

Abstract
Hepatitis C virus (HCV) is a global epidemic manifested mainly by chronic infection. One strategy that HCV employs to establish chronic infection is to use the viral Ser protease NS3/4A to cleave some unknown cellular targets involved in innate immunity. Here we show that the target of NS3/4A is the mitochondrial antiviral signaling protein, MAVS, that activates NF-κB and IFN regulatory factor 3 to induce type-I interferons. NS3/4A cleaves MAVS at Cys-508, resulting in the dislocation of the N-terminal fragment of MAVS from the mitochondria. Remarkably, a point mutation of MAVS at Cys-508 renders MAVS resistant to cleavage by NS3/4A, thus maintaining the ability of MAVS to induce interferons in HCV replicon cells. NS3/4A binds to and colocalizes with MAVS in the mitochondrial membrane, and it can cleave MAVS directly in vitro. These results provide an example of host–pathogen interaction in which the virus evades innate immunity by dislodging a pivotal antiviral protein from the mitochondria and suggest that blocking the cleavage of MAVS by NS3/4A may be applied to the prevention and treatment of HCV.
Keywords: IFN regulatory factor 3, NF-κB, retinoic acid-induced gene I, IκB kinase
Hepatitis C virus (HCV) infects >170 million people in the world, and ≈80% of the infected individuals develop persistent infection (1, 2). HCV is an enveloped single-strand RNA virus belonging to the Flaviviridae family. It contains a 9.6-kb RNA genome that encodes a large polyprotein (>3,000 aa), which is cleaved into 10 structural and nonstructural (NS) proteins through the action of cellular peptidases as well as the viral-encoded proteases including NS3/4A. NS3 contains Ser protease and RNA helicase activities that require its cofactor NS4A, which tethers the holoenzyme complex to an intracellular membrane compartment. The NS3/4A protease is not only essential for generating mature viral proteins required for viral replication, but also can suppress the host antiviral immune system by cleaving putative cellular targets involved in the induction of type-I interferons (IFNs), such as IFN-α and IFN-β (3).
The induction of IFNs is regulated by the transcription factors NF-κB and IFN regulatory factor 3 (IRF3), which are activated by a signaling cascade emanating from dsRNA generated during the replication of viral RNA (4, 5). The activation of NF-κB requires the phosphorylation of its inhibitor IκB by IκB kinase (IKK) complex, which can be activated by a large variety of agents, including proinflammatory cytokines and microbial pathogens (6). The phosphorylation of IκB targets this inhibitor for ubiquitination and subsequent degradation by the proteasome, thereby allowing NF-κB to enter the nucleus to regulate downstream genes. The activation of IRF3 requires its phosphorylation by two IKK-related kinases, TBK1 and IKKε (7, 8). After phosphorylation, IRF3 is dimerized and translocated into the nucleus where it forms an enhanceosome complex together with NF-κB and other transcription factors to turn on the expression of targets genes such as IFN-β (4). IFNs then induce a large array of antiviral genes through the activation of the Janus tyrosine kinase/signal transducer and activator of transcription pathway.
The receptor for intracellular viral dsRNA has recently been identified as retinoic acid-induced gene I (RIG-I), which contains a RNA helicase domain that binds to dsRNA (9). RIG-I also contains tandem N-terminal caspase recruitment domains (CARDs) that interact with another CARD domain protein, mitochondrial antiviral signaling (MAVS; also known as IPS-1, VISA, and CARDIF) (10–13). MAVS contains a C-terminal transmembrane (TM) domain that targets it to the mitochondrial outer membrane (10). Importantly, the mitochondrial localization of MAVS is essential for its signaling function, because the removal of the mitochondrial-targeting domain of MAVS abolishes its ability to induce IFNs. Epistasis experiments have shown that MAVS functions downstream of RIG-I and upstream of IκB kinase and TBK1 in the viral signaling pathway.
