

Abstract
Signaling pathways interact to integrate and regulate information flow in evoking complex cellular responses. We have studied the mechanisms and consequences of interactions between the Gq and Wnt/β-catenin pathways. In human colon carcinoma SW480 cells, activation of the Gq pathway inhibits β-catenin signaling as determined by transcriptional reporter and cell proliferation assays. Ca2+ release from internal stores results in nuclear export and calpain-mediated degradation of β-catenin in the cytoplasm. Gαq does not inhibit the effects of constitutively activated ΔN-XTCF3-VP16 chimera in SW480 cells. Similarly, in HEK293 cells the Gq pathway suppresses β-catenin–T cell factor/lymphocyte enhancer factor-1 transcriptional activity induced by Wnt/Frizzled interaction or glycogen synthase kinase-3β-resistant β-catenin, but not ΔN-XTCF3-VP16. We conclude that Gq signaling promotes nuclear export and calpain-mediated degradation of β-catenin, which therefore contributes to the inhibition of Wnt/β-catenin pathway.
Interactions between signaling pathways balance information flow within a cell, allowing the cell to decide when and how to mount a response. Identifying cellular components that serve as junctions between signaling pathways is essential to our understanding of how regulatory signaling networks are constructed (1). We have been interested in understanding how signaling pathways interact with one another to regulate diverse biological functions, including cell proliferation (2, 3) and long-term potentiation of synaptic responses (4). It has become apparent that multiple signaling pathways such as the mitogen-activated protein kinase (5), Src-Stat3 (6), and Wnt/β-catenin (7) pathways regulate cell proliferation.
β-Catenin is a multifunctional protein serving both as a structural component and a signaling component of the Wnt pathway in regulating embryogenesis and tumorigenesis (8). In nonstimulated cells, β-catenin is largely associated with cadherin. There is very little β-catenin in the cytoplasm or nucleus because of its rapid degradation by the proteasome promoted by the destruction complex consisting of Axin, glycogen synthase kinase-3β (GSK-3β), and adenomatous polyposis coli (APC) protein (9, 10). In this complex, β-catenin is phosphorylated by casein kinase I α (CKIα) and GSK-3β (11), then recognized by the F-box protein β-TrCP in the ubiquitin-ligase complex and targeted for degradation in the proteasome (12–14). By an as yet unknown mechanism, Wnt through its coreceptors LRP (low-density-lipoprotein receptor-related protein) and Frizzled (15–17), involving a downstream component Dishevelled, blocks proteasome-mediated degradation, therefore β-catenin translocates into the nucleus, forms a complex with T cell factor/lymphocyte enhancer factor-1 (TCF/LEF-1) family transcription factors, and regulates Wnt target genes (8). It is important to keep the activation of this pathway tightly controlled, because mutations in APC or β-catenin, which result in stabilization of β-catenin, are detected in different types of cancer (18).
Here we have analyzed how heterotrimeric G protein pathways may interact with the canonical Wnt pathway. Specifically we have focused on the interactions between the Gαq and Wnt/β-catenin pathways and investigate how the Gq pathway could inhibit β-catenin signaling.
Materials and Methods
Cell Lines, Plasmids, and Reagents.
Detailed information about the culturing of the SW480 and HEK293 cells, plasmids, and reagents are in Supporting Materials and Methods, which is published as supporting information on the PNAS web site, www.pnas.org.
TOPFLASH/FOPFLASH Reporter Assay.
TCF/LEF-1 transcriptional activity was measured by using the TOPFLASH and FOPFLASH reporter assays as described in detail in Supporting Materials and Methods.
Cell Proliferation Assay.
Cells were plated on 96-well plates in DMEM supplemented with 10% FBS for 24 h. Viable cells were quantified with [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sul-fophenyl)-2H-tetrazolium (MTS) substrate at 490 nm, according to the manufacturer's instructions (Promega). Relative cell proliferation was shown compared with the corresponding control cells.
Whole-Cell Extract, Cell Fractionation, and Immunoblotting.
Whole-cell extract and cell fractionation for immunoblotting were prepared as described in Supporting Materials and Methods.
In Vitro Degradation by μ-Calpain.
HEK293 whole-cell extracts (1 mg/ml) were prepared and incubated with μ-calpain (10 μg/ml, Calbiochem) and an appropriate amount of CaCl2 in the buffer (30 mM Tris⋅HCl, pH 7.5/1.5 mM DTT) at 37°C for 30 min. Reaction was stopped by adding 4 × SDS sample buffer. The samples were resolved by SDS/PAGE followed by immunoblotting.
Immunofluorescence Microscopy.
Visualization of protein subcellular localization by immunofluorescence staining was performed as described in Supporting Materials and Methods.
Results
Effect of Gq Signaling Pathway on β-Catenin-Dependent TCF/LEF-1 Reporter and Cell Proliferation.
