

Abstract
Dendritic cells (DC) are of central importance in the initiation of T cell-mediated adaptive immunity because these professional phagocytes internalize, process, and present microbial antigens to T lymphocytes. T lymphocytes have a pivotal role in controlling and clearing infection with intracellular pathogens through cytokine production. T lymphocytes also can mediate direct lysis of infected cells or activate B and T cells. In this article, we report that DC, when cocultured with Salmonella, fail to efficiently stimulate T cells for proliferation. We show that the failure of T lymphocytes to respond to Salmonella-infected DC is not simply due to Salmonella-induced programmed DC death or interference with up-regulation of costimulatory molecules CD80 and CD86. We cocultured bacteria with purified T lymphocytes, and we demonstrate here that Salmonella have a direct, contact-dependent inhibitory effect on the T cells, even in the absence of DC. This direct, Salmonella-induced inhibitory effect reduces the ability of T cells to proliferate and produce cytokines in response to stimulation and appears to require live bacteria. Cumulatively, these results are evidence that Salmonella may interfere with the development of acquired immunity, providing insights into the complex nature of this hostpathogen interaction.
Keywords: bacterial interference, adaptive immunity
Protection against invading microbes is coordinately regulated by innate and adaptive host defense mechanisms (1, 2). However, many microbial pathogens appear to have evolved successfully to combat, exploit, or evade host immunity (3). For example, naïve mice that are challenged with the enteric bacterium Salmonella enterica serovar Typhimurium fail to control and clear infection and succumb to an acute typhoid-like systemic illness (4). In contrast, mice previously immunized with a live-attenuated Salmonella vaccine strain are protected from lethal challenge (5), demonstrating that acquired resistance against Salmonella can develop in these hosts. It takes up to 6 wk to eradicate the vaccine strain and 60-90 days to acquire immunological memory (5). These observations suggest that adaptive immunity against Salmonella, which critically depends on the successful engagement of T lymphocytes (5), develops rather slowly.
A critical step in the development of acquired resistance to intracellular bacterial pathogens is the stimulation of T lymphocytes by infected cells. The following two major subsets of T cells exist among the total population of T lymphocytes: CD4+ T cells, which recognize exogenous antigens and typically are stimulated during infection with vacuolar pathogens, and CD8+ T cells, which recognize cytosolic antigens and are often required to resolve infection with pathogens that escape from the vacuole and enter the host cell cytoplasm. Intriguingly, optimal protection against Salmonella, which reside in a membrane-bound compartment within infected cells, depends on both CD4+ and CD8+ T lymphocytes (5).
To monitor the priming of T lymphocytes during Salmonella infection, we inserted a well characterized T cell epitope into a secreted Salmonella protein. Our results demonstrate that epitope-specific T cells are not primed in mice infected with epitope-expressing bacteria. We also studied the stimulation of T lymphocytes by Salmonella-infected dendritic cells (DC) in vitro and found that these infected DC do not efficiently stimulate T cells for proliferation. In further characterizing this phenotype, we discovered that Salmonella directly affect the ability of T lymphocytes to proliferate in response to stimulus.
Materials and Methods
Bacterial Strains and Growth Conditions. Bacteria were grown as described in ref. 6. Heat-killed bacteria were obtained by incubating bacterial suspensions at 65°C for 20 min. Escherichia coli DH5α; WT S. enterica serovar Typhimurium ATCC 14028; an isogenic virulence plasmid-cured derivative of ATCC 14028; and a set of isogenic aroA, invA, spiB, phoP, and phoPc mutant strains have been described (7-12) or were generated by using P22HTint transduction. sti-deficient Salmonella were a generous gift from John Mekalanos (Harvard Medical School).
To generate a Salmonella strain expressing chromosomally encoded SptP-OVA, a 1,650-bp sptP fragment was amplified from the Salmonella chromosome by PCR using the primers 5′-ACCGCTCGAGCTGCAGGAATATGCTAA-3′ and 5′-CTTAGGATCCTATGTTTTTATCAGCTTGC-3′ and cloned into a suicide plasmid. A double-stranded oligonucleotide encoding the ovalbumin-(257-264) octapeptide (OVA257-264) (5′-CTAGCATAATTAACTTCGAAAAGCTTG-3′) was then inserted into the unique PvuII site of sptP, and the resulting construct was returned to the Salmonella chromosome through biparental mating and homologous recombination.
