the beads in the chain just touch their neighbors, which essentially ensures the noncrossability of the chain, thus preserving the chain topology. Our nondimensionalized simulation parameters are chosen as follows: A = 500, B = C = 20, lB = 0.284, lD = 0.328, q = −16.675, σ = 1, vHC = 20, Rcap = 5, vcap = 20, ξ = 1, and ξR = 0.213. We note that our capsid radius Rcap = 5 (12.5 nm) is substantially smaller than the typical bacteriophage capsid (Purohit et al., 2004). As in Kindt et al. (2001), the reduced capsid size makes it possible to achieve a packaged volume fraction ΦP of the typical bacteriophage (ΦP ∼ 0.45; Purohit et al., 2004) while performing the computationally taxing simulations in a reasonable time. For our choice of parameters, the volume fraction is given by ΦP = 0.001N. We choose to maintain the material properties of the polymer to match those of DNA. Although one alternative possibility is to rescale the persistence length to match the new capsid size, it is not clear whether this accurately captures the actual system. Rescaling the bending rigidity would alter the balance between the energetic contributions, and we do not know how to properly rescale all of the simulation parameters to maintain the proper balance. Furthermore, during the later stages of the DNA packaging in the actual system, the newly fed DNA would in effect feel a confinement that is smaller than the capsid size, and we want to capture those energetics correctly.
). Choosing the equilibrium bond length to be l0 = 2.5 nm, the beads in the chain just touch their neighbors, which essentially ensures the noncrossability of the chain, thus preserving the chain topology. Our nondimensionalized simulation parameters are chosen as follows: A = 500, B = C = 20, lB = 0.284, lD = 0.328, q = −16.675, σ = 1, vHC = 20, Rcap = 5, vcap = 20, ξ = 1, and ξR = 0.213. We note that our capsid radius Rcap = 5 (12.5 nm) is substantially smaller than the typical bacteriophage capsid (Purohit et al., 2004). As in Kindt et al. (2001), the reduced capsid size makes it possible to achieve a packaged volume fraction ΦP of the typical bacteriophage (ΦP ∼ 0.45; Purohit et al., 2004) while performing the computationally taxing simulations in a reasonable time. For our choice of parameters, the volume fraction is given by ΦP = 0.001N. We choose to maintain the material properties of the polymer to match those of DNA. Although one alternative possibility is to rescale the persistence length to match the new capsid size, it is not clear whether this accurately captures the actual system. Rescaling the bending rigidity would alter the balance between the energetic contributions, and we do not know how to properly rescale all of the simulation parameters to maintain the proper balance. Furthermore, during the later stages of the DNA packaging in the actual system, the newly fed DNA would in effect feel a confinement that is smaller than the capsid size, and we want to capture those energetics correctly.
We perform packaging dynamics in a similar manner as in Kindt et al. (2001); beads are fed into the spherical confinement one at a time while tracking the interior bead motion by numerically solving their equations of motion (Eqs. 8 and 9).
However, we have two important differences from Kindt et al. (2001). First, the entering bond vector  is clamped such that the direction of insertion is held fixed in the negative z-direction. Second, the opposite end of the chain is clamped and held fixed opposite the portal of entry, thus
is clamped such that the direction of insertion is held fixed in the negative z-direction. Second, the opposite end of the chain is clamped and held fixed opposite the portal of entry, thus  and
and  We test the effect of twist on the packaged conformation by performing simulations where the feeding chain end is rotated as the chain enters and comparing with simulations where the feeding chain enters without rotation.
We test the effect of twist on the packaged conformation by performing simulations where the feeding chain end is rotated as the chain enters and comparing with simulations where the feeding chain enters without rotation.
The chain topology is characterized by the linking number ΔLk, the twist ΔTw, and the writhe Wr of the curve (White, 1969). These quantities are defined for curves that are closed either by joining the ends into a circle or by extending the ends of the chain toward infinity in the direction of the end tangents. We use the latter method in this work because our chain is open with ends that are clamped and pointed in fixed directions. The linking number satisfies ΔLk = ΔTw + Wr, where these two contributions are determined from the chain conformation and twist angles. For our discrete representation, the twist ΔTw is given by
 |
(10) |
The writhe Wr of the conformation is given by a Gauss integral over the curve (White, 1969); the calculation of Wr for our discrete chain representation makes use of techniques in Klenin and Langowski (2000) with only slight alterations upon extending the chain ends to infinity. The calculation of the Gauss integral for two straight segments is performed analytically using “Method 1b” in Klenin and Langowski (2000). We sum the Gauss integral over the polymer chain segments to find the chain/chain contribution to the writhe Wr. In addition, the Gauss integral of the polymer chain segments with the chain end segments are found by taking the limit as the chain end segment length goes to infinity. The total writhe Wr is the sum of the chain/chain and the chain/end-segment contributions. Performing the calculation of Tw and Wr separately provides both a method of characterizing the topology of the chain as well as a test of whether the chain has crossed itself by checking that Tw and Wr sum to the fixed linking number Lk.