Intestinal epithelium cell is one of the cell types that proliferates in response to activation of the Wnt/β-catenin pathway. Disregulation of Wnt/β-catenin caused by mutations in β-catenin, APC, and Axin has been identified in colorectal and hepatocellular cancers (18–22). In particular, about 85% of all sporadic and hereditary colorectal tumors have been found to contain an APC mutation (23). Mutational loss of APC function in colon cancer cells activates the Wnt transcriptional response by stabilizing β-catenin (24) and is thus critical for tumorigenesis (18, 19). One of the extensively studied human colon cancer cell lines is SW480, which carries a truncation mutation at amino acid 1337. The mutant APC proteins in this cell line do not bind to Axin and lose most β-catenin binding sites (25). In initial experiments we screened to see whether the constitutively active QL-Gα mutants lacking intrinsic GTPase activity modulate β-catenin-dependent TCF/LEF-1 transcriptional activity in SW480 cells. SW480 cells were transfected with the various activated Gα subunits and TOPFLASH or FOPFLASH, and then assayed for luciferase activity. Q209L–Gαq inhibited TCF/LEF-1 transcriptional activity by about 80% compared with the control, whereas the other constitutively active Gα subunits, namely, Q213L–Gαs, Q204L–Gαi-1, and Q231L–Gα12, had little or no effect (Fig. 1a). We next determined whether activation of Gαq by the muscarinic acetylcholine type 3 receptor (M3R) had the same effect. Carbachol-induced activation of Gq in cells transfected with M3R could repress TCF/LEF-1 transcriptional activity as effectively as Q209L–Gαq (Fig. 1b). Carbachol activation of the Gi-coupled muscarinic acetylcholine type 2 receptor (M2R) had no effect on TCF/LEF-1 activity (Fig. 1b). Similarly, in HEK293 cells Q209L–Gαq, Q209L–Gα11, or agonist-activated M3R blocked transcriptional activity induced by mutant β-catenin (S33Y), which is resistant to phosphorylation by GSK-3β and thus to degradation by proteasome (Fig. 6, which is published as supporting information on the PNAS web site). These results indicate that the activation of the Gq pathway through Gαq selectively inhibits β-catenin–TCF/LEF-1-dependent transcription.
Figure 1.

Effect of the Gq signaling pathway on TCF/LEF-1-mediated transcriptional activity and proliferation. (a) Effects of various Gα subunits on β-catenin-dependent TCF/LEF-1 transcriptional activity. SW480 cells were transfected with 025 μg of empty vector as control or cDNAs encoding various constitutively activated Gα subunits, and relative TCF/LEF-1 activity was measured. The results are expressed as mean ± SD of duplicates. (b) Activation of Gq-coupled receptor inhibits β-catenin-dependent TCF/LEF-1 transcriptional activity. SW480 cells were transfected with empty vector as control or cDNAs expressing WT or mutant activated Gαq, M2R, or M3R as indicated. Carbachol (1 mM) was added 24 h after transfection, and transcriptional activity was measured 48 h after transfection. (c) Q209L–Gαq down-regulates the cyclin D1 protein level. SW480 cells were transfected with empty plasmid pIRES2-EGFP as control or pQ209L-Gαq-IRES2-EGFP expression plasmid. EGFP-positive cells were enriched by fluorescence-activated cell sorting. Cell extracts were prepared, electrophoretically resolved, and immunoblotted with cyclin D1 antibody. β-actin was used for loading control. WB, Western blot. (d) Q209L–Gαq inhibits proliferation of SW480 cells. Cells were prepared as in c. Indicated number of cells was plated on 96-well plates and assayed for proliferation by using a colorimeteric assay. Cell proliferation was normalized with corresponding control cells transfected with empty vector and shown as mean ± SD.
We tested whether the Gq pathway affected target genes of TCF/LEF-1. One of the endogenous targets involved in proliferation, cyclin D1 (26, 27), was examined at the protein level (Fig. 1c). In Q209L–Gαq-transfected SW480 cells, the cyclin D1 level was significantly lower than in the control (Fig. 1c). As persistent β-catenin–TCF/LEF-1 activity is critical for the proliferation of transformed colon epithelial cells (7), we determined whether the Q209L–Gαq signaling pathway had any effect on cell proliferation. In SW480 cells transfected with Q209L–Gαq, proliferation was inhibited by about 70% with different starting cell densities (Fig. 1d). These observations indicate that when β-catenin–TCF/LEF-1 transcriptional activity is inhibited by Gq signaling, colon epithelial cell proliferation is negatively affected.
Gq Pathway Triggers Calpain-Mediated Degradation of β-Catenin.
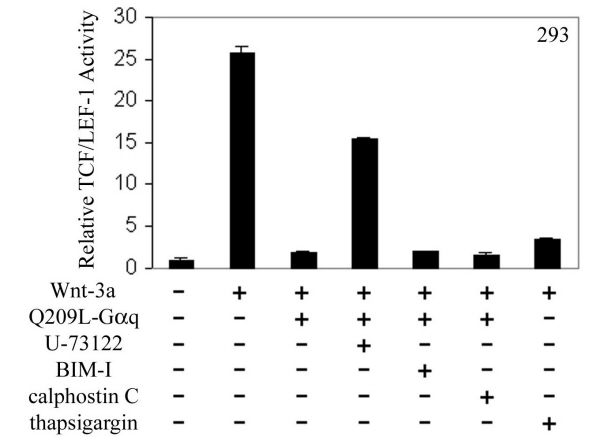
Gαq stimulates phospholipase C β (PLCβ) to form inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). IP3 causes calcium release from internal stores, whereas DAG activates PKC. To ask whether PKC mediated the Gq effect, pan PKC inhibitor bisindolylmaleimide I (BIM-I) was used in the TCF/LEF-1 reporter assay. Also, to determine whether calcium mediated the Gq effect, we tested whether thapsigargin, which elevates cytosolic calcium by inhibiting endoplasmic Ca2+-ATPase (28), mimicked the Gq effect. In SW480 cells, the inhibitory effect of Q209L–Gαq or agonist-activated M3R was not reversed by BIM-I (Fig. 2a), whereas thapsigargin was almost as effective as Q209L–Gαq in suppressing TCF/LEF-1 activity (Fig. 2b). In HEK293 cells transfected with Wnt-3a and human Frizzled-1, the TCF/LEF-1 transcriptional activity was similarly blocked by Q209L–Gαq and thapsigargin. This inhibition was relieved by PLCβ blocker U-73122 but not the PKC blockers BIM-I or calphostin C (Fig. 7, which is published as supporting information on the PNAS web site). These results suggest that calcium release, but not PKC, mediates the effect of Gαq signaling pathway on β-catenin–TCF/LEF-1 activity.