DC Culture. DC were cultured from C57BL/6J mice (The Jackson Laboratory) or caspase-1-/- mice (13) as described in ref. 6. To prepare cells for infection, cultures were grown overnight in antibiotic-free media. CD11c+ DC were purified by using CD11c-conjugated MACS microbeads and magnetic separation columns (Miltenyi Biotec, Auburn, CA) and, where indicated, labeled exogenously with synthetic OVA257-264 (5 nM) to ensure DC surface display of peptide-MHC-I complex and eliminate dependence on intracellular antigen-processing pathways. Cells were washed three times, infected as described in ref. 6, and either analyzed by flow cytometry or cocultured with fluorescently labeled OT-I T cells (see below). Where noted, DC maturation was induced with LPS as described previously (6).
T Cell Proliferation Assay. T cells were isolated from 8- to 12-wk-old OT-I mice or C57BL/6J mice (both from The Jackson Laboratory), purified by using CD90.2-conjugated MACS microbeads and magnetic separation columns (Miltenyi Biotec), and labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE) as described (14). T cells were then either added to CD11c+ DC or infected with bacteria. When added to CD11c+ DC, T cells were resuspended in RP-10 [RPMI medium 1640 (Invitrogen)/10% FBS/l-glutamine/Hepes/50 μM 2-mercaptoethanol], supplemented with 2% penicillin/streptomycin and 50 μg/ml gentamicin to kill all bacteria. In preparation for coculture with T lymphocytes, bacteria were washed extensively in RP-10. Next, T cells were resuspended in RP-10 and infected for 3 h at a multiplicity of infection (moi) of 12.5 (unless indicated otherwise). Cells were then washed once; resuspended in RP-10 supplemented with 1 μg/ml anti-CD28, 2% penicillin/streptomycin, and 50 μg/ml gentamicin; and reseeded in wells that had been coated previously with 2.5 μg/ml anti-CD3ε. After 3 days of incubation, cells were stained with anti-TCRβ (BD Biosciences), a pan-T cell marker, and proliferation of TCRβ+ cells (including both CD4+ and CD8+ T lymphocytes) was then monitored by flow cytometry using a FACSCalibur flow cytometer (BD Biosciences).
Immunization Studies and Culture of OVA257-264-Specific T Cells. Groups of four 8-wk-old female C57BL/6J mice were immunized orally with 109 colony-forming units of Salmonella or i.p. with 106 plaque-forming units of Vac-OVA. Splenocytes were obtained 7 wk after immunization and stimulated for 5 d on irradiated syngeneic spleen cells (2,000 rad) and pulsed with OVA257-264. These cells were then tested in a 51Cr-release assay. T cell cultures were maintained by weekly restimulation, first in RP-10 and after 2 wk in RP-10 supplemented with IL-2.
The 51Cr-Release Assay. EL4 or 1308.1 targets were incubated for 1 h with 100 μCi (1 Ci = 37 GBq) of sodium [51Cr]chromate and then either coated with OVA257-264 or infected with Salmonella at an moi of 100. Targets were then washed and seeded at 5 × 104 cells per well (96-well plate). Serial dilutions of T cell cultures were then added, and cell suspensions were incubated for 4 h. Cytotoxic activity of T cells was quantified by measuring 51Cr release from targets by using a Wallac (Gaithersburg, MD) 1470 Wizard γ counter. The percentage of cytotoxicity was calculated by using the following formula: 100% × [(E - S)/(M - S)]. E and S are amounts of 51C released from experimental and uninfected (UI) cells, respectively, whereas M is the amount of 51C released from UI cells after lysis with 1% Triton X-100.
Results and Discussion
SptP, a Substrate for the Salmonella Pathogenicity Island (SPI)-1-Encoded Type III Secretion System (TTSS), Gains Access to the MHC-I Pathway of Antigen Processing and Presentation to CD8+ T Cells. CD8+ T cells appear to contribute to the control and clearance of infection with Salmonella (5), and they are likely to recognize Salmonella antigens that are introduced into the host-cell cytosol. Upon contact with host cells, Salmonella induce the expression of two TTSSs, sophisticated protein export machineries that facilitate direct translocation of bacterial proteins into the cytoplasm of host cells (15). Studies have shown that CD8+ T cells can recognize peptides embedded within type III secreted effector proteins (16) and that TTSS can be exploited for delivery of heterologous vaccine antigens into the MHC-I pathway of antigen processing and presentation to CD8+ T cells (17, 18). For example, recent evidence indicates that mice immunized with a live-attenuated Salmonella vaccine strain expressing viral antigens fused to SptP, a SPI-1-encoded type III secreted effector protein, are protected against viral challenge (17). The use of Salmonella to deliver heterologous vaccine antigens via its TTSS led us to investigate whether CD8+ T cells directed against type III secreted proteins can contribute to controlling and clearing Salmonella infection. We constructed a Salmonella strain expressing a chimeric form of SptP carrying the well characterized CD8+ T cell epitope OVA257-264. Upon returning the sptP::OVA257-264 construct to the chromosome of WT Salmonella, virulence of the resulting strain and single-copy expression of the fusion protein were confirmed (data not shown). Next, we demonstrated by using a 51Cr-release assay that OVA257-264-specific T cells recognize and lyse target cells infected with recombinant Salmonella expressing SptP-OVA but not cells infected with WT bacteria (Fig. 1A).