References
- Allison, S., R. Austin, and M. Hogan. 1989. Bending and twisting dynamics of short linear DNAs: analysis of the triplet anisotropy decay of a 209-basepair fragment by Brownian simulation. J. Chem. Phys. 90:3843–3854. [Google Scholar]
- Arsuaga, J., R. K. Z. Tan, M. Vazquez, D. W. Sumners, and S. C. Harvey. 2002. Investigation of viral DNA packaging using molecular mechanics models. Biophys. Chem. 101:475–484. [DOI] [PubMed] [Google Scholar]
- Aubrey, K. L., S. R. Casjens, and G. J. Thomas. 1992. Studies of virus structure by laser Raman-spectroscopy. 38. Secondary structure and interactions of the packaged dsDNA genome of bacteriophage-P22 investigated by Raman difference spectroscopy. Biochemistry. 31:11835–11842. [DOI] [PubMed] [Google Scholar]
- Baase, W. A., and W. C. Johnson. 1979. Circular-dichroism and DNA secondary structure. Nucleic Acids Res. 6:797–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednar, J., P. Furrer, A. Stasiak, J. Dubochet, E. H. Egelman, and A. D. Bates. 1994. The twist, writhe and overall shape of supercoiled DNA change during counterion-induced transition from a loosely to a tightly interwound superhelix: possible implications for DNA structure in-vivo. J. Mol. Biol. 235:825–847. [DOI] [PubMed] [Google Scholar]
- Black, L. W., W. W. Newcomb, J. W. Boring, and J. C. Brown. 1985. Ion etching of bacteriophage-T4: support for a spiral-fold model of packaged DNA. Proc. Natl. Acad. Sci. USA. 82:7960–7964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casjens, S. 1989. In Chromosomes: Eucaryotic, Procaryotic and Viral, Vol. III. K. Adolph, editor. CRC Press, Baca Raton, FL. 241–261.
- Cerritelli, M. E., N. Q. Cheng, A. H. Rosenberg, C. E. McPherson, F. P. Booy, and A. C. Steven. 1997. Encapsidated conformation of bacteriophage T7 DNA. Cell. 91:271–280. [DOI] [PubMed] [Google Scholar]
- Chan, A., R. Kilkuskie, and S. Hanlon. 1979. Correlations between the duplex winding angle and the circular-dichroism spectrum of calf thymus DNA. Biochemistry. 18:84–91. [DOI] [PubMed] [Google Scholar]
- Chirico, G., and J. Langowski. 1994. Kinetics of DNA supercoiling studied by Brownian dynamics simulation. Biopolymers. 34:415–433. [Google Scholar]
- de Gennes, P. G. 1971. Reptation of a polymer chain in presence of fixed obstacles. J. Chem. Phys. 55:572–579. [Google Scholar]
- de Gennes, P. G. 1982. In Polymer Liquid Crystals. A. Ciferri, W. R. Krigbaum, and R. B. Meyer, editors. Academic Press, New York, NY. Chap. 5.
- Doi, M., and S. F. Edwards. 1986. The Theory of Polymer Dynamics. Oxford University Press, New York, NY.