Recent studies have shown that NS3/4A inhibits IFN induction by RIG-I; however, RIG-I is not cleaved by NS3/4A (14, 15). NS3/4A also was found to cleave TIR domain-containing adapter-inducing IFN-β (TRIF) (16), an adaptor protein that binds to Toll-like receptors such as TLR3 and TLR4 (17). However, TRIF is not essential for IFN induction by viruses (18). Thus, the target of NS3/4A in the viral pathway remains to be identified. In this work, we present evidence that MAVS is the proteolytic target of NS3/4A. We found that NS3/4A cleaves MAVS at Cys-508, resulting in the dislocation of the N-terminal fragment of MAVS from the mitochondria, thereby suppressing the induction of IFN-β. A point mutation at Cys-508 prevents the cleavage of MAVS by NS3/4A and restores IFN-β induction in a HCV replicon cell line, suggesting that the cleavage of MAVS is responsible for the suppression of IFN induction in HCV replicating cells. We also show that NS3/4A binds to and colocalizes with MAVS in the mitochondria. Finally, we show that NS3/4A cleaves MAVS at Cys-508 in vitro, providing the direct biochemical evidence that MAVS is the target of NS3/4A.
Materials and Methods
Plasmids and Antibodies (Abs). Expression constructs for MAVS, MAVS (CARD-TM), TBK1, RIG-I(N), IFN-β-luciferase (Luc), NF-κB-Luc, UAS-Luc, and Gal4-IRF3 are described in ref. 10. pcDNA3-Flag-MAVS-hemagglutinin (HA) was constructed by subcloning the DNA fragment encoding MAVS-HA into the XhoI and XbaI sites of the pcDNA3-Flag vector such that the N-terminal Flag epitope was fused in frame with MAVS-HA. NS3/4A was amplified by RT-PCR from the RNA of HCV replicon cell line K2040 (19) and then cloned into pcDNA3 in frame with an N-terminal Flag or Myc tag. The S139A mutant of NS3/4A and the Cys mutants of MAVS (C435R, C452R, and C508R) were generated by site-directed mutagenesis using the QuikChange kit (Stratagene). pET14b-NS3/4A was constructed by subcloning NS3/4A into the XhoI and BamH1 sites of pET14b (Novagen). All plasmids were verified by automatic DNA sequencing.
The polyclonal Ab against MAVS was generated and purified by antigen column as described in ref. 10. The HCV NS3 Ab was purchased from NovoCastra (Newcastle, U.K.). The monoclonal Abs against Flag (M2, Sigma), Myc (9E10, Santa Cruz) and HA (HA.11, Covance) were purchased from the indicated suppliers.
Cell Culture, Transfection, and Luc Reporter Assays. HEK293 and HeLa cells were from American Type Culture Collection. The Huh7 and HCV replicon cell line K2040 were provided by Michael Gale (University of Texas Southwestern) (19). Transfection of HEK293 cells was carried out by using the calcium phosphate precipitation method. HeLa cells were transfected by using the PolyFect reagent (Qiagen, Valencia, CA). Huh7 and replicon cells were transfected by using lipofectamine 2000 (Invitrogen) or Superfect (Qiagen) reagents. Luc reporter assays were performed as described in ref. 10.
Subcellular Fractionation. HEK293 cells were transfected with expression vectors for Flag-MAVS-HA or the C508R mutant together with Myc-tagged NS3/4A or its inactive mutant S139A. Cells were washed 36 h after transfection in hypotonic buffer (10 mM Tris·HCl, pH 7.5/10 mM KCl/1.5 mM MgCl2/protease inhibitors) and then homogenized in the same buffer by douncing 20 times. The homogenate was centrifuged at 500 × g for 5 min to remove nuclei and unbroken cells. The supernatant was centrifuged again at 5,000 × g for 10 min to generate membrane pellets (P; containing mostly mitochondria) and cytosolic supernatant (S). The same procedure was used for subcellular fractionation of Huh7 and HCV replicon cells.
Cleavage of MAVS by NS3/4A Protease in Vitro. pcDNA3-Flag-NS3/4A or its mutant S139A was transfected into HEK293 cells to express the protease, which then was purified by binding to the Flag-Sepharose (M2-Sepharose, Sigma) followed by elution with the Flag peptide (0.2 mg/ml). The eluted NS3/4A or S139A mutant was concentrated by using Amicon microconcentrator (Millipore, 10,000 Da cut-off) and diluted in buffer A (20 mM Tris·HCl/150 mM NaCl/0.1% Triton X-100/5 mM DTT, pH 7.5). This procedure was repeated three times to reduce the concentration of the Flag peptide. For expression of NS3/4A in Escherichia coli, pET14b-NS3/4A was transfected into the E. coli strain BL21/pLysS, and the transformant was induced to express His-6-NS3/4A after the addition of IPTG. His-6-NS3/4A was purified by using nickel affinity column and dialyzed against buffer B (20 mM Tris·HCl, pH 7.5/100 mM NaCl/10% glycerol).