Figure 2.

Activation of the Gαq signaling pathway promotes calpain-mediated β-catenin proteolysis in a Ca2+-dependent manner. (a) The inhibitory effect of the Gq signaling pathway on TCF/LEF-1-mediated transcription is not mediated by PKC. SW480 cells were transfected with empty plasmid as control, Q209L-Gαq, or M3R expression plasmids. PKC inhibitor bisindolylmaleimide I (1 μM) was added on transfection and carbachol (1 mM) was added 24 h posttransfection. Relative TCF/LEF-1 activity was assayed 48 h after transfection. (b) Thapsigargin mimics the effect of Q209L–Gαq. SW480 cells were transfected with indicated expression plasmids. Thapsigargin (50 nM) was added on transfection. Relative TCF/LEF-1 activity was assayed 48 h after transfection. (c) Q209L–Gαq induces reduction in cytosolic β-catenin levels. SW480 cells were transfected with empty plasmid as control or Q209L–Gαq expression plasmid. Cycloheximide (25 μg/ml) was added 24 h after transfection. Cells were harvested at the indicated times after cycloheximide addition. Cytosolic fraction was prepared, resolved by SDS/PAGE, and blotted with antibody specific against the C terminus of β-catenin. Proteolytic products of β-catenin are indicated by arrowheads. (d) Q209L–Gαq induces cleavage of cytosolic β-catenin. Cytosolic fractions of SW480 cells were prepared at the indicated time points after cycloheximide treatment, electrophoretically resolved, and blotted with β-catenin antibody specific for the C terminus. Proteolytic products of β-catenin are indicated by arrowheads. (e) Thapsigargin induces calpain-mediated proteolysis of β-catenin in a calcium-dependent manner. SW480 cells were pretreated with DMSO or inhibitors for 10 min, and then treated with thapsigargin (50 nM) for 30 min. The untreated cells were used as control. The concentrations of the various inhibitors were BAPTA/AM (40 μM), NH4Cl (1 mM), calpeptin (50 μM), calpastatin (10 μM), lactacystin (10 μM), and MG-132 (10 μM). Whole-cell extract was prepared, resolved by SDS/PAGE, and blotted with the C-terminal β-catenin antibody. Proteolytic products of β-catenin are indicated by arrowheads. (f) Activation of Gq-coupled M3R induces calpain-mediated proteolysis of β-catenin. SW480 cells stably expressing M3R were treated with carbachol (1 mM), lactacystin (10 μM), or calpeptin (10 μM) as indicated. Whole-cell lysates were prepared at the indicated time points, and β-catenin fragments were detected by antibody specific for the C terminus of β-catenin. Proteolytic products of β-catenin are indicated by arrowheads. (g and h) Calpain-mediated cleavage occurs at the N-terminal region of β-catenin. As described in Materials and Methods, HEK293 cell extracts were incubated at 37°C for 30 min alone as control or with CaCl2 (0.1 mM) and μ-calpain in the presence of the indicated reagents: EGTA (1 mM), ALLN (10 μM), ALLM (10 μM), E-64 (25 μM), calpastatin (10 μM), lactacystin (10 μM), and MG-132 (10 μM). Cleavage of β-catenin was assessed with antibodies specific for the N-terminal or C-terminal regions of β-catenin. Proteolytic products of β-catenin are indicated by arrowheads. WB, Western blot; NT, N terminus; CT, C terminus.
To investigate whether Q209L–Gαq regulated cytosolic β-catenin levels posttranslationally, SW480 cells were treated with cycloheximide to inhibit protein synthesis 24 h after the cells were transfected with control or Q209L–Gαq expression vectors. Cytosolic β-catenin was examined over time. Initially, the cytosolic β-catenin level was almost the same in control and Q209L–Gαq-transfected cells. However, at 8 h, cytosolic β-catenin in Q209L–Gαq-transfected cells was significantly lower than control cells, and this difference was even more dramatic at 24 h (Fig. 2b), indicating that Q209L–Gαq reduces cytosolic β-catenin levels in SW480 cells. In the presence of protein synthesis inhibitor, many more profound proteolytic fragments in Q209L–Gαq-transfected cells were observed compared with the control when antibody specific for the C-terminal region of β-catenin was used (Fig. 2 c and d).
Because thapsigargin mimics the Q209L–Gαq effect in the TCF/LEF-1 reporter assay, we determined whether thapsigargin stimulated β-catenin proteolysis in SW480 cells. In cells treated with thapsigargin, β-catenin fragments of about 90 kDa and 75 kDa were detected by β-catenin antibody specific for the C-terminal region (Fig. 2e, DMSO lane). To determine the identity of the protease(s) involved in β-catenin cleavage, SW480 cells were pretreated with the cell-permeable calcium chelator 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate acetoxymethyl ester (BAPTA/AM) or various protease inhibitors before β-catenin proteolysis was induced by thapsigargin (Fig. 2d, lanes BAPTA/AM to MG-132). The β-catenin proteolysis was calcium-dependent, because it was completely blocked by the calcium chelator BAPTA/AM. Among the different protease inhibitors, β-catenin cleavage was inhibited by calpain inhibitors calpeptin, calpastatin, and MG-132, but not by other protease inhibitors, such as lysosomal protease inhibitor NH4Cl or proteasome-specific inhibitor lactacystin. These data demonstrate that thapsigargin triggers calpain-mediated cleavage of β-catenin in a calcium-dependent manner. To examine whether the Gαq pathway could trigger calpain-mediated proteolysis of β-catenin, M3R-expressing SW480 cells were treated with carbachol in the absence or presence of protease inhibitor lactacystin or calpeptin. Carbachol treatment led to the appearance of proteolytic fragments of β-catenin, which was completely blocked by the calpain inhibitor calpeptin, but not by the proteasome inhibitor lactacystin (Fig. 2f). Thus, activation of Gαq pathway in SW480 cells promotes calpain-mediated cleavage of β-catenin.