Fig. 1.

OVA-specific CD8+ T cells are not primed during infection with recombinant Salmonella expressing SptP-OVA. E:T ratio, ratio of effector to target cells. (A) OVA-specific T cells lyse 1308.1 epithelial cells infected with recombinant Salmonella expressing SptP-OVA (Salmonella sptP::OVA, □), but not cells infected with WT bacteria (Salmonella WT, ▪). (B and C) Robust lytic activity was detected in T cell cultures obtained from mice immunized with vaccinia virus expressing OVA (Vac-OVA) but not in cultures obtained from mice vaccinated with live-attenuated Salmonella expressing WT SptP (Salmonella aroA). (D) No cytolytic activity was detected in T cell cultures obtained from mice immunized with a Salmonella strain expressing chromosomally encoded SptP-OVA (Salmonella aroA sptP::OVA). (B-D) Squares indicate untreated EL4 target cells (□) or EL4 cells coated with OVA peptide (▪). Data in A are representative of three independent experiments, and data in B-D are representative of groups of four mice.
OVA257-264-Specific CD8+ T Lymphocytes Are Not Stimulated During Infection with Recombinant Salmonella Expressing SptP-OVA. We then examined the ability of SptP-OVA-expressing Salmonella to stimulate OVA257-264-specific CD8+ T cells in vivo. Not surprisingly, mice infected with virulent Salmonella expressing SptP-OVA rapidly succumbed to infection such that stimulation of OVA257-264-specific CD8+ T cells could not be monitored (data not shown). Instead, mice were immunized orally with an attenuated Salmonella strain expressing chromosomally encoded SptP-OVA (Salmonella aroA sptP::OVA257-264). Splenocytes from vaccinated animals were obtained 7 wk after immunization and stimulated on irradiated syngeneic spleen cells pulsed with commercially synthesized OVA257-264. After 5 days of in vitro culture, these cells were assayed for their ability to lyse OVA257- 264-coated EL4 target cells in a 51Cr-release assay. As predicted, robust lytic activity was detected in T cell cultures that were obtained from control mice immunized with vaccinia virus expressing OVA (Vac-OVA) but not in cultures obtained from mice vaccinated with Salmonella aroA expressing WT SptP (Fig. 1 B and C). Surprisingly, no CD8+ T cell-mediated lysis of peptide-pulsed targets was detected in T cell cultures obtained from mice immunized with Salmonella aroA sptP::OVA257-264 (Fig. 1D), suggesting that OVA257-264-specific CD8+ T lymphocytes were not primed during infection. These results differ from the vaccine study in which similar SptP fusion proteins did stimulate CD8+ T cells (17), and they may reflect differences in the level of chimeric SptP protein resulting from chromosomal (this study) vs. plasmid (17) expression. Another explanation for the unresponsiveness of T cells to the OVA tag in vivo is that temporal and spatial expression of SptP during Salmonella infection may not result in detectable T cell stimulation. Rather than proceeding with an exhaustive investigation as to why SptP-OVA-expressing Salmonella did not stimulate T cells or whether other type III secreted effector proteins, when tagged, might stimulate OVA-specific T cells, we explored the hypothesis that Salmonella actively interfere with the development of T cell-mediated immunity. Studies showing that Salmonella infection of mice is accompanied by immunosuppression (see refs. 19-21, ref. 22 and references therein, and ref. 23) support this hypothesis.
DC Infected with Salmonella Do Not Efficiently Stimulate T Cells for Proliferation. One of the hypotheses that may explain why OVA257-264-specific CD8+ T cells are not primed during infection with OVA257-264-expressing Salmonella is that Salmonella have evolved one or more mechanisms to interfere with the ability of professional phagocytes to process and present antigens to T lymphocytes. To determine whether Salmonella interfere with antigen presentation to T cells, we investigated whether Salmonella-infected DC were capable of stimulating naïve T lymphocytes for proliferation. Proliferation of T cells was monitored by flow cytometry using an assay based on dilution of the fluorescent vital dye CFSE during sequential rounds of cell division (14).