Flag-MAVS or its mutant C508R was translated in vitro by using the TNT rabbit reticulocyte lysate (Promega) supplemented with [35S]methionine. The 35S-labeled Flag-MAVS or C508R was purified by using Flag-Sepharose as described above for NS3/4A. To measure the cleavage of MAVS by NS3/4A, 0.5 μl of 35S-labeled MAVS or C508R was incubated with 0.5 μl of NS3/4A or S139A in a 10-μl reaction containing buffer A. After incubation at 30°C for 2 h, the reaction products were resolved by SDS/PAGE and analyzed by using PhosphorImager (Molecular Dynamics).
The experimental procedures for viral infection, immunoblotting, immunoprecipitation, and confocal microscopy are described in ref. 10.
Results
NS3/4A Blocks IFN-β Induction by MAVS. To determine whether NS3/4A inhibits IFN-β induction by MAVS, we transfected expression vectors encoding NS3/4A and MAVS into HEK293 cells together with a Luc reporter driven by the IFN-β promoter (IFN-β-Luc). As controls, we infected cells with Sendai virus (SeV) or transfected cells with an expression vector encoding the N-terminal CARD domains of RIG-I [RIG-I(N)], which has previously been shown to induce IFN-β (9). In each case, NS3/4A inhibited the induction of IFN-β (Fig. 1A). In contrast, the induction of IFN-β by TBK1 was not affected by NS3/4A, suggesting that NS3/4A inhibits the RIG-I pathway at the level of MAVS or at a step downstream of MAVS but upstream of TBK1. To determine whether the protease activity of NS3/4A was required for this inhibition, we mutated the active site Ser-139 of NS3/4A (equivalent to Ser-1165 of the HCV polyprotein) to Ala (20, 21). This mutant migrated more slowly than wild-type (WT) NS3 (Fig. 1D Bottom), because it could no longer be cleaved at the junction between NS3 and NS4A. The protease-dead mutant completely lost its ability to inhibit IFN-β induction by SeV, RIG-I(N), or MAVS. Thus, it is likely that NS3/4A inhibits IFN induction by cleaving MAVS or a target downstream of MAVS. NS3/4A also prevented the MAVS-induced activation of NF-κB (Fig. 1B) and IRF3 reporters (Fig. 1C), as well as the phosphorylation and dimerization of IRF3 (Fig. 1D), further supporting the notion that NS3/4A inactivates a common target required for both NF-κB and IRF3 activation.
Fig. 1.

NS3/4A blocks IFN induction by MAVS. (A) HEK293 cells were transfected with IFN-β-Luc and the expression vector for either the WT or S139A mutant of NS3/4A. The next day, cells were transfected with expression vectors encoding MAVS, TBK1, or RIG-I(N), or infected with SeV. The Luc activity was measured 24 h later and normalized for transfection efficiency. The error bar represents standard deviation from the mean value of duplicated experiments. (B) Similar to A, except that NF-κB Luc reporter was used in lieu of IFN-β-Luc. (C) Similar to A, except that cells were transfected with the UAS-Luc reporter together with the Gal4-IRF3 expression vector. (D) HEK293 cells were transfected with the WT or S139A mutant of NS3/4A together with the expression vectors for MAVS (lanes 5–7) or TBK1 (lanes 8–10). In lanes 2–4, cells were infected with SeV for 24 h. Cell lysates were resolved by electrophoresis under native (Top) or denaturing condition (Middle) and then immunoblotted with an Ab against IRF3. The expression of NS3/4A and TBK1 also was analyzed by immunoblotting with a Flag Ab (Bottom).
MAVS Is the Proteolytic Target of NS3/4A. To determine whether MAVS is the proteolytic target of NS3/4A, we transfected the expression vector encoding FLAG-NS3/4A or FLAG-NS3/4A (S139A) into HEK293 or Huh7 (a human hepatoma cell line). Because MAVS is a mitochondrial membrane protein, the cell lysates were separated into cytosolic fraction (S) and membrane pellets (P) by centrifugation, and the endogenous MAVS protein analyzed by immunoblotting with an affinity-purified MAVS Ab (Fig. 2A). In both cell lines, the expression of NS3/4A, but not the protease-dead mutant of NS3/4A, led to the generation of a faster-migrating fragment of MAVS that was ≈3–5 kDa shorter than the full-length MAVS (Fig. 2A Upper; compare lanes 3 with 4 and 9 with 10). Interestingly, unlike the full-length MAVS that was present in the membrane pellet, the truncated fragment of MAVS was mainly present in the soluble cytosolic fraction, suggesting that the truncated fragment did not contain the C-terminal TM domain. Immunoblotting experiments also showed that the majority of NS3 is in the membrane pellet, presumably because of its association with NS4A (Fig. 2A Lower).