We also examined whether calpain was able to promote cleavage of β-catenin in vitro. For this, HEK293 cell lysate was incubated with exogenously μ-calpain and Ca2+, and then assayed for β-catenin fragments with antibodies directed against either the N-terminal or C-terminal regions of β-catenin. When the N-terminal region-specific antibody was used, calpain treatment led to the disappearance of the full-length β-catenin. Degradation could be inhibited by EGTA and calpain inhibitors ALLN, ALLM, E-64, calpastatin, and MG-132, but not by the irreversible proteasome-specific inhibitor lactacystin (Fig. 2g). When the antibody for C-terminal β-catenin was used, μ-calpain incubation revealed two distinct fragments of β-catenin, which was blocked by EGTA or the calpain inhibitors (Fig. 2h). These results suggest that β-catenin can be cleaved by calpain at two potential sites in the N-terminal region of β-catenin.
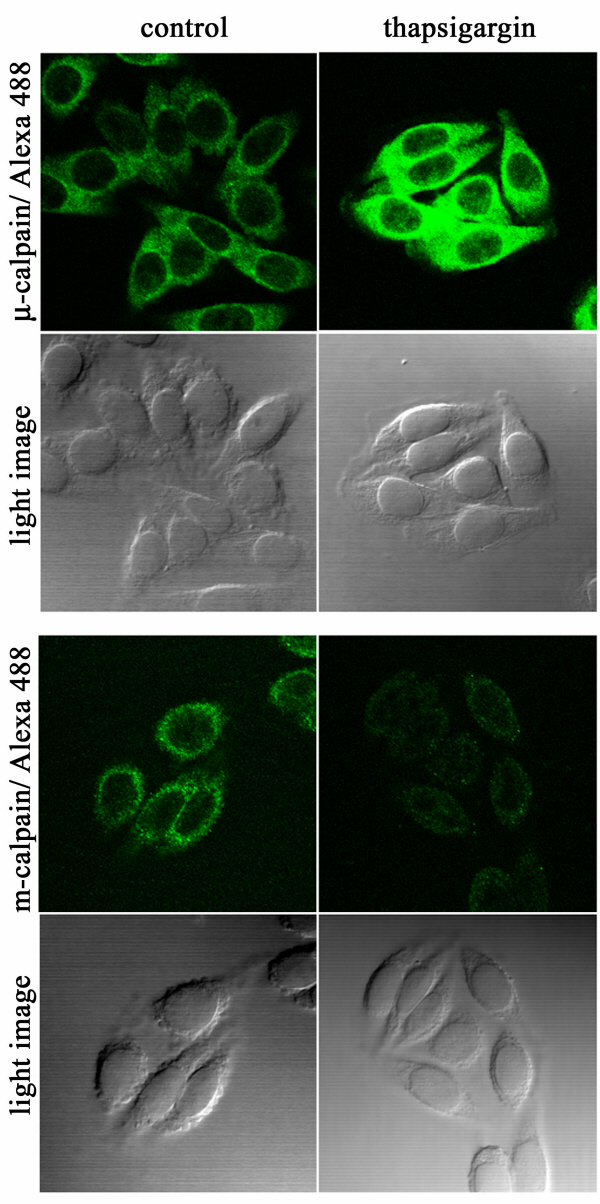
Calcium-Dependent Export of Nuclear β-Catenin.
The majority of β-catenin is localized in the nuclei in SW480 cells because of the lack of functional APC (29, 30). This finding was confirmed by immunostaining (Fig. 3 aI and aIV). However, the major isoforms of calpain, m- and μ-calpains, are localized in the cytoplasm of SW480 cells without or with treatment of thapsigargin (Fig. 8, which is published as supporting information on the PNAS web site). This finding raises the question of how nuclear localized β-catenin is cleaved by calpain in the cytoplasm on calcium release from internal stores in SW480 cells. When cells were treated with thapsigargin for 30 min, β-catenin could be visualized in both nucleus and cytoplasm (Fig. 3 aII and aV). However, when the cells were pretreated with the cell-permeable calcium chelator BAPTA/AM, the effect of thapsigargin on β-catenin localization could be reversed and predominant nuclear localization was observed again (Fig. 3 aIII and aVI). These observations suggest that calcium mobilization can promote nuclear export of β-catenin in the absence of functional APC.
Figure 3.

Ca2+ release promotes nuclear export of β-catenin. (a) Ca2+-dependent nuclear export of β-catenin in SW480 cells. Subcellular localization of endogenous β-catenin was detected by immunofluorescence staining with anti-β-catenin (I–III). (IV–VI) Light images corresponding to I–III. (I and IV) Untreated SW480 cells as a control. (II and V) Cells that were treated with thapsigargin (50 nM) for 30 min. (III and VI) Cells that were pretreated with BAPTA/AM (50 μM) for 10 min and then incubated with thapsigargin (50 nM) for 30 min. (b) Ca2+-dependent proteolysis of β-catenin occurs in the cytoplasm. Total lysate, cytosolic, and nuclear fractions were prepared as described in Materials and Methods and incubated with the indicated amount of Ca2+ at 37°C for 30 min. β-Catenin cleavage products were detected by antibody specific for the C-terminal region after being resolved by SDS/PAGE. Proteolytic products of β-catenin are indicated by arrowheads. WB, Western blot; CT, C terminus. (c) Nuclear β-catenin is down-regulated by Q209L–Gαq. Q209L–Gαq-transfected cells were labeled by EGFP expression (I). Q209L–Gαq-expressing cells are shown by arrowheads (I–III). (Scale bars: 5 μm.)