- Earnshaw, W. C., and S. C. Harrison. 1977. DNA arrangement in isometric phage heads. Nature. 268:598–602. [DOI] [PubMed] [Google Scholar]
- Evilevitch, A., L. Lavelle, C. M. Knobler, E. Raspaud, and W. M. Gelbart. 2003. Osmotic pressure inhibition of DNA ejection from phage. Proc. Natl. Acad. Sci. USA. 100:9292–9295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes, S., and D. Anderson. 1997. The bacteriophage phi 29 packaging proteins supercoil the DNA ends. J. Mol. Biol. 266:901–914. [DOI] [PubMed] [Google Scholar]
- Hall, S. B., and J. A. Schellman. 1982a. Flow dichroism of capsid DNA phages. 1. Fast and slow T4b. Biopolymers. 21:1991–2010. [DOI] [PubMed] [Google Scholar]
- Hall, S. B., and J. A. Schellman. 1982b. Flow dichroism of capsid DNA phages. 2. Effect of DNA deletions and intercalating dyes. Biopolymers. 21:2011–2031. [DOI] [PubMed] [Google Scholar]
- Holzwarth, G., D. G. Gordon, J. E. McGinnes, B. P. Dorman, and M. F. Maestre. 1974a. Mie scattering contributions to optical density and circular-dichroism of T2-bacteriophage. Abstracts of Papers of the American Chemical Society. 10. [DOI] [PubMed]
- Holzwarth, G., D. G. Gordon, J. E. McGinnes, B. P. Dorman, and M. F. Maestre. 1974b. Mie scattering contributions to optical density and circular-dichroism of T2-bacteriophage. Biochemistry. 13:126–132. [DOI] [PubMed] [Google Scholar]
- Hud, N. V. 1995. Double-stranded DNA organization in bacteriophage heads: an alternative toroid-based model. Biophys. J. 69:1355–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jendrejack, R. M., E. T. Dimalanta, D. C. Schwartz, M. D. Graham, and J. J. de Pablo. 2003. DNA dynamics in a microchannel. Phys. Rev. Lett. 91:038102. [DOI] [PubMed] [Google Scholar]
- Kindt, J., S. Tzlil, A. Ben-Shaul, and W. M. Gelbart. 2001. DNA packaging and ejection forces in bacteriophage. Proc. Natl. Acad. Sci. USA. 98:13671–13674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenin, K., and J. Langowski. 2000. Computation of writhe in modeling of supercoiled DNA. Biopolymers. 54:307–317. [DOI] [PubMed] [Google Scholar]
- Krasilnikov, A. S., A. Podtelezhnikov, A. Vologodskii, and S. M. Mirkin. 1999. Large-scale effects of transcriptional DNA supercoiling in vivo. J. Mol. Biol. 292:1149–1160. [DOI] [PubMed] [Google Scholar]
- Kratky, O., and G. Porod. 1949. Rontgenuntersuchung geloster fadenmolekule. Recl. Trav. Chim. Pay. B. 68:1106–1122. [Google Scholar]
- LaMarque, J. C., T. V. L. Le, and S. C. Harvey. 2004. Packaging double-helical DNA into viral capsids. Biopolymers. 73:348–355. [DOI] [PubMed] [Google Scholar]
- Leikin, S., D. C. Rau, and V. A. Parsegian. 1991. Measured entropy and enthalpy of hydration as a function of distance between DNA double helices. Phys. Rev. A. 44:5272–5278. [DOI] [PubMed] [Google Scholar]
- Lepault, J., J. Dubochet, W. Baschong, and E. Kellenberger. 1987. Organization of double-stranded DNA in bacteriophages: a study by cryoelectron microscopy of vitrified samples. EMBO J. 6:1507–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love, A. E. H. 1944. A Treatise on the Mathematical Theory of Elasticity. Dover Publications, New York, NY.
- Mahadevan, L., and J. B. Keller. 1996. Coiling of flexible ropes. Proc. R. Soc. Lond. Ser. A Math. Phys. Eng. Sci. 452:1679–1694. [Google Scholar]
- Marenduzzo, D., and C. Micheletti. 2003. Thermodynamics of DNA packaging inside a viral capsid: the role of DNA intrinsic thickness. J. Mol. Biol. 330:485–492. [DOI] [PubMed] [Google Scholar]
- Marko, J. F., and E. D. Siggia. 1994. Fluctuations and supercoiling of DNA. Science. 265:506–508. [DOI] [PubMed] [Google Scholar]
- Meiners, J. C., and S. R. Quake. 2000. Femtonewton force spectroscopy of single extended DNA molecules. Phys. Rev. Lett. 84:5014–5017. [DOI] [PubMed] [Google Scholar]
- Müller, J., S. Oehler, and B. Müller-Hill. 1996. Repression of lac promoter as a function of distance, phase and quality of an auxiliary lac operator. J. Mol. Biol. 257:21–29. [DOI] [PubMed] [Google Scholar]
- North, A. C., and A. Rich. 1961. X-ray diffraction studies of bacterial viruses. Nature. 191:1242–1245. [DOI] [PubMed] [Google Scholar]
- Odijk, T. 1998. Hexagonally packed DNA within bacteriophage T7 stabilized by curvature stress. Biophys. J. 75:1223–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odijk, T., and F. Slok. 2003. Nonuniform Donnan equilibrium within bacteriophages packed with DNA. J. Phys. Chem. B. 107:8074–8077. [Google Scholar]
- Olson, N. H., M. Gingery, F. A. Eiserling, and T. S. Baker. 2001. The structure of isometric capsids of bacteriophage T4. Virology. 279:385–391. [DOI] [PubMed] [Google Scholar]
- Purohit, P. K., M. Inamdar, P. Grayson, T. Squires, J. Kondev, and R. Phillips. 2005. Forces during bacteriophage DNA packaging and ejection. Biophys. J. 88:851–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit, P. K., J. Kondev, and R. Phillips. 2003a. Force steps during viral DNA packaging? J. Mech. Phys. Solids. 51:2239–2257. [Google Scholar]
- Purohit, P. K., J. Kondev, and R. Phillips. 2003b. Mechanics of DNA packaging in viruses. Proc. Natl. Acad. Sci. USA. 100:3173–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, K. E., R. C. Williams, and R. Calendar. 1973. Mode of DNA packing within bacteriophage heads. J. Mol. Biol. 78:255–259. [DOI] [PubMed] [Google Scholar]
- Rose, M. E. 1957. Elementary Theory of Angular Momentum. Wiley, New York, NY.