Fig. 2.

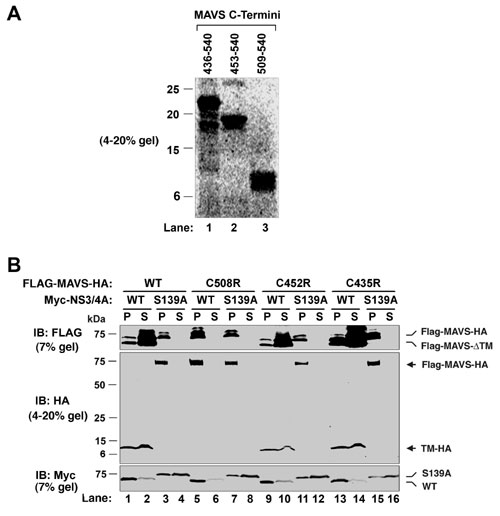
NS3/4A cleaves MAVS at Cys-508. (A) HEK293 (lanes 1–6) or Huh7 (lanes 7–12) cells were transfected with expression vectors for the WT (lanes 3,4, 9, and 10) or S139A mutant (lanes 5, 6, 11, and 12) of NS3/4A or vector (lanes 1, 2, 7, and 8). Cell lysates were separated into membrane pellet (P) or cytosolic supernatant (S) and then immunoblotted with an Ab against MAVS or Flag. The cleavage product of MAVS was indicated as MAVS*. (B) HEK293 cells were transfected with the expression vectors for NS3/4A together with a MAVS mutant containing only the CARD and TM domains (miniMAVS; Top). The activation of IFN-β by the CARD-TM fragment of MAVS in the presence of WT or S139A mutant of NS3/4A was determined by measuring the expression of the IFN-β-Luc reporter (Middle). Cell lysates were analyzed by immunoblotting with the Flag Ab that detects both NS3/4A and MAVS proteins (Bottom). (C) Alignment of the junction sequences of NS proteins of HCV and the putative cleavage site at the C terminus of MAVS from different species. (D) HEK293 cells were transfected with expression vectors encoding the WT or C508R mutant of MAVS together with those encoding the WT or S139A mutant of NS3/4A. Cell lysates were separated by differential centrifugation into the membrane pellet and cytosolic supernatant, which were then resolved by 7% (Top and Bottom) or 4–20% (Middle) SDS/PAGE, followed by immunoblotting with the indicated Ab that detects the N terminus (Flag; Top) or C terminus (HA; Middle) of MAVS, or NS3/4A (Myc; Bottom). (E) HEK293 cells were transfected with MAVS and NS3/4A expression vectors as in D, except that IFN-β-Luc was also cotransfected to measure the induction of IFN-β by Luc assay.
MAVS contains an N-terminal CARD domain, a middle Prorich domain, and a C-terminal TM domain. We have previously shown that a truncated MAVS protein containing only the CARD and TM domains (hereafter referred to as miniMAVS) is sufficient to induce IFN-β (10). To further delineate the NS3/4A cleavage site of MAVS, we tested whether the miniMAVS was also sensitive to NS3/4A inhibition. As shown in Fig. 2B, IFN-β induction by the miniMAVS was also inhibited by NS3/4A (Middle). Furthermore, the miniMAVS was cleaved to a shorter fragment by the WT, but not the S139A mutant, of NS3/4A (Fig. 2B Bottom). The truncated fragment was also ≈4 kDa shorter than the miniMAVS (Fig. 2B, compare lanes 4 and 5), suggesting that this small MAVS protein of only 140 residues in length contains a cleavage site of NS3/4A, most likely at a residue near the TM domain.