To obtain T cells of a single specificity, CD90.2-expressing lymphocytes were isolated and purified from OT-I mice. Most of the T lymphocytes in these TCR transgenic mice are CD8+ and specific for OVA257-264 in the context of H-2Kb (24). Purified T cells were subsequently labeled with CFSE and added to H-2Kb-restricted DC cultured from C57BL/6J mice. As expected, no T cell proliferation was observed when naïve, CFSE-labeled OT-I T lymphocytes were cocultured with untreated DC (Fig. 2A). In contrast, substantial T cell clonal expansion was detected when fluorescently labeled OT-I T lymphocytes were added to UI DC pulsed with commercially synthesized OVA257-264 (Fig. 2B). Intriguingly, these peptide-coated DC, when cocultured with WT Salmonella, were no longer capable of stimulating OT-I T cells to proliferate (Fig. 2C), demonstrating that Salmonella can block peptide presentation or later steps required for T cell expansion.
Fig. 2.

Salmonella-infected DC do not efficiently stimulate T cells for proliferation. (A and B) Clonal expansion of OVA-specific CD8+ T cells depended on presentation of OVA peptide by DC. (C) However, when peptide-coated DC were cocultured with Salmonella (Stm), these cells were no longer capable of stimulating OVA-specific CD8+ T cells for proliferation. Data are representative of at least three independent experiments.
The Lack of T Cell Proliferation Is Not Simply Due to Salmonella-Induced, Caspase-1-Dependent Programmed DC Death. We published evidence demonstrating that Salmonella are capable of killing DC via a caspase-1-dependent mechanism and that sipB-deficient Salmonella, which are unable to express a fully functional SPI1-encoded TTSS, are not cytotoxic (6). On the basis of these findings, we hypothesized that Salmonella-induced death of the antigen-presenting cell may be the reason why T cells, when cocultured with Salmonella-infected DC, fail to proliferate.
To test this hypothesis, fluorescently labeled OVA257-264-specific CD8+ T cells were added to peptide-pulsed DC previously infected with nontoxic, sipB-deficient Salmonella (6). As shown in Fig. 3A, these DC remained unable to stimulate T cells for proliferation, suggesting that the failure of T cells to respond to Salmonella-infected DC is not simply due to Salmonella-induced DC death. Importantly, similar results were obtained when T lymphocytes were added to Salmonella-infected, OVA257-264-coated, caspase-1-deficient DC (Fig. 3B), which are resistant to Salmonella-induced cytotoxicity (6). These results are evidence that the lack of T cell clonal expansion is not simply due to Salmonella-induced DC death and suggest that the failure of T lymphocytes to proliferate in response to Salmonella-infected DC may be due to a lack of costimulation. Experiments designed to test this hypothesis revealed that Salmonella- or E. coli-infected DC, like LPS-treated DC, express cell surface MHC-I and up-regulate the costimulatory molecules CD80 and CD86 to a similar extent (data not shown), demonstrating that Salmonella-infected DC express molecules required for T cell activation. Collectively, these results suggest that the failure of T lymphocytes to proliferate may be due to a direct effect of Salmonella on the T cell.
Fig. 3.

The failure of T cells to respond to Salmonella-infected DC is not simply due to Salmonella-induced programmed DC death. (A) OVA-specific CD8+ T cells remained incapable of proliferating in response to OVA-coated DC cocultured with sipB-deficient Salmonella, which cannot kill these professional phagocytes (6). (B) OVA-specific CD8+ T cells also failed to proliferate when added to WT Salmonella-infected OVA-coated caspase-1-deficient DC, which are resistant to Salmonella-induced toxicity (6). Data are representative of at least three independent experiments.
A Direct, Contact-Dependent Effect of Salmonella on T Lymphocytes Is Responsible for the Inhibitory Phenotype. To determine whether Salmonella act directly on T lymphocytes to prevent these cells from proliferating, we tested the ability of Salmonella to block T cell clonal expansion after CD3ε/CD28 ligation (25). A polyclonal population of CD90.2-expressing T cells was isolated and purified from C57BL/6J mice, labeled with CFSE, and infected with Salmonella. After 3 h, cells were washed once, resuspended in media supplemented with antibiotics and anti-CD28, and seeded in wells that were previously coated with anti-CD3ε. T cell division was analyzed after 3 days of in vitro culture by flow cytometry. As expected, proliferation of UI T lymphocytes depended on the presence of anti-CD3ε (Fig. 4A). Intriguingly, Salmonella-infected T cells failed to proliferate in response to anti-CD3ε treatment (Fig. 4B). This inhibitory phenotype required live bacteria (Fig. 4B) and depended on the moi (Fig. 4C). Importantly, comparable results were obtained when T cells were stimulated with phorbol 12-myristate 13-acetate (PMA) and ionomycin (data not shown).