So far 14 isoforms of calpain have been identified (31). It is possible that some of them are nuclear localized and activated in response to calcium (32, 33). To determine whether this occurred, calcium-dependent protease activity for β-catenin was assayed for the cytosolic and nuclear fractions from SW480 cells. In the presence of an increasing amount of calcium, protease activity for β-catenin could be detected in the total lysate and cytosolic fraction, but not in the nuclear fraction (Fig. 3b). Even though we cannot completely rule out the possibility that some isoforms of calpain are imported into the nucleus and activated in response to calcium release, this result suggests β-catenin can be exported out of the nucleus and cleaved by calpain in the cytoplasm in a calcium-dependent manner.
Cellular localization of β-catenin is determined by the balance of nuclear import and export facilitated by its associated proteins (34, 35). If Gq signaling enhances nuclear export of β-catenin and promotes calpain-mediated degradation, we expected that nuclear β-catenin would be down-regulated by Q209L–Gαq in SW480 cells. To test this notion, Q209L–Gαq was subcloned into a bicistronic construct coexpressing enhanced GFP (EGFP). Q209L–Gαq-transfected cells could be detected by EGFP expression (shown by arrowheads in Fig. 3 cI–cIII). As predicted, there was much weaker nuclear β-catenin staining in Q209L–Gαq-transfected cells compared with untransfected cells (Fig. 3cII).
Lack of Gq Effects on Constitutively Active TCF Activities.
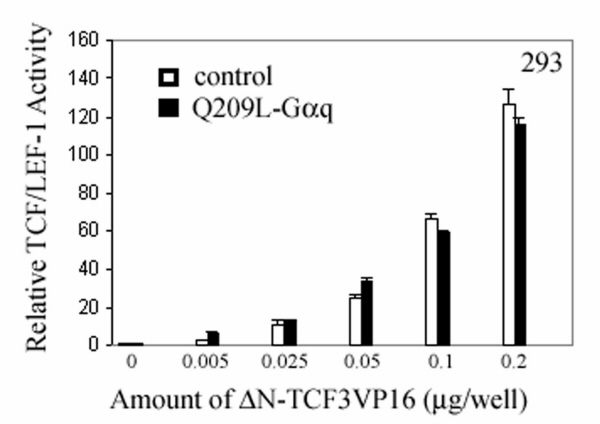
Because β-catenin does not contain a DNA-binding motif, it exerts its transcriptional activity by forming a complex with members of the TCF/LEF-1 family, which contain the HMG box DNA binding domain (36). As the data in Fig. 1 indicated that the Gαq signaling pathway inhibited β-catenin-dependent TCF/LEF-1 transcriptional activity, we asked whether constitutively active TCF/LEF-1 could block the effect of the Gαq signaling pathway. For this work, we constructed a chimera of Xenopus TCF3 (XTCF3) to the transactivation domain of VP16 protein (37). To avoid the possible interference by endogenous β-catenin, the N-terminal β-catenin interaction domain was deleted from the fusion protein ΔN-XTCF3-VP16 (36). In SW480 cells, ΔN-XTCF3-VP16 alone had little effect on TCF/LEF-1 reporter activity, but it was able to rescue the inhibitory effect of Q209L–Gαq (Fig. 4a). Similarly, in HEK293 cells ΔN-XTCF3-VP16 showed dose-dependent stimulation of TCF/LEF-1 reporter activity, which was not affected even in the presence of Q209L–Gαq (Fig. 9, which is published as supporting information on the PNAS web site). Consistently, for SW480 cells stably expressing M3R, agonist treatment led to down-regulation of the cyclin D1 level, and this effect was not seen in cells expressing ΔN-XTCF3-VP16 (Fig. 4b).
Figure 4.

Constitutively active TCF3 rescues effect of Gq signaling pathway on TCF/LEF-1-mediated transcriptional activity and cell proliferation in SW480 cells. (a) A constitutively activated form of TCF3, ΔN-XTCF3-VP16, rescues the effect of Gq signaling on TCF/LEF-1-mediated transcriptional activity in SW480 cells. SW480 cells were transfected with empty plasmid as a control or indicated expression plasmids. Relative TCF/LEF-1 transcriptional activity was assayed as described. (b) ΔN-XTCF3-VP16 rescues the effect of Gq signaling on cyclin D1 expression in SW480 cells. SW480 cells stably expressing M3R alone or together with ΔN-XTCF3-VP16 were treated without or with carbachol (1 mM) for 12 h. Total lysates were prepared and resolved by SDS/PAGE. Immunoblotting was performed with antibody against cyclin D1. The untreated M3R-expressing cells were used as control and β-actin was the loading control. WB, Western blot. (c) ΔN-XTCF3-VP16 rescues the effect of Gq signaling on SW480 cell proliferation. A total of 2.5 × 103 cells stably expressing M3R alone or together with ΔN-XTCF3-VP16 were plated on 96-well plates. Cells were treated with carbachol (1 mM) for 12 h as indicated. Cell proliferation was assessed colorimeterically. The untreated M3R-expressing cells were used as control. Data shown are mean ± SD.
We also determined whether expression of ΔN-XTCF3-VP16 could counteract the inhibitory effect of Gq signaling on SW480 cell proliferation. As shown in Fig. 4c, carbachol treatment of SW480 cells expressing M3R inhibited proliferation by about 50%. In contrast, when these cells stably expressed ΔN-XTCF3-VP16, cell proliferation was not inhibited by the agonist occupied M3R (Fig. 4c). These results indicate that the inhibitory effect of Gq pathway on SW480 cell proliferation is upstream of the TCF/LEF-1.
