

Abstract
Lipoxin A4 (LXA4) and aspirin-triggered 15-epi-LXA4 (ATL) are emerging as endogenous braking signals for neutrophil-mediated tissue injury. Recent studies indicate that peroxynitrite (ONOO−) may function as an intracellular signal for the production of IL-8, a potent proinflammatory cytokine in human leukocytes. In this study, we evaluated the impact of the metabolically stable analogues of LXA4/ATL on lipopolysaccharide (LPS)-induced ONOO− formation and ONOO−-mediated IL-8 gene expression in human leukocytes. At nanomolar concentrations, LXA4 analogues markedly reduced LPS-stimulated superoxide formation, evoked increases in intracellular diamino-fluorescein fluorescence (an indicator of NO formation), and consequently reduced ONOO− formation in isolated neutrophils, as well as in neutrophils, monocytes, and lymphocytes, in whole blood. LXA4/ATL analogues attenuated nuclear accumulation of activator protein-1 and nuclear factor-κB in both polymorphonuclear and mononuclear leukocytes and inhibited IL-8 mRNA expression and IL-8 release by 50–65% in response to LPS. The LXA4 inhibitory responses were concentration dependent and were not shared by 15-deoxy-LXA4. None of the LXA4 analogues studied affected neutrophil survival, nor reversed the apoptosis delaying action of LPS in neutrophils. In addition, LXA4 analogues had no significant effect on exogenous ONOO−-induced IL-8 gene and protein expression. These findings suggest that by attenuating ONOO− formation, LXA4 and ATL can oppose ONOO− signaling in leukocytes and provide a rationale for using stable synthetic analogues as antiinflammatory compounds in vivo.
Lipoxins, formed by leukocytes during cell–cell interactions under a variety of conditions including inflammation, represent a unique class of lipid mediators with potent antiinflammatory actions (1). The cellular mechanisms by which they reduce inflammation are of interest. Native lipoxin A4 (LXA4) and its stable analogues bind to a G protein-coupled receptor, and inhibit recruitment of neutrophils by attenuating their chemotaxis, adhesion, and transmigration across vascular endothelial and epithelial cells (for a review, see ref. 1), and by diminishing chemokine production in intestinal mucosa (2) and fibroblasts (3). In addition, LXA4 may further facilitate resolution of inflammation by inhibiting superoxide formation (4), by nonphlogistic activation of monocytes and macrophages (5, 6) and by inhibiting colonocyte apoptosis (2). Intriguingly, aspirin can also trigger the transcellular biosynthesis of a series of 15-epimer (aspirin-triggered 15-epi-LXA4, ATL), which share many antiinflammatory activities with the native LXA4 (7). Thus, LXA4 and ATL appear to serve as endogenous “stop signals” for neutrophil-mediated tissue injury (1, 8).
Enhanced production of peroxynitrite (ONOO−), formed in the reaction of nitric oxide (NO) with superoxide anion (O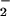 ), is a hallmark of a variety of inflammatory pathologies (9, 10). The cytotoxic effects of ONOO− have been well documented. Recent results suggest a role for ONOO− in the regulation of intracellular signaling pathways that modulate inflammatory reactions by leukocytes. In particular, ONOO− activates ERK in neutrophils, leading to up-regulation of surface expression of CD11b/CD18 and increased polymorphonuclear leukocytes (PMN) adhesion to endothelial cells (11). Scavenging of ONOO− reduces the influx of leukocytes into inflamed tissues, and prevents the development of tissue injury (12, 13). ONOO− functions as an intracellular messenger to mediate IL-8 gene expression in lipopolysaccharide (LPS)-, TNF-α-, or IL-1β-stimulated human leukocytes (14, 15) through activation of the transcription factors nuclear factor-κB (NF-κB) and activator protein-1 (AP-1) (15, 16). IL-8 plays a pivotal role in recruitment and activation of neutrophils (17) and monocytes (18) in various experimental models of inflammation (19, 20). Agents that modulate ONOO− formation and subsequent IL-8 production may, therefore, possess potent antiinflammatory properties.
), is a hallmark of a variety of inflammatory pathologies (9, 10). The cytotoxic effects of ONOO− have been well documented. Recent results suggest a role for ONOO− in the regulation of intracellular signaling pathways that modulate inflammatory reactions by leukocytes. In particular, ONOO− activates ERK in neutrophils, leading to up-regulation of surface expression of CD11b/CD18 and increased polymorphonuclear leukocytes (PMN) adhesion to endothelial cells (11). Scavenging of ONOO− reduces the influx of leukocytes into inflamed tissues, and prevents the development of tissue injury (12, 13). ONOO− functions as an intracellular messenger to mediate IL-8 gene expression in lipopolysaccharide (LPS)-, TNF-α-, or IL-1β-stimulated human leukocytes (14, 15) through activation of the transcription factors nuclear factor-κB (NF-κB) and activator protein-1 (AP-1) (15, 16). IL-8 plays a pivotal role in recruitment and activation of neutrophils (17) and monocytes (18) in various experimental models of inflammation (19, 20). Agents that modulate ONOO− formation and subsequent IL-8 production may, therefore, possess potent antiinflammatory properties.