Fig. 4.

Salmonella inhibit T cell proliferation through a direct immunosuppressive effect on T cells. (A and B) Anti-CD3ε-dependent T cell clonal expansion was suppressed after infection with live Salmonella but not heat-killed bacteria, an inhibitory phenotype that was blocked when Salmonella and T cells were separated by using a transwell system. Also, the transfer of culture supernatant from Salmonella-infected T cells onto UI T cells partially reduced the ability of these cells to proliferate in response to stimulation. (C and D) Full inhibition of T cell expansion depended on the moi, required 3 h of infection, and was observed when T cells were infected with WT Salmonella, but not E. coli. T cells were infected at an moi of 12.5 unless indicated otherwise. Data are representative of at least three independent experiments.
Next, we determined whether direct contact is required for Salmonella to inhibit T cell proliferation. As shown in Fig. 4B, T cells proliferated in response to anti-CD3ε cross-linking when a transwell system was used to separate these cells from Salmonella through a semipermeable membrane. These data demonstrate that direct contact is required for Salmonella to suppress T cell clonal expansion. Interestingly, our further exploration of the system led us to discover that when culture supernatant from stimulated, Salmonella-infected T lymphocytes was added to UI T cells, these cells had a partially reduced ability to proliferate in response to anti-CD3ε (Fig. 4B). Collectively, these results suggest that direct contact between Salmonella and the T cell may trigger the release of a soluble inhibitory molecule(s).
To further characterize the block in T cell proliferation, the surface expression of CD69, CD25α, CD44, and CD62L was monitored 1 day after stimulation. No statistically significant differences between UI and Salmonella-infected T lymphocytes were detected with respect to the surface levels of these T cell activation markers (data not shown). However, Salmonella-infected T cells, in contrast to UI T cells, failed to produce IL-2 and IFN-γ after anti-CD3ε cross-linking (data not shown). As expected, activation-induced cytokine production by Salmonella-infected T lymphocytes was restored when bacteria were separated from T cells by using a transwell system (data not shown).
When bacterial impairment of T cell proliferation was studied over time, it was revealed that full inhibition by Salmonella required 3 h of infection, whereas E. coli over the same period were incapable of blocking T cell expansion (Fig. 4D). Interestingly, this Salmonella-induced inhibitory effect was not restricted to a particular T cell subset, because both CD4+ and CD8+ T lymphocytes were affected similarly (data not shown).
Last, in light of reports showing that Salmonella-induced, natural killer (NK)-cell-regulated NO production by macrophages, and possibly DC, can mediate T cell immunosuppression (ref. 22 and references therein, see also refs. 26-29), we monitored the ability of Salmonella-infected T cells to proliferate in the presence of up to 2 mM NG-monomethyl-l-arginine. This competitive inhibitor of all three isoforms of NO synthase (iNOS, eNOS, and nNOS) did not reduce the antiproliferative effects of contact with Salmonella (data not shown).
In summary, these data are evidence that T cells, when cocultured with live Salmonella, fail to proliferate in response to anti-CD3ε cross-linking, thus demonstrating that viable Salmonella have an inhibitory effect on T lymphocytes, even in the absence of DC. This inhibitory effect, which is induced by a NO-independent mechanism, requires direct contact between Salmonella and the T cell and is further characterized by a lack of cytokine production.
SPI1, SPI2, phoP, sti, and the Virulence Plasmid Are Not Required for Salmonella to Inhibit T Cell Proliferation. As soon as we established that Salmonella are capable of directly affecting the ability of T lymphocytes to proliferate, we initiated experiments to identify specific Salmonella genes that are required for this inhibitory phenotype. To this end, a number of previously described Salmonella mutants were tested for their ability to block T cell clonal expansion.
To determine whether the SPI1-encoded invasion machinery is required for Salmonella to block T cell proliferation, we tested a Salmonella strain mutated in the invA gene (7), encoding an essential structural component of the SPI1-encoded TTSS (15). As shown in Fig. 5, invA-deficient bacteria and WT Salmonella blocked T cell proliferation to a similar extent, suggesting that the SPI1-encoded invasion apparatus is not required for Salmonella to inhibit T cell clonal expansion. Similar results were obtained when T cells were infected with spiB-deficient Salmonella (Fig. 5), which are unable to assemble a functional SPI2-encoded TTSS (8). Interestingly, recent evidence indicates that intracellular Salmonella can suppress MHC-II-restricted antigen presentation by professional antigen-presenting cells by means of a SPI2-dependent mechanism (30, 31), suggesting that Salmonella may have evolved several independent strategies to subvert adaptive immune defense mechanisms. The observation that some type III secreted effector proteins can be translocated by either SPI1 or SPI2 TTSS (32, 33) led us to construct and test an invA spiB (SPI1 SPI2) double mutant, which expresses neither TTSS. As shown in Fig. 5, T cells infected with the SPI1 SPI2 double mutant still failed to proliferate in response to anti-CD3ε treatment, further showing that Salmonella can suppress T cell expansion by means of a TTSS-independent mechanism.