Discussion
Even though Ca2+ signaling has been implicated in regulation of the Wnt/β-catenin pathway in different animal models (38–42), the molecular mechanisms underlying these interactions are largely not well understood. Our studies showed that the Gq pathway interacts with the Wnt/β-catenin signaling pathway at the level of β-catenin, the key molecule in the canonical Wnt signaling pathway. Surprisingly, our data demonstrate that calpain-mediated proteolysis of β-catenin can be triggered by the activation of the Gq signaling pathway. This finding reveals a mode of control over β-catenin, which is extensively regulated in diverse cellular and developmental contexts. It appears to be a common theme that both ATP-dependent and Ca2+-dependent pathways can contribute to degradation of important regulators of cellular functions, such as p53, i-κB (43–46), and now β-catenin. Our data indicate that the Gq pathway uses calpain as a regulator of β-catenin to control signal flow through Wnt/β-catenin pathway. The model in Fig. 5 summarizes the key observations from this study.
Figure 5.

Schematic representation of the interactions between the Gq and Wnt/β-catenin signaling pathways. As shown in the model, APC promotes β-catenin nuclear export and facilitates proteasome-mediated degradation of β-catenin after sequential phosphorylation by CKIα and GSK-3β on the Axin-based complex. Wnt binding to coreceptors LRP and Frizzled blocks proteasome-mediated degradation, which further leads to nuclear localization of β-catenin, gene expression, and cell proliferation. As an alternative degradation pathway, Ca2+ activation by Gq signaling pathway promotes nuclear export of β-catenin into cytoplasm, where it is further degraded by calpain in a Ca2+-dependent manner. Consequently, β-catenin–TCF/LEF-dependent cell proliferation is inhibited. LRP, low-density-lipoprotein receptor-related protein; UPS, ubiquitin-proteasome system; PLCβ, phospholipase C β; PIP2, phosphatidylinositol 4,5-bisphosphate; IP3R, IP3 receptor.
The role of proteases as signaling components within apoptotic signaling pathways is well documented. Here we find that regulated proteases such as calpain have a role in serving as junctions between signaling pathways. It is noteworthy that instead of protein kinases and phosphatases that typically serve as junctions between G protein pathways and other signaling pathways, the junction here is a downstream protease. Thus proteases can be added to protein kinases and phosphatases as effectors of G protein pathways. In this system we have demonstrated that the target of calpain stimulated by the Gq pathway is β-catenin, but in other cell types it is likely that calpain may have many other substrates. Thus G protein-coupled receptors that couple members of the Gq family to phospholipase C may have the ability to regulate a number of processes in which calpain substrates are involved. This hypothesis will have to be experimentally tested in multiple systems.
It is generally assumed that cytosolic accumulation of β-catenin on Wnt induction or by overexpression can automatically lead to its nuclear localization. However, recent studies in oocytes and semipermeabilized cells show that β-catenin itself can freely shuttle between cytoplasm and nucleoplasm independently of classical localization signals and RanGTP (34, 47–49). Therefore, subcellular localization of β-catenin is predominantly determined by the balance of its cytoplasmic/nuclear retention and nuclear import/export facilitated by its binding partners. For example, when β-catenin is associated with E-cadherin, it is tethered at the plasma membrane (50). It has been reported that Gα12 and Gα13 may cause the dissociation of β-catenin from cadherins (51). In normal cells, newly synthesized β-catenin in the cytoplasm is captured by APC or Axin and targeted for destruction by proteasome, which leads to very low levels of β-catenin in cytoplasm and nucleus. In tumor cells where the normal β-catenin degradation pathway is blocked, β-catenin accumulates in both cytoplasm and nucleus. High levels of nuclear β-catenin can be retained in the nucleus caused by APC mutations (29, 30) or by TCF/LEF that is up-regulated by positive feedback in tumor cells (52, 53). Our studies show that calcium release from internal stores enhances the movement of nuclear β-catenin back into the cytoplasm and promotes degradation by a calpain-mediated mechanism in SW480 cells. Thus therapeutic agents that trigger calcium mobilization can be valuable in the treatment of colon cancers.
Previous studies from other laboratories and ours have shown that Gq-coupled receptors (54) or activated Gαq stimulated the proliferation of NIH 3T3 cells (55). In NIH 3T3 fibroblast cells, we had found that activated Gαq potentiated the effects of platelet-derived growth factor in confluent cells to stimulate their proliferation (56). By contrast, in SW480 colon cancer cells, the major proliferative pathway appears to be the β-catenin–TCF/LEF-1 pathway, and here because of calcium-dependent nuclear export and calpain-mediated degradation of β-catenin, activated Gαq inhibits cell proliferation (Fig. 5). Therefore, depending on the cell type and the signaling pathways that drive the proliferation, the Gq pathway can either stimulate or inhibit cell proliferation. These seemingly opposing cellular effects of the same G protein signaling pathway vividly highlight the necessity of understanding the context within which these signaling pathways operate and exert their effects.
Supplementary Material
Acknowledgments
We thank Dr. H. Clevers for providing the pTOPFLASH, pFOPFLASH, ΔN-XTCF, and β-catenin constructs, Dr. J. Kitajewski for reagents and useful early discussions, Drs. S. Wilk, M. Diverse-Pierluissi, D. Weinstein, H. Qi, H. Xiong, Y. Chen, C. He, D. Jordan, M. Yang, Y. Deng, D. Aaronson, and B. Yoo for helpful discussions and technical support, S. Neves for drawing of the model, and Dr. P. Ram for critical comments on the manuscript. This study was supported by National Institutes of Health Grants GM-54508 and CA 81050. Confocal laser scanning microscopy was performed at the Mount Sinai School of Medicine-Microscopy Shared Resource Facility, supported with funding from National Institutes of Health-National Cancer Institute Shared Resources Grant CA095823, National Science Foundation Major Research Instrumentation Grant DBI-9724504, and National Institutes of Health Shared Instrumentation Grant RR0 9145. This research is part of G.L.'s doctoral thesis.