In the present experiments, we studied the impact of ATL and LXA4 on LPS-stimulated ONOO− formation, NF-κB and AP-1 activation, and IL-8 gene expression in human leukocytes. We used two distinct metabolically stable analogues of ATL, 15(R/S)-methyl LXA4 and 15-epi-16-p-fluorophenoxy-LXA4, here denoted ATL1 and ATL2, respectively, and 16-phenoxy-LXA4, a stable analogue of LXA4. These analogues were designed to retain the bioactivity of native lipoxins and ATL by resisting their rapid enzymatic inactivation in tissues (21, 22).
Materials and Methods
Materials.
Stable analogs of LXA4 and 15-epi-LXA4 were prepared by total organic synthesis as in ref. 21. Structures were confirmed by reversed phase-HPLC, NMR, and mass spectral analysis. Stock solutions were stored at −80°C in 99% ethanol. Vehicle controls comprised dilutions of the solvent to the highest concentration of lipoxins used in the experiments (maximal ethanol concentration 0.01%). ONOO− was obtained from Alexis (San Diego).
Cell Stimulation.
Venous blood (anticoagulated with sodium heparin, 50 units/ml) was obtained from healthy volunteers who had denied taking any medication for at least 2 weeks. The Clinical Research Committee approved the experimental protocols. PMN were isolated as described (23). Whole blood aliquots or isolated PMN (5 × 106 cells per ml) in sterile microcentrifuge tubes were placed on a rotator, incubated for 15 min with lipoxins, and challenged with LPS (1 μg/ml) or ONOO− (80 μM) at 37°C in 5% CO2 atmosphere. At the designated time points, the plasma was harvested and stored at −20°C for later cytokine analysis. Cells were processed as described below.
ONOO− Formation.
For detection of intracellular ONOO− formation, NO-dependent fluorescence of rhodamine, an oxidation product of dihydrorhodamine 123 (DHR 123), was monitored as a marker of exposure of DHR 123 to ONOO− (24). The NO synthase blocker-inhibitable proportion of DHR 123 oxidation can be attributed to ONOO−, because ONOO− readily oxidizes DHR 123, whereas NO does not (24). DHR 123 (20 μM, Molecular Probes) was added to some samples during the last 60 min of incubation in the presence or absence of l-NAME (1 mM), an inhibitor of NO synthases. Following lysis of erythrocytes, PMN, monocytes, and lymphocytes were gated by their forward and side scatter characteristics, and fluorescence was analyzed by a flow cytometer (FACScan, Becton Dickinson) (14).
PMN extracts (prepared as in ref. 11) were analyzed for the presence of nitrotyrosine, a “fingerprint” of ONOO− (10), by Western blotting using a horseradish peroxidase-conjugated goat antinitrotyrosine antibody (NY20H-G1a, Academy Bio-Medical Co., Houston). Nitrated proteins were quantified by an enzyme immunoassay (Cayman Chemicals, Ann Arbor, MI), using nitrotyrosine as standard. The detection limit of the assay was 2 ng/ml. Intra- and interassay coefficients of variation were typically <6%.
NO and Superoxide Production.
Intracellular formation of NO was monitored by flow cytometry following incorporation of diaminofluorescein diacetate (DAF, 5 μM, CLONTECH) into PMN in accordance with the supplier's protocol. Superoxide production was determined as superoxide dismutase-inhibitable reduction of ferricytochrome c (23).
IL-8 Protein and RNase Protection Assay.
Plasma concentrations of IL-8 were determined by a selective ELISA (BioSource International, Camarillo, CA). For RNase protection assays, 50 μl of blood was mixed with an equal volume of lysis/denaturation solution (Ambion, Austin, TX). Labeled probes for human IL-8 and GAPDH were synthesized and the assay was performed with the Direct Protect kit (Ambion) as described (14).
NF-κB and AP-1.
Intranuclear, DNA bound NF-κB/p65 and AP-1/c-Fos were measured with a flow cytometric assay (15) and were used as an estimate of NF-κB and AP-1 activity in the cell, respectively. This assay allows simultaneous detection of DNA-bound transcription factors in PMN and mononuclear leukocyte populations in whole blood. Leukocyte nuclei were prepared using the Cycletest Plus DNA reagent kit (Becton Dickinson), first stained with rabbit polyclonal anti-human NF-κB/p65 or c-Fos antibodies, or with normal rabbit IgG (to assess nonspecific binding of IgG to nuclei) and then with FITC-conjugated anti-rabbit IgG antibody (all from Santa Cruz Biotechnology) and propidium iodide. Singlet cell nuclei were gated using the doublet-discrimination module and fluorescence intensity for both PMN and mononuclear cell nuclei was analyzed with a FACScan flow cytometer using the LYSIS II and CELLFIT software.
Neutrophil Viability and Apoptosis.
PMN viability was assessed by flow cytometry immediately after staining with propidium iodide (0.5 μg/ml). The binding of phycoerythrin-labeled annexin V was used as a sensitive marker of PMN apoptosis (25).
Statistical Analysis.
Results are expressed as means ± SEM. Statistical comparisons were made by ANOVA using ranks (Kruskal–Wallis test) followed by Dunn's multiple contrast hypothesis tests to identify differences between various treatments. Values of P < 0.05 were considered significant.