Fig. 5.
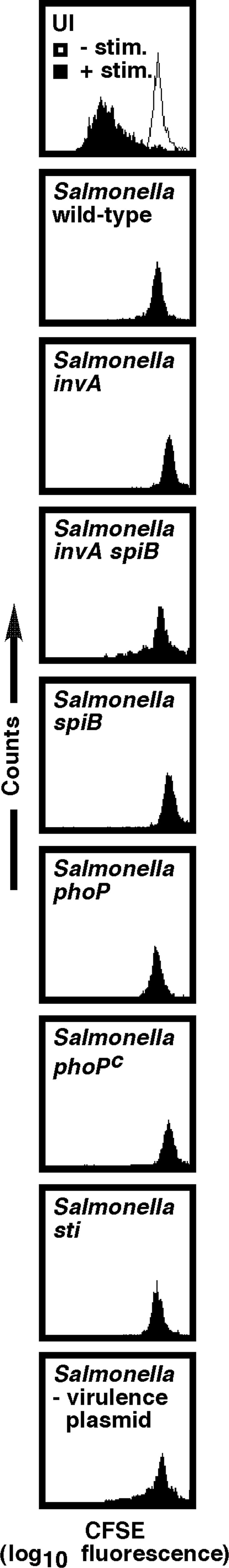
SPI1, SPI2, phoP, sti, and the virulence plasmid are not required for Salmonella to inhibit T cell proliferation. T cells, when infected with invA, spiB, phoP, phoPc, or sti mutant Salmonella or a Salmonella strain cured of the virulence plasmid, failed to proliferate in response to anti-CD3ε/CD28 treatment, demonstrating that these loci are not required for the direct, contact-dependent inhibitory effect of Salmonella on T cells. Data are representative of at least three independent experiments.
Evidence that expression of a large number of Salmonella virulence genes is controlled by the two-component regulatory system PhoP/Q is documented extensively (34). In addition to controlling genes required for survival within macrophages, PhoP (possibly by regulation of SPI2; ref. 35) is required for Salmonella to inhibit MHC-II-restricted antigen presentation by professional phagocytes (36). To determine whether the ability of Salmonella to inhibit T cell proliferation is phoP-dependent, we infected fluorescently labeled T cells with either phoP-null Salmonella (phoP) (9) or bacteria that constitutively express PhoP (phoPc) (10). As shown in Fig. 5, both Salmonella mutants were fully capable of inhibiting T cell proliferation after CD3ε/CD28 ligation, suggesting that the ability of Salmonella to block T cell clonal expansion does not require the phoP gene.
Next, we tested whether a functional sti gene is required for Salmonella to inhibit T cell proliferation. It has been reported that purified Sti has an immunosuppressive effect on T lymphocytes (37). As shown in Fig. 5, sti-deficient Salmonella, like WT bacteria, were fully capable of inhibiting CD3ε/CD28-induced T cell proliferation, demonstrating that the ability of Salmonella to block T cell clonal expansion does not require a functional sti gene.
Last, to determine whether the Salmonella virulence plasmid, which has been implicated in causing immunosuppression during Salmonella infection of mice (19), is required for Salmonella to inhibit anti-CD3ε-induced T cell proliferation, we tested a virulence plasmid-cured Salmonella strain. As shown in Fig. 5, purified T cells infected with a Salmonella strain lacking the virulence plasmid still failed to proliferate in response to CD3ε/CD28 ligation, suggesting that, in this system, the virulence plasmid is not required for Salmonella to block T cell proliferation.