Abbreviations
- APC
adenomatous polyposis coli
- BAPTA/AM
1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate acetoxymethyl ester
- CKIα
casein kinase I α
- EGFP
enhanced GFP
- GSK-3β
glycogen synthase kinase-3β
- IP3
inositol 1,4,5-triphosphate
- M3R
muscarinic acetylcholine type 3 receptor
- M2R
muscarinic acetylcholine type 2 receptor
- TCF/LEF-1
T cell factor/lymphocyte enhancer factor-1
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Jordan J D, Landau E M, Iyengar R. Cell. 2000;103:193–200. doi: 10.1016/s0092-8674(00)00112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen J, Iyengar R. Science. 1994;263:1278–1281. doi: 10.1126/science.8122111. [DOI] [PubMed] [Google Scholar]
- 3.Ram P T, Horvath C M, Iyengar R. Science. 2000;287:142–144. doi: 10.1126/science.287.5450.142. [DOI] [PubMed] [Google Scholar]
- 4.Blitzer R D, Connor J H, Brown G P, Wong T, Shenolikar S, Iyengar R, Landau E M. Science. 1998;280:1940–1942. doi: 10.1126/science.280.5371.1940. [DOI] [PubMed] [Google Scholar]
- 5.Cowley S, Paterson H, Kemp P, Marshall C J. Cell. 1994;77:841–852. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 6.Yu C L, Meyer D J, Campbell G S, Larner A C, Carter-Su C, Schwartz J, Jove R. Science. 1995;269:81–83. doi: 10.1126/science.7541555. [DOI] [PubMed] [Google Scholar]
- 7.Polakis P. Curr Opin Genet Dev. 1999;9:15–21. doi: 10.1016/s0959-437x(99)80003-3. [DOI] [PubMed] [Google Scholar]
- 8.Bienz M, Clevers H. Cell. 2000;103:311–320. doi: 10.1016/s0092-8674(00)00122-7. [DOI] [PubMed] [Google Scholar]
- 9.Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. EMBO J. 1998;17:1371–1384. doi: 10.1093/emboj/17.5.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakanaka C, Weiss J B, Williams L T. Proc Natl Acad Sci USA. 1998;95:3020–3023. doi: 10.1073/pnas.95.6.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu C, Li Y, Semenov M, Han C, Baeg G H, Tan Y, Zhang Z, Lin X, He X. Cell. 2002;108:837–847. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- 12.Orford K, Crockett C, Jensen J P, Weissman A M, Byers S W. J Biol Chem. 1997;272:24735–24738. doi: 10.1074/jbc.272.40.24735. [DOI] [PubMed] [Google Scholar]
- 13.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. EMBO J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kitagawa M, Hatakeyama S, Shirane M, Matsumoto M, Ishida N, Hattori K, Nakamichi I, Kikuchi A, Nakayama K. EMBO J. 1999;18:2401–2410. doi: 10.1093/emboj/18.9.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pinson K I, Brennan J, Monkley S, Avery B J, Skarnes W C. Nature (London) 2000;407:535–538. doi: 10.1038/35035124. [DOI] [PubMed] [Google Scholar]
- 16.Tamai K, Semenov M, Kato Y, Spokony R, Liu C, Katsuyama Y, Hess F, Saint-Jeannet J P, He X. Nature (London) 2000;407:530–535. doi: 10.1038/35035117. [DOI] [PubMed] [Google Scholar]
- 17.Wehrli M, Dougan S T, Caldwell K, O'Keefe L, Schwartz S, Vaizel-Ohayon D, Schejter E, Tomlinson A, DiNardo S. Nature (London) 2000;407:527–530. doi: 10.1038/35035110. [DOI] [PubMed] [Google Scholar]
- 18.Morin P J, Sparks A B, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler K W. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 19.Korinek V, Barker N, Morin P J, van Wichen D, de Weger R, Kinzler K W, Vogelstein B, Clevers H. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 20.Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, Kawasoe T, Ishiguro H, Fujita M, Tokino T, et al. Nat Genet. 2000;24:245–250. doi: 10.1038/73448. [DOI] [PubMed] [Google Scholar]
- 21.Clevers H. Nat Genet. 2000;24:206–208. doi: 10.1038/73396. [DOI] [PubMed] [Google Scholar]
- 22.Taniguchi K, Roberts L R, Aderca I N, Dong X, Qian C, Murphy L M, Nagorney D M, Burgart L J, Roche P C, Smith D I, et al. Oncogene. 2002;21:4863–4871. doi: 10.1038/sj.onc.1205591. [DOI] [PubMed] [Google Scholar]
- 23.Kinzler K W, Vogelstein B. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 24.Smits R, Kielman M F, Breukel C, Zurcher C, Neufeld K, Jagmohan-Changur S, Hofland N, van Dijk J, White R, Edelmann W, et al. Genes Dev. 1999;13:1309–1321. doi: 10.1101/gad.13.10.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kishida S, Yamamoto H, Ikeda S, Kishida M, Sakamoto I, Koyama S, Kikuchi A. J Biol Chem. 1998;273:10823–10826. doi: 10.1074/jbc.273.18.10823. [DOI] [PubMed] [Google Scholar]