NS3/4A Cleaves MAVS at Cys-508. NS3/4A is a Ser protease that cleaves at the C terminus of a Cys or Thr residue within a loosely defined consensus sequence [(E/D)xxxx(C/T)(S/A), where x denotes any amino acid], which is found at the junction of HCV NS proteins (Fig. 2C) (22). Inspection of the miniMAVS amino acid sequence revealed a potential NS3/4A cleavage sequence, 503ER-EVPCH509. The potential cleavage site, Cys-508, is located 6 aa from the beginning of the TM domain and 32 aa from the C terminus of MAVS. Cleavage at Cys-508 is predicted to generate an ≈32-residue C-terminal fragment, consistent with the size difference between the miniMAVS and the miniMAVS lacking the TM domain (Fig. 2B Bottom; compare lanes 4 and 5). Indeed, when NS3/4A was cotransfected together with an expression construct encoding a MAVS protein that is tagged with FLAG at the N terminus and HA at the C terminus, the MAVS protein was cleaved into a soluble FLAG-tagged N-terminal fragment that was smaller than the intact protein (Fig. 2D Top; compare lanes 1 and 4). Moreover, an ≈7-kDa C-terminal fragment was detectable in both the membrane pellet and cytosolic fractions with a HA Ab (Fig. 2D Middle; lanes 3 and 4). This cleavage product of MAVS was not detected in cells expressing the S139A mutant of NS3/4A. Although the apparent molecular mass of the C-terminal cleavage fragment was greater than the theoretical molecular mass of the fragment containing the C-terminal 32 residues of MAVS plus a HA epitope (≈5 kDa), this result is likely because of the abnormal migration of small peptides on SDS/PAGE. Indeed, when we synthesized the C-terminal 32 residues of MAVS (residue 509–540) plus the HA epitope by in vitro translation, this fragment also migrated as an ≈7-kDa band (see Fig. 5A, which is published as supporting information on the PNAS web site), identical to the C-terminal fragment of MAVS after cleavage by NS3/4A.
To demonstrate that Cys-508 is indeed the cleavage site, we mutated Cys-508 to Arg, which is predicted to disrupt NS3/4A cleavage based on the analysis of sequence requirement for HCV polyprotein cleavage (23). Remarkably, this single amino acid substitution completely blocked the cleavage of MAVS by NS3/4A (Fig. 2D, lanes 7–12). Furthermore, the protease-resistant mutant of MAVS was fully capable of inducing IFN-β, and this induction was no longer inhibited by NS3/4A (Fig. 2E). In contrast to Cys-508, mutations at two adjacent Cys residues, Cys-435 and -452, did not prevent the cleavage of MAVS by NS3/4A (Fig. 5B). Together, these results indicate that NS3/4A cleaves MAVS at Cys-508 when these proteins are coexpressed in the same cells.
NS3/4A Binds to and Cleaves MAVS Directly in Vitro. To determine whether NS3/4A could cleave MAVS directly in vitro, we expressed FLAG-NS3/4A or FLAG-NS3/4A (S139A) protein in HEK293 cells and immunopurified the protein using the FLAG Ab followed by elution with a FLAG peptide. We also synthesized 35S-labeled FLAG-MAVS or FLAG-MAVS-C508R protein in vitro using rabbit reticulocyte lysates and purified the proteins using the FLAG Ab. The purified MAVS proteins were incubated with NS3/4A protease or lysates containing the protease. As shown in Fig. 3A, the WT MAVS was cleaved by the active NS3/4A protease, but not the inactive NS3/4A mutant (lanes 1–5). In contrast, a point mutation at Cys-508 completely blocked the cleavage of MAVS by NS3/4A in vitro (lanes 6–10). The in vitro cleavage product represents the N-terminal fragment of MAVS, because the C-terminal 32-aa fragment does not contain any Met that can be radioactively labeled. Similar results were obtained by using NS3/4A protease expressed and purified from E. coli (see Fig. 6A, which is published as supporting information on the PNAS web site). These results indicate that NS3/4A directly cleaves MAVS at Cys-508 in vitro.
Fig. 3.

NS3/4A binds to and colocalizes with MAVS in the mitochondria, and it cleaves MAVS in vitro.(A) Flag-tagged WT or S139A mutant of NS3/4A was expressed in HEK293 cells and then purified by using Flag-Sepharose. The purified NS3/4A proteins (lanes 4, 5, 9, and 10) or lysates containing NS3/4A (lanes 1–3 and 6–8) were incubated with 35S-labeled MAVS or C508R mutant of MAVS, which was synthesized by in vitro translation and purified by using Flag-Sepharose. After incubation at 