In conclusion, our studies demonstrate that purified T lymphocytes, when infected with Salmonella, fail to proliferate in response to stimulation. This inhibitory phenotype is due to a direct, contact-dependent effect of Salmonella on the T cell that does not require SPI1, SPI2, phoP, sti, or the virulence plasmid. It would be interesting to focus not only on the identification and characterization of the Salmonella gene product(s) that are required for this process but also on the elucidation of the molecular mechanism of this inhibitory phenotype at a cellular level. Our findings contribute to an emerging picture of Salmonella as an enteric pathogen that has evolved numerous coordinated strategies to evade immune defense mechanisms. These strategies include the ability to (i) survive and multiply within macrophages and DC (9, 38, 39); (ii) induce both programmed macrophage and DC death (6, 40); (iii) inhibit antigen processing and presentation by professional antigen presenting cells (30, 31, 36, 41); and (iv), as shown in this study, directly inhibit the ability of T lymphocytes to proliferate in response to stimulation. Some of these strategies appear to be similar to strategies used by other enteric bacteria and may provide insights into how pathogens have evolved to overcome innate and adaptive host defense mechanisms.
Acknowledgments
We thank Abbott Bioresearch Center (Worcester, MA) for caspase-1-deficient mice, and John Mekalanos (Harvard Medical School) for sti mutant Salmonella. We also thank members of our laboratory for stimulating discussions and constructive comments on the manuscript. This work was supported in part by National Institutes of Health Grant AI055962 (to M.N.S.). A.W.M.v.d.V. was supported by a grant from Philip Morris USA.
Author contributions: A.W.M.v.d.V., M.K.C., and M.N.S. designed research and analyzed data; A.W.M.v.d.V. and M.K.C. performed research; A.W.M.v.d.V. contributed new reagents/analytic tools; and A.W.M.v.d.V. and M.N.S. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: CFSE, carboxyfluorescein diacetate succinimidyl ester; DC, dendritic cell(s); moi, multiplicity of infection; OVA, ovalbumin; OVA257-264, ovalbumin-(257-264) octapeptide; SPI, Salmonella pathogenicity island; TTSS, type III secretion system; UI, uninfected.
References
- 1.Rescigno, M. (2002) Trends Microbiol. 10, 425-461. [DOI] [PubMed] [Google Scholar]
- 2.Rescigno, M. & Borrow, P. (2001) Cell 106, 267-270. [DOI] [PubMed] [Google Scholar]
- 3.Hornef, M. W., Wick, M. J., Rhen, M. & Normark, S. (2002) Nat. Immunol. 3, 1033-1040. [DOI] [PubMed] [Google Scholar]
- 4.Santos, R. L., Zhang, S., Tsolis, R. M., Kingsley, R. A., Adams, L. G. & Baumler, A. J. (2001) Microbes Infect. 3, 1335-1344. [DOI] [PubMed] [Google Scholar]
- 5.Mittrucker, H. W. & Kaufmann, S. H. (2000) J. Leukocyte Biol. 67, 457-463. [DOI] [PubMed] [Google Scholar]
- 6.van der Velden, A. W. M., Velasquez, M. & Starnbach, M. N. (2003) J. Immunol. 171, 6742-6749. [DOI] [PubMed] [Google Scholar]
- 7.Galán, J. E. & Curtiss, R., III (1989) Proc. Natl. Acad. Sci. USA 86, 6383-6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsolis, R. M., Townsend, S. M., Ficht, T. A., Adams, L. G. & Bäumler, A. J. (1999) Infect. Immun. 67, 4879-4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fields, P. I., Swanson, R. V., Haidaris, C. G. & Heffron, F. (1986) Proc. Natl. Acad. Sci. USA 83, 5189-5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller, S. I. & Mekalanos, J. J. (1990) J. Bacteriol. 172, 2485-2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsolis, R., Bäumler, A. J., Stoljiljkovic, I. & Heffron, F. (1995) J. Bacteriol. 177, 4628-4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmer, B. M., Tran, M. & Heffron, F. (1999) J. Bacteriol. 181, 1364-1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li, P., Allen, H., Banerjee, S., Franklin, S., Herzog, L., Johnston, C., McDowell, J., Paskind, M., Rodman, L., Salfeld, J., et al. (1995) Cell 80, 401-411. [DOI] [PubMed] [Google Scholar]