Results
LXA4/ATL Analogues Inhibit LPS-Induced IL-8 Production and IL-8 mRNA Expression.
Incubation of human whole blood with LPS evoked increases in IL-8 production that were markedly reduced by LXA4 analogues in a concentration-dependent manner (EC50, 16–26 nM) assessed at 4 and 24 h post-LPS administration (Fig. 1). Apparent maximum inhibition was achieved at ≈500 nM with 16-phenoxy-LXA4, ATL1, and ATL2 being virtually equally potent inhibitors, whereas 15-deoxy-LXA4 was without effect (Fig. 1). None of the LXA4 analogues by themselves affected baseline IL-8 production (data not shown).
Figure 1.

LXA4 analogues attenuate IL-8 production. Human whole blood samples were incubated with vehicle (unstimulated) or 16-phenoxy-LXA4, ATL1, ATL2, or 15-deoxy-LXA4 for 15 min and then challenged with 1 μg/ml LPS at 37°C for 4 h (A) or 24 h (B). (Left) Concentration-dependent inhibition of LPS-stimulated IL-8 production by 16-phenoxy-LXA4. (Right) Effects of LXA4/ATL analogues (all at 500 nM). Values are the mean ± SEM for six experiments with different donor cell preparations. *, P < 0.05; **, P < 0.01 (vs. LPS).
Next, we examined whether LXA4/ATL actions involved pre- or posttranscriptional events. Results shown in Fig. 2 confirmed those obtained for protein synthesis, with LPS inducing an approximately 2-fold increase of IL-8 mRNA that was attenuated by ATL1, ATL2, and 16-phenoxy-LXA4, but not by 15-deoxy-LXA4. ONOO− (80 μM)-induced increases in IL-8 production and IL-8 mRNA were only slightly suppressed in the presence of LXA4/ATL analogues (P > 0.1; Fig. 3).
Figure 2.

LXA4 analogues inhibit LPS-stimulated IL-8 mRNA expression. Blood samples were incubated with LXA4 analogues for 15 min then challenged with LPS (1 μg/ml) for 4 h at 37°C. (A) Representative RNase protection assay using probes for IL-8 and GAPDH. (B) Densitometric analysis of autoradiographs of the samples probed for IL-8 and GAPDH. The IL-8 results were adjusted based on the GAPDH values to represent equivalent RNA loading in each lane, and the results were then expressed as a percentage of the band obtained from blood incubated with vehicle (control). The results represent means ± SEM of blots from five experiments with different blood donors. *, P < 0.05; **, P < 0.01 (vs. LPS).
Figure 3.

Lack of effect of LXA4 analogues on ONOO−-induced IL-8 protein and mRNA expression. Blood samples were incubated with LXA4 analogues (500 nM) for 15 min, and then challenged with ONOO− (80 μM) for 4 h at 37°C. (A) Plasma levels of IL-8. (B) Representative RNase protection assay using probes for IL-8 and GAPDH. (C) Densitometry analysis of autoradiographs of the samples probed for IL-8 and GAPDH. The results represent means ± SEM for four experiments with different blood donors. *, P < 0.05 (vs. untreated).
Inhibition of ONOO− Formation.
We assessed intracellular rhodamine fluorescence at 4 h post-LPS administration when the highest level of NO-dependent DHR 123 oxidation can be detected (14). LPS evoked 4- to 6-fold increases in rhodamine fluorescence in PMN, monocytes, and lymphocytes, which were prevented by ATL1 in a concentration-dependent fashion (Fig. 4A). Similar inhibition was obtained with ATL2 and 16-phenoxy-LXA4, but not with 15-deoxy-LXA4 (Fig. 4B). LPS-evoked increases in rhodamine fluorescence were also reduced by l-NAME (Fig. 4B), indicating that a large portion of DHR 123 oxidation was due to ONOO− formation (14, 24). No additive inhibition was observed with a combination of LXA4 analogues and l-NAME (Fig. 4B). Furthermore, addition of the NO donor spermine NONOate (0.5 mM) to l-NAME-treated PMN restored DHR 123 oxidation [rhodamine fluorescence in relative fluorescence units (RFU); control, 13 ± 1; LPS, 56 ± 6; l-NAME+LPS, 25 ± 2; spermine NONOate+l-NAME+LPS, 54 ± 5; n = 5, P > 0.1 compared with LPS].
Figure 4.

LXA4 analogues inhibit LPS-induced oxidation of DHR 123 to rhodamine in human leukocytes. Blood samples were preincubated with vehicle or LXA4 analogues in the absence or presence of l-NAME (1 mM) as indicated, and then challenged with LPS at 37°C for 4 h. DHR 123 (20 μM) was added during the last 60 min of the incubation period. Intracellular rhodamine fluorescence is expressed as RFU. (A) Concentration-dependent effects of ATL1. (B) Comparison of the effects of LXA4 analogues (all at 500 nM) in the absence or presence of l-NAME. Values are means ± SEM (n = 5). *, P < 0.05; **, P < 0.01 (vs. LPS).