- Schvartzman, J. B., and A. Stasiak. 2004. A topological view of the replicon. EMBO Rep. 5:256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serwer, P. 1986. Arrangement of double-stranded DNA packaged in bacteriophage capsids: an alternative model. J. Mol. Biol. 190:509–512. [DOI] [PubMed] [Google Scholar]
- Serwer, P., S. J. Hayes, and R. H. Watson. 1992. Conformation of DNA packaged in bacteriophage-T7: analysis by use of ultraviolet light-induced DNA-capsid cross-linking. J. Mol. Biol. 223:999–1011. [DOI] [PubMed] [Google Scholar]
- Simpson, A. A., Y. Z. Tao, P. G. Leiman, M. O. Badasso, Y. N. He, P. J. Jardine, N. H. Olson, M. C. Morais, S. Grimes, D. L. Anderson, T. S. Baker, and M. G. Rossmann. 2000. Structure of the bacteriophage phi 29 DNA packaging motor. Nature. 408:745–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, D. E., S. J. Tans, S. B. Smith, S. Grimes, D. L. Anderson, and C. Bustamante. 2001. The bacteriophage phi 29 portal motor can package DNA against a large internal force. Nature. 413:748–752. [DOI] [PubMed] [Google Scholar]
- Spakowitz, A. J., and Z.-G. Wang. 2003. Semiflexible polymer solutions. I. Phase behavior and single-chain statistics. J. Chem. Phys. 119:13113–13128. [Google Scholar]
- Thomen, P., U. Bockelmann, and F. Heslot. 2002. Rotational drag on DNA: a single molecule experiment. Phys. Rev. Lett. 88:248102. [DOI] [PubMed] [Google Scholar]
- Tunis-Schneider, M. J., and M. F. Maestre. 1970. Circular dichroism spectra of oriented and unoriented deoxyribonucleic acid films: a preliminary study. J. Mol. Biol. 52:521–541. [DOI] [PubMed] [Google Scholar]
- Tzlil, S., J. T. Kindt, W. M. Gelbart, and A. Ben-Shaul. 2003. Forces and pressures in DNA packaging and release from viral capsids. Biophys. J. 84:1616–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virrankoski-Castrodeza, V., M. J. Fraser, and J. H. Parish. 1982. Condensed DNA structures derived from bacteriophage heads. J. Gen. Virol. 58:181–190. [DOI] [PubMed] [Google Scholar]
- Wang, J. C. 1978. DNA: bihelical structure, supercoiling, and relaxation. Cold Spring Harb. Symp. Quant. Biol. 43:29–33. [DOI] [PubMed] [Google Scholar]
- Wang, J. C. 1979. Helical repeat of DNA in solution. Proc. Natl. Acad. Sci. USA. 76:200–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., and N. R. Cozzarelli. 1986. Meeting on Biological Effects of DNA Topology. Cold Spring Harbor Laboratory, NY.
- Wang, J. C., and G. N. Giaever. 1988. Action at a distance along a DNA. Science. 240:300–304. [DOI] [PubMed] [Google Scholar]
- White, J. H. 1969. Self-linking and Gauss-integral in higher dimensions. Am. J. Math. 91:693–728. [Google Scholar]
- Widom, J., and R. L. Baldwin. 1983. Tests of spool models for DNA packaging in phage lambda. J. Mol. Biol. 171:419–437. [DOI] [PubMed] [Google Scholar]
- Yamakawa, H. 1997. Helical Wormlike Chains in Polymer Solutions. Springer-Verlag, Berlin, Germany.
- Zhang, Z. X., B. Greene, P. A. Thuman-Commike, J. Jakana, P. E. Prevelige, J. King, and W. Chiu. 2000. Visualization of the maturation transition in bacteriophage P22 by electron cryomicroscopy. J. Mol. Biol. 297:615–626. [DOI] [PubMed] [Google Scholar]
- Zhu, Y. X., and S. Granick. 2001. Viscosity of interfacial water. Phys. Rev. Lett. 87:096104. [DOI] [PubMed] [Google Scholar]