- 14.Lyons, A. B., Hasbold, J. & Hodgkin, P. D. (2001) Methods Cell Biol. 63, 375-398. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh, P. (2004) Microbiol. Mol. Biol. Rev. 68, 771-795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Starnbach, M. N. & Bevan, M. J. (1994) J. Immunol. 153, 1603-1612. [PMC free article] [PubMed] [Google Scholar]
- 17.Russmann, H., Shams, H., Poblete, F., Fu, Y., Galan, J. E. & Donis, R. O. (1998) Science 281, 565-568. [DOI] [PubMed] [Google Scholar]
- 18.Russmann, H., Weissmuller, A., Geginat, G., Igwe, E. I., Roggenkamp, A., Bubert, A., Goebel, W., Hof, H. & Heesemann, J. (2000) Eur. J. Immunol. 30, 1375-1384. [DOI] [PubMed] [Google Scholar]
- 19.Hoertt, B. E., Ou, J., Kopecko, D. J., Baron, L. S. & Warren, R. L. (1989) Plasmid 21, 48-58. [DOI] [PubMed] [Google Scholar]
- 20.Deschenes, M., Guenounou, M. & Nauciel, C. (1989) Res. Immunol. 140, 55-65. [DOI] [PubMed] [Google Scholar]
- 21.Matsui, K. & Arai, T. (1995) FEMS Immunol. Med. Microbiol. 10, 227-234. [DOI] [PubMed] [Google Scholar]
- 22.Eisenstein, T. K. (2001) Microbes Infect. 3, 1223-1231. [DOI] [PubMed] [Google Scholar]
- 23.Mittrucker, H. W., Kohler, A. & Kaufmann, S. H. (2002) Infect. Immun. 70, 199-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hogquist, K. A., Jameson, S. C., Heath, W. R., Howard, J. L., Bevan, M. J. & Carbone, F. R. (1994) Cell 76, 17-27. [DOI] [PubMed] [Google Scholar]
- 25.Kruisbeek, A. M., Shevach, E. & Thornton, A. M. (2001) in Current Protocols in Immunology, eds. Bierer, B. E., Coligan, J. E., Margulies, D. H., Shevach, E. M., Strober, W. & Kruisbeek, A. M. p. E. (Wiley, New York), pp. 3.12.1-14.
- 26.Huang, D., Schwacha, M. G. & Eisenstein, T. K. (1996) Infect. Immun. 64, 3786-3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.MacFarlane, A. S., Schwacha, M. G. & Eisenstein, T. K. (1999) Infect. Immun. 67, 891-898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.al-Ramadi, B. K., Meissler, J. J., Jr., Huang, D. & Eisenstein, T. K. (1992) Eur. J. Immunol. 22, 2249-2254. [DOI] [PubMed] [Google Scholar]
- 29.Schwacha, M. G., Meissler, J. J., Jr., & Eisenstein, T. K. (1998) Infect. Immun. 66, 5862-5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheminay, C., Mohlenbrink, A. & Hensel, M. (2005) J. Immunol. 174, 2892-2899. [DOI] [PubMed] [Google Scholar]
- 31.Mitchell, E. K., Mastroeni, P., Kelly, A. P. & Trowsdale, J. (2004) Eur. J. Immunol. 34, 2559-2567. [DOI] [PubMed] [Google Scholar]
- 32.Miao, E. A., Scherer, C. A., Tsolis, R. M., Kingsley, R. A., Adams, L. G., Bäumler, A. J. & Miller, S. I. (1999) Mol. Microbiol. 34, 850-864. [DOI] [PubMed] [Google Scholar]
- 33.Miao, E. A. & Miller, S. I. (2000) Proc. Natl. Acad. Sci. USA 97, 7539-7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Groisman, E. A. (2001) J. Bacteriol. 183, 1835-1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deiwick, J., Nikolaus, T., Erdogan, S. & Hensel, M. (1999) Mol. Microbiol. 31, 1759-1773. [DOI] [PubMed] [Google Scholar]
- 36.Wick, M.-J., Harding, C. V., Twesten, N. J., Normark, S. J. & Pfeifer, J. D. (1995) Mol. Microbiol. 16, 465-476. [DOI] [PubMed] [Google Scholar]
- 37.Matsui, K., Nagano, K., Arai, T., Hirono, I. & Aoki, T. (1998) FEMS Immunol. Med. Microbiol. 22, 341-349. [DOI] [PubMed] [Google Scholar]
- 38.Niedergang, F., Sirard, J. C., Blanc, C. T. & Kraehenbuhl, J. P. (2000) Proc. Natl. Acad. Sci. USA 97, 14650-14655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marriott, I., Hammond, T. G., Thomas, E. K. & Bost, K. L. (1999) Eur. J. Immunol. 29, 1107-1115. [DOI] [PubMed] [Google Scholar]
- 40.Hueffer, K. & Galan, J. E. (2004) Cell. Microbiol. 6, 1019-1025. [DOI] [PubMed] [Google Scholar]
- 41.Qimron, U., Madar, N., Mittrucker, H. W., Zilka, A., Yosef, I., Bloushtain, N., Kaufmann, S. H., Rosenshine, I., Apte, R. N. & Porgador, A. (2004) Cell. Microbiol. 6, 1057-1070. [DOI] [PubMed] [Google Scholar]