LPS gave increases, as expected, in both O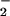 and NO production with isolated PMN (Fig. 5). LXA4 analogues, with the exception of 15-deoxy-LXA4, attenuated LPS-stimulated O
and NO production with isolated PMN (Fig. 5). LXA4 analogues, with the exception of 15-deoxy-LXA4, attenuated LPS-stimulated O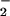 generation (Fig. 5). LPS-induced increases in the fluorescence of the NO-sensitive dye DAF were significantly higher in the presence of ATL1, ATL2, or 16-phenoxy-LXA4 (Fig. 5). These analogues reduced LPS-induced, NO-dependent DHR 123 oxidation parallel with inhibition of O
generation (Fig. 5). LPS-induced increases in the fluorescence of the NO-sensitive dye DAF were significantly higher in the presence of ATL1, ATL2, or 16-phenoxy-LXA4 (Fig. 5). These analogues reduced LPS-induced, NO-dependent DHR 123 oxidation parallel with inhibition of O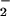 generation (Fig. 5).
generation (Fig. 5).
Figure 5.

Effects of LXA4 analogues on O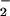 , NO, and ONOO− formation by LPS-stimulated isolated neutrophils. PMN were incubated with vehicle (control) or one of the LXA4 analogues (all at 500 nM) for 15 min and then with LPS (1 μg/ml) for 4 h. Superoxide formation was measured as reduction of ferricytochrome c in the culture medium. Intracellular NO formation was monitored using diaminofluorescein (DAF) and is expressed as RFU. DAF fluorescence was reduced 98% by l-NAME. ONOO− formation was assessed as l-NAME-inhibitable oxidation of DHR 123 to rhodamine and is expressed as RFU. Values are means ± SEM (n = 5–8). *, P < 0.05; **, P < 0.01 (vs. LPS).
, NO, and ONOO− formation by LPS-stimulated isolated neutrophils. PMN were incubated with vehicle (control) or one of the LXA4 analogues (all at 500 nM) for 15 min and then with LPS (1 μg/ml) for 4 h. Superoxide formation was measured as reduction of ferricytochrome c in the culture medium. Intracellular NO formation was monitored using diaminofluorescein (DAF) and is expressed as RFU. DAF fluorescence was reduced 98% by l-NAME. ONOO− formation was assessed as l-NAME-inhibitable oxidation of DHR 123 to rhodamine and is expressed as RFU. Values are means ± SEM (n = 5–8). *, P < 0.05; **, P < 0.01 (vs. LPS).
To provide further evidence for ONOO− formation, we quantified PMN nitrotyrosine content. PMN exposed to LPS contained significantly higher amounts of nitrotyrosine than unchallenged cells (Fig. 6), and the increases were comparable to those evoked by 80 μM ONOO−. Prior treatment of PMN with ATL1, ATL2, or 16-phenoxy-LXA4, but not with 15-deoxy-LXA4, resulted in marked reductions in LPS-induced tyrosine nitration (Fig. 6A).
Figure 6.

Nitrotyrosine content in neutrophils. (A) Isolated PMN were incubated with vehicle (control) or one of the LXA4 analogues for 15 min and then with LPS (1 μg/ml) for 4 h at 37°C. Cells were lysed and nitrotyrosine content was determined by an enzyme immunoassay. Values are means ± SEM (n = 7). *, P < 0.05; **, P < 0.01 (vs. LPS). (B) Nitrotyrosine Western blots from neutrophil extracts. Note the increased tyrosine nitration of several proteins by LPS and inhibition with LXA4 analogues. Tyrosine nitration by ONOO− (80 μM) treatment is shown as a positive control. Blot is representative of five separate experiments.
Western blotting of proteins in lysates of LPS-treated PMN revealed the appearance of nitrotyrosine-positive staining of proteins with 16, 20, 26, 28–30, 36, and 48 kDa molecular mass, corresponding to those nitrated by exogenous ONOO− (Fig. 6B). LPS-induced increases in tyrosine nitration were attenuated by ATL1, ATL2, and 16-phenoxy-LXA4, but not by 15-deoxy-LXA4 (Fig. 6B). Immunospecificity of the primary antibody for nitrotyrosine was confirmed when immunoreactivity of PMN extracts was abolished by absorption of antinitrotyrosine activity with soluble 10 mM nitrotyrosine, but was unaffected by 10 mM aminotyrosine (data not shown).
Inhibition of LPS-Stimulated Nuclear Accumulation of AP-1 and NF-κB.
LPS, as expected, mobilized NF-κB/p65 and c-Fos to the nucleus (i.e., the increased fluorescence represents increased amounts of NF-κB or AP-1 bound to DNA), and this mobilization was attenuated by ATL1 (Fig. 7 A and B). Immunostaining was completely blocked by preincubation of the antibodies with the appropriate blocking peptides. Stimulation of leukocytes in the presence of ATL1, ATL2, or 16-phenoxy-LXA4 gave quantitatively similar results with respect to the extent of inhibition of LPS-evoked nuclear accumulation of NF-κB/p65 and AP-1/c-Fos in both PMN and mononuclear cells (Fig. 7C). None of the LXA4 analogues affected immunostaining of nuclei for NF-κB/p65 and c-Fos in unchallenged cells (data not shown).
Figure 7.

LXA4 analogues inhibit nuclear accumulation of NF-κB/p65 and AP-1/c-Fos in PMN and mononuclear leukocytes. Whole blood samples were incubated with vehicle (control) or one of the LXA4 analogues (500 nM) for 15 min, and then with LPS (1 μg/ml) for 30 min at 37°C. Leukocyte nuclei were prepared for immunostaining as described in Materials and Methods. (A) Acquisition of leukocyte nuclei. (B) Representative fluorescence histograms of singlet nuclei of PMN after staining for NF-κB/p65 (Left) or c-Fos (Right). Also shown is the staining of nuclei prepared from blood incubated with vehicle as controls (curves labeled C) and staining with normal rabbit IgG followed by FITC-labeled anti-rabbit IgG antibody (curves labeled IgG). As a negative control, antibodies were preincubated for 2 h with the appropriate blocking peptide (curves labeled BP) before addition to nuclei. (C) Comparison of the inhibitory effects of LXA4 analogues (all at 500 nM). The results are means ± SEM of five to six experiments with different blood donors. *, P < 0.05; **, P < 0.01 (vs. LPS).
LXA4 Analogues and Neutrophil Viability and Apoptosis.
Because increased NO and ONOO− formation may be associated with changes in cell survival (10), we studied the impact of LXA4 analogues on neutrophil survival. Consistent with previous studies (26), after 24 h in vitro, LPS increased the percentage of viable PMN and reduced the percentage of annexin V-positive cells. However, none of the LXA4 analogues tested significantly affected the ability of LPS to prolong neutrophil survival and delay apoptosis (Fig. 8).
Figure 8.

Lack of effect of LXA4 analogues on viability and apoptosis of neutrophils. PMN were maintained in suspension culture at 37°C with and without LPS (1 μg/ml) in the absence and presence of ATL1, ATL2, and 15-deoxy-LXA4 (all at 500 nM). After 24 h of incubation, aliquots of neutrophils were stained with propidium iodide or phycoerythrin-labeled annexin V. Data are reported as percentage of viable and apoptotic cells, respectively, and represent means ± SEM (n = 4). *, P < 0.05 (vs. untreated).
Discussion
The present results provide evidence for a mechanism by which LXA4 and ATL may affect the inflammatory process by inhibiting ONOO− formation by leukocytes. This is associated with attenuation of nuclear accumulation of transcription factors NF-κB and AP-1, and subsequent reduction of IL-8 gene expression and production. Because recent studies have increasingly pointed to the pivotal role of IL-8 in directing leukocyte traffic into inflamed areas, our observations bear directly on the mechanism for the antiinflammatory effects of LXA4 and, in particular, the aspirin-triggered epimer, ATL.
Bacterial LPS is one of the most potent stimuli for IL-8 expression in most cell types, including human leukocytes (27). In addition to the available in vitro data (2, 3, 28), here we have shown that incubation of human whole blood samples with ATL1, ATL2, and 16-phenoxy-LXA4 at nanomolar concentrations results in concentration-dependent attenuation of LPS-induced IL-8 release. The higher concentrations required in whole blood to effectively inhibit IL-8 production than in isolated cells might reflect interactions of LXA4 analogues with serum components such as albumin. Nevertheless, our results demonstrate that LXA4/ATL analogues are potent inhibitors of key responses of leukocytes in amplifying the inflammatory response in whole blood and in a culture milieu that is biologically relevant. Thus, our results enhance earlier findings with intestinal epithelial cells (2, 28) and fibroblasts (3) and confirm the capability of LXA4 analogues to regulate proinflammatory cytokine production in leukocytes. Furthermore, these LXA4/ATL analogues continue to demonstrate bioactivity for at least 24 h after their bolus addition to whole blood samples.
The present results indicate that metabolically stable LXA4/ATL analogues markedly attenuate ONOO− formation by human leukocytes. To monitor intracellular ONOO− formation, we monitored NO-dependent oxidation of DHR 123 to rhodamine and quantified nitration of protein tyrosine residues, a specific “fingerprint” of ONOO− (10), and obtained consistent results with these two assays. A significant portion of rhodamine fluorescence in LPS-stimulated leukocytes can be attributed to ONOO−, because it depends on NO-related species—it can be inhibited by l-NAME—whereas NO per se does not oxidize DHR 123 (24). Further, the NO donor spermine NONOate restored DHR 123 oxidation in l-NAME-treated PMN, indicating that the effect of l-NAME in the DHR 123 assay was not due to inhibition of assembly of the NADPH oxidase. The lack of additive inhibitory actions of LXA4 analogues and l-NAME on DHR 123 oxidation would suggest that these compounds inhibited the same reaction—i.e., the ONOO−-dependent oxidation. The nitration reactions have been attributed to the interaction of •NO with O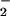 at a rate constant of 6.7 × 109 M−1⋅s−1 at pH 7.4 to form ONOO− (10) and have been observed in circumstances of LPS-induced inflammation. There is evidence that the nitration yield may be increased via the formation of the nitrosoperoxycarbonate anion from physiologic concentrations of CO2 and bicarbonate, thereby enhancing the reactivity of ONOO− (29). Although nitrotyrosine formation is considered as a specific “fingerprint” of ONOO− (10), some studies proposed additional pathways of tyrosine nitration (30), indicating that nitrotyrosine may rather serve as an indicator of “reactive nitrogen species” (31).
at a rate constant of 6.7 × 109 M−1⋅s−1 at pH 7.4 to form ONOO− (10) and have been observed in circumstances of LPS-induced inflammation. There is evidence that the nitration yield may be increased via the formation of the nitrosoperoxycarbonate anion from physiologic concentrations of CO2 and bicarbonate, thereby enhancing the reactivity of ONOO− (29). Although nitrotyrosine formation is considered as a specific “fingerprint” of ONOO− (10), some studies proposed additional pathways of tyrosine nitration (30), indicating that nitrotyrosine may rather serve as an indicator of “reactive nitrogen species” (31).
The inhibitory action of LXA4 is most likely due to inhibition of O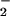 production. Levy et al. (4) have reported that LXA4 reduces TNF-α-induced O
production. Levy et al. (4) have reported that LXA4 reduces TNF-α-induced O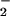 formation in human PMN. Activation of the LXA4 receptor leads to agonist-induced accumulation of the polyisoprenoid presqualene diphosphate, which inhibits phospholipase D directly, and thereby blocks assembly of the NADPH oxidase (32). LXA4, when added to PMN, does not alter NADPH utilization by the NADPH oxidase. Our results to date, however, do not exclude the possibility that lipoxins also have an impact on cytoskeleton assembly of the NADPH oxidase. In our experiments, inhibition of O
formation in human PMN. Activation of the LXA4 receptor leads to agonist-induced accumulation of the polyisoprenoid presqualene diphosphate, which inhibits phospholipase D directly, and thereby blocks assembly of the NADPH oxidase (32). LXA4, when added to PMN, does not alter NADPH utilization by the NADPH oxidase. Our results to date, however, do not exclude the possibility that lipoxins also have an impact on cytoskeleton assembly of the NADPH oxidase. In our experiments, inhibition of O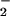 formation might have shifted the ratio of O
formation might have shifted the ratio of O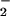 to NO, resulting in decreased ONOO− formation and consequently DHR 123 oxidation. Indeed, the highest yield of ONOO− formation (detected as 3-nitrotyrosine) can be obtained for similar fluxes of NO and O
to NO, resulting in decreased ONOO− formation and consequently DHR 123 oxidation. Indeed, the highest yield of ONOO− formation (detected as 3-nitrotyrosine) can be obtained for similar fluxes of NO and O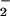 (33). Although a controversy exists as to whether ONOO− is capable of tyrosine nitration at physiological pH (reviewed in ref. 34), our results with exogenous ONOO− clearly indicate the existence of such reaction in human PMN. Intriguingly, immunoblots of tyrosine-nitrated proteins from LPS-challenged PMN resemble a pattern similar to those observed with exogenous ONOO−. Tyrosine nitration of these proteins was attenuated by both ATL1 and ATL2 parallel with decreases in the total amount of nitrotyrosine per milligram of protein. The identity and biological function of these proteins remain to be investigated. Also, the actions of LXA4 were unlikely to be due to an ONOO− scavenging effect, because neither 15-deoxy-LXA4 nor LXA4/ATL analogues affected significantly PMN responses to exogenous ONOO−.
(33). Although a controversy exists as to whether ONOO− is capable of tyrosine nitration at physiological pH (reviewed in ref. 34), our results with exogenous ONOO− clearly indicate the existence of such reaction in human PMN. Intriguingly, immunoblots of tyrosine-nitrated proteins from LPS-challenged PMN resemble a pattern similar to those observed with exogenous ONOO−. Tyrosine nitration of these proteins was attenuated by both ATL1 and ATL2 parallel with decreases in the total amount of nitrotyrosine per milligram of protein. The identity and biological function of these proteins remain to be investigated. Also, the actions of LXA4 were unlikely to be due to an ONOO− scavenging effect, because neither 15-deoxy-LXA4 nor LXA4/ATL analogues affected significantly PMN responses to exogenous ONOO−.
We were surprised to observe increased DAF fluorescence in the presence of LXA4 analogues plus LPS as compared with LPS alone. However, it is unlikely that such increases might mirror elevated NO formation, because LXA4 analogues by themselves did not enhance DAF fluorescence. Indeed, when NO is produced in relative excess to superoxide (i.e., unchanged NO synthesis and reduced superoxide generation) NO2 from ONOO− decomposition may react with NO to form more nitrosating intermediates, leading to increased DAF fluorescence. These competing reactions might represent important pathways by which the oxidative chemistry of NO, O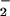 , and ONOO− may be modulated.
, and ONOO− may be modulated.
Consistent with previous reports, we detected NF-κB and AP-1 activation in response to LPS in both PMN and mononuclear leukocytes by using a flow-cytometric assay. Although this system permits simultaneous detection of nucleus-bound transcription factors in both PMN and mononuclear leukocytes without prior isolation of these cells, nuclei from monocytes and lymphocytes cannot be analyzed separately. Therefore, the quantities of transcription factors present in PMN and monocytes cannot be compared.
Despite their predominantly cytoplasmic localization in resting cells, low amounts of NF-κB/p65 and c-Fos DNA-binding activities can be observed in the nuclei of unstimulated PMN and mononuclear cells (refs. 35 and 36, and the present study), and this was not attributable to cytosolic contamination of the nuclei. Although the significance of these observations is unclear, constitutive nuclear NF-κB has been proposed to contribute to constitutive expression of transcripts encoding κB-dependent genes. Of interest, none of the LXA4 analogues affected the nuclear presence of NF-κB in unchallenged cells, indicating the existence of a mechanism distinct from that inducing NF-κB translocation after cell stimulation. On the other hand, LXA4/ATL analogues effectively suppressed LPS-evoked nuclear accumulation of NF-κB and AP-1. Because inhibition of NF-κB activation results in about 75% inhibition of IL-8 release, and exogenous ONOO− can increase nuclear binding of NF-κB and AP-1 in both PMN (15) and mononuclear cells (15, 16), it is likely that AP-1 may act in concert with NF-κB to induce IL-8 production. The mechanism by which ONOO− increases nuclear binding of NF-κB is not fully understood. The positive correlation of phosphorylation and nitration of tyrosine residues in IκB-α with NF-κB translocation (16) would suggest that LXA4/ATL analogues might have protected IκB-α from nitration by ONOO−, thereby preventing activation of NF-κB. Inhibition of NF-κB has been suggested to contribute considerably to the antiinflammatory actions of aspirin (37). Extending these observations, our results suggest another mechanism to inhibit NF-κB activation in leukocytes—i.e., through induction of 15-epi-LXA4.
PMN survival is contingent on rescue from apoptosis by signals from the environment. LPS can prolong PMN survival by delaying apoptosis (26). However, LXA4 analogues did not affect these actions of LPS, indicating that the signaling pathway that mediates the apoptosis delaying action of LPS is not sensitive to LXA4, nor does it involve ONOO−.
The present observations coupled with the inhibitory actions of LXA4 and ATL on the expression of leukocyte adhesion molecules (1, 7) may represent important components of the “stop signaling” for leukocyte trafficking into inflamed tissues. Decreased ONOO− formation is also consistent with a reduced cytotoxicity. The observations that LXA4 analogues are potent inhibitors of ONOO− formation and IL-8 production by human leukocytes are important additions to the bioactivity profile of these compounds and suggest a role for LXA4/ATL analogues as potential therapeutic modifiers of diseases associated with enhanced ONOO− formation.
Acknowledgments
This work was supported by grants to J.G.F. from the Canadian Institutes of Health Research (MOP-12573) and the Heart and Stroke Foundation of Canada, and a grant to N.A.P. and C.N.S. from the National Institutes of Health (PO1 DE13499).
Abbreviations
- LXA4
lipoxin A4 (5S,6R,15S-trihydorxy-7,9,13-trans-11-cis-eicosatetraenoic acid)
- ATL
aspirin-triggered 15-epi-LXA4
- LPS
lipopolysaccharide
- DHR 123
dihydrorhodamine 123
- DAF
diaminofluorescein diacetate
- RFU
relative fluorescence units
- PMN
polymorphonuclear leukocytes
- NF-κB
nuclear factor-κB
- AP-1
activator protein-1
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Serhan C N. Prostaglandins. 1997;53:107–137. doi: 10.1016/s0090-6980(97)00001-4. [DOI] [PubMed] [Google Scholar]
- 2.Goh J, Baird A W, O'Keane C, Watson R W G, Cottell D, Bernasconi G, Petasis N A, Godson C, Brady H R, MacMathuna P. J Immunol. 2001;167:2772–2780. doi: 10.4049/jimmunol.167.5.2772. [DOI] [PubMed] [Google Scholar]
- 3.Sodin-Semrl S, Taddeo B, Tseng D, Varga J, Fiore S. J Immunol. 2000;164:2660–2666. doi: 10.4049/jimmunol.164.5.2660. [DOI] [PubMed] [Google Scholar]
- 4.Levy B D, Fokin V V, Clark J M, Wakelam M J O, Petasis N A, Serhan C N. FASEB J. 1999;13:903–911. doi: 10.1096/fasebj.13.8.903. [DOI] [PubMed] [Google Scholar]
- 5.Serhan C N. Biochim Biophys Acta. 1994;1212:1–25. doi: 10.1016/0005-2760(94)90185-6. [DOI] [PubMed] [Google Scholar]
- 6.Godson C, Mitchell S, Harvey K, Petasis N A, Hogg N, Brady H R. J Immunol. 2000;164:1663–1667. doi: 10.4049/jimmunol.164.4.1663. [DOI] [PubMed] [Google Scholar]
- 7.Clària J, Serhan C N. Proc Natl Acad Sci USA. 1995;92:9475–9479. doi: 10.1073/pnas.92.21.9475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Godson C, Brady H R. Curr Opin Investig Drugs. 2000;1:380–385. [PubMed] [Google Scholar]
- 9.Beckman J S, Beckman T W, Chen J, Marshall P M, Freeman B M. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beckman J S, Koppenol W H. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 11.Zouki C, Zhang S L, Chan J S D, Filep J G. FASEB J. 2001;15:25–27. doi: 10.1096/fj.00-0521fje. [DOI] [PubMed] [Google Scholar]
- 12.Hooper D C, Scott G S, Zborek A, Mikheeva T, Kean R B, Koprowski H, Spitsin S V. FASEB J. 2000;14:691–698. doi: 10.1096/fasebj.14.5.691. [DOI] [PubMed] [Google Scholar]
- 13.Cuzzocrea S, Misko T P, Costantino G, Mazzon E, Micali A, Caputi A P, MacArthur H, Salvemini D. FASEB J. 2000;14:1061–1072. doi: 10.1096/fasebj.14.9.1061. [DOI] [PubMed] [Google Scholar]
- 14.Filep J G, Beauchamp M, Baron C, Paquette Y. J Immunol. 1998;161:5656–5662. [PubMed] [Google Scholar]
- 15.Zouki C, József L, Ouellet S, Paquette Y, Filep J G. J Leukocyte Biol. 2001;69:815–824. [PubMed] [Google Scholar]
- 16.Matat B M, Galinanes M. J Biol Chem. 2002;277:2330–2335. doi: 10.1074/jbc.M106393200. [DOI] [PubMed] [Google Scholar]
- 17.Luster A D. N Engl J Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- 18.Gerszten R E, Garcia-Zepeda E A, Lim Y C, Yoshida M, Ding H A, Gimbrone M J, Jr, Luster A D, Luscinskas F W, Rosenzweig A. Nature (London) 1999;398:718–723. doi: 10.1038/19546. [DOI] [PubMed] [Google Scholar]
- 19.Harada A, Sekido N, Akahoshi T, Wada T, Mukaida N, Matsushima K. J Leukocyte Biol. 1994;56:559–564. [PubMed] [Google Scholar]
- 20.Mulligan M S, Jones M L, Bolanowski M A, Baganoff M P, Deppeler C L, Meyers D M, Ryan U S, Ward P A. J Immunol. 1993;150:5585–5595. [PubMed] [Google Scholar]
- 21.Serhan C N, Maddox J F, Petasis N A, Akritopoulou-Zanze I, Papayianni A, Brady H R, Colgan S P, Madara J L. Biochemistry. 1995;34:14609–14615. doi: 10.1021/bi00044a041. [DOI] [PubMed] [Google Scholar]
- 22.Clish C B, O'Brien J A, Gronert K, Stahl G L, Petasis N A, Serhan C N. Proc Natl Acad Sci USA. 1999;96:8247–8252. doi: 10.1073/pnas.96.14.8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Filep J, Földes-Filep E. Life Sci. 1989;44:517–524. doi: 10.1016/0024-3205(89)90613-9. [DOI] [PubMed] [Google Scholar]
- 24.Kooy N W, Royall J A, Ischiropoulos H, Beckman J S. Free Radical Biol Med. 1994;16:149–156. doi: 10.1016/0891-5849(94)90138-4. [DOI] [PubMed] [Google Scholar]
- 25.Homburg C H E, de Haas M, von dem Borne A E G Kr, Verhoeven A J, Reutelingsperger C P M, Roos D. Blood. 1995;85:532–540. [PubMed] [Google Scholar]
- 26.Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A. Blood. 1992;80:2012–2020. [PubMed] [Google Scholar]
- 27.DeForge L, Kenney J S, Jones M L, Warren J S, Remick D G. J Immunol. 1992;148:2133–2141. [PubMed] [Google Scholar]
- 28.Gewirtz A T, McCormick B, Neish A S, Petasis N A, Gronert K, Serhan C N. J Clin Invest. 1998;101:1860–1869. doi: 10.1172/JCI1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gow A, Duran D, Thom S R, Ischiropoulos H. Arch Biochem Biophys. 1996;333:42–48. doi: 10.1006/abbi.1996.0362. [DOI] [PubMed] [Google Scholar]
- 30.Eiserich J P, Hristova M, Cross C E, Jones A D, Freeman B A, Halliwell B, van der Vliet A. Nature (London) 1998;391:393–397. doi: 10.1038/34923. [DOI] [PubMed] [Google Scholar]
- 31.Halliwell B. FEBS Lett. 1997;411:157–160. doi: 10.1016/s0014-5793(97)00469-9. [DOI] [PubMed] [Google Scholar]
- 32.Levy B D, Petasis N A, Serhan C N. Nature (London) 1997;389:985–990. doi: 10.1038/40180. [DOI] [PubMed] [Google Scholar]
- 33.Goldstein S, Czapski G, Lind J, Merényi G. J Biol Chem. 2000;275:3031–3036. doi: 10.1074/jbc.275.5.3031. [DOI] [PubMed] [Google Scholar]
- 34.Reiter C D, Teng R J, Beckman J S. J Biol Chem. 2000;275:32460–32466. doi: 10.1074/jbc.M910433199. [DOI] [PubMed] [Google Scholar]
- 35.McDonald P P, Bald A, Cassatella M A. Blood. 1997;89:3421–3433. [PubMed] [Google Scholar]
- 36.Griffin G E, Leung K, Folks T M, Kunkel S, Nabel G J. Nature (London) 1990;339:70–73. doi: 10.1038/339070a0. [DOI] [PubMed] [Google Scholar]
- 37.Kopp E, Gosh S. Science. 1994;265:956–959. doi: 10.1126/science.8052854. [DOI] [PubMed] [Google Scholar]