- 26.Tetsu O, McCormick F. Nature (London) 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 27.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben-Ze'ev A. Proc Natl Acad Sci USA. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tepel M, Ruess C, Mehring N, Neusser M, Zidek W. Clin Exp Hypertens. 1994;16:493–506. doi: 10.3109/10641969409067958. [DOI] [PubMed] [Google Scholar]
- 29.Rosin-Arbesfeld R, Townsley F, Bienz M. Nature (London) 2000;406:1009–1012. doi: 10.1038/35023016. [DOI] [PubMed] [Google Scholar]
- 30.Henderson B R. Nat Cell Biol. 2000;2:653–660. doi: 10.1038/35023605. [DOI] [PubMed] [Google Scholar]
- 31.Huang Y, Wang K K. Trends Mol Med. 2001;7:355–362. doi: 10.1016/s1471-4914(01)02049-4. [DOI] [PubMed] [Google Scholar]
- 32.Ma H, Shih M, Fukiage C, Azuma M, Duncan M K, Reed N A, Richard I, Beckmann J S, Shearer T R. Invest Ophthalmol Visual Sci. 2000;41:4232–4239. [PubMed] [Google Scholar]
- 33.Ma H, Fukiage C, Kim Y H, Duncan M K, Reed N A, Shih M, Azuma M, Shearer T R. J Biol Chem. 2001;276:28525–28531. doi: 10.1074/jbc.M100603200. [DOI] [PubMed] [Google Scholar]
- 34.Eleftheriou A, Yoshida M, Henderson B R. J Biol Chem. 2001;276:25883–25888. doi: 10.1074/jbc.M102656200. [DOI] [PubMed] [Google Scholar]
- 35.Henderson B R, Galea M, Schuechner S, Leung L. J Biol Chem. 2002;277:24258–24264. doi: 10.1074/jbc.M110602200. [DOI] [PubMed] [Google Scholar]
- 36.Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destree O, Clevers H. Cell. 1996;86:391–399. doi: 10.1016/s0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]
- 37.Aoki M, Hecht A, Kruse U, Kemler R, Vogt P K. Proc Natl Acad Sci USA. 1999;96:139–144. doi: 10.1073/pnas.96.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ault K T, Durmowicz G, Galione A, Harger P L, Busa W B. Development (Cambridge, UK) 1996;122:2033–2041. doi: 10.1242/dev.122.7.2033. [DOI] [PubMed] [Google Scholar]
- 39.Hedgepeth C M, Conrad L J, Zhang J, Huang H C, Lee V M, Klein P S. Dev Biol. 1997;185:82–91. doi: 10.1006/dbio.1997.8552. [DOI] [PubMed] [Google Scholar]
- 40.Kume S, Muto A, Inoue T, Suga K, Okano H, Mikoshiba K. Science. 1997;278:1940–1943. doi: 10.1126/science.278.5345.1940. [DOI] [PubMed] [Google Scholar]
- 41.Slusarski D C, Yang-Snyder J, Busa W B, Moon R T. Dev Biol. 1997;182:114–120. doi: 10.1006/dbio.1996.8463. [DOI] [PubMed] [Google Scholar]
- 42.Torres M A, Yang-Snyder J A, Purcell S M, DeMarais A A, McGrew L L, Moon R T. J Cell Biol. 1996;133:1123–1137. doi: 10.1083/jcb.133.5.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pariat M, Carillo S, Molinari M, Salvat C, Debussche L, Bracco L, Milner J, Piechaczyk M. Mol Cell Biol. 1997;17:2806–2815. doi: 10.1128/mcb.17.5.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kubbutat M H, Vousden K H. Mol Cell Biol. 1997;17:460–468. doi: 10.1128/mcb.17.1.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phillips R J, Ghosh S. Mol Cell Biol. 1997;17:4390–4396. doi: 10.1128/mcb.17.8.4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miyamoto S, Seufzer B J, Shumway S D. Mol Cell Biol. 1998;18:19–29. doi: 10.1128/mcb.18.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yokoya F, Imamoto N, Tachibana T, Yoneda Y. Mol Biol Cell. 1999;10:1119–1131. doi: 10.1091/mbc.10.4.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fagotto F, Gluck U, Gumbiner B M. Curr Biol. 1998;8:181–190. doi: 10.1016/s0960-9822(98)70082-x. [DOI] [PubMed] [Google Scholar]
- 49.Wiechens N, Fagotto F. Curr Biol. 2001;11:18–27. doi: 10.1016/s0960-9822(00)00045-2. [DOI] [PubMed] [Google Scholar]
- 50.Orsulic S, Huber O, Aberle H, Arnold S, Kemler R. J Cell Sci. 1999;112:1237–1245. doi: 10.1242/jcs.112.8.1237. [DOI] [PubMed] [Google Scholar]
- 51.Meigs T E, Fields T A, McKee D D, Casey P J. Proc Natl Acad Sci USA. 2001;98:519–524. doi: 10.1073/pnas.021350998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hovanes K, Li T W, Munguia J E, Truong T, Milovanovic T, Lawrence Marsh J, Holcombe R F, Waterman M L. Nat Genet. 2001;28:53–57. doi: 10.1038/ng0501-53. [DOI] [PubMed] [Google Scholar]
- 53.Korinek W S, Copeland M J, Chaudhuri A, Chant J. Science. 2000;287:2257–2259. doi: 10.1126/science.287.5461.2257. [DOI] [PubMed] [Google Scholar]
- 54.Julius D, Livelli T J, Jessell T M, Axel R. Science. 1989;244:1057–1062. doi: 10.1126/science.2727693. [DOI] [PubMed] [Google Scholar]
- 55.De Vivo M, Chen J, Codina J, Iyengar R. J Biol Chem. 1992;267:18263–18266. [PubMed] [Google Scholar]
- 56.De Vivo M, Iyengar R. J Biol Chem. 1994;269:19671–19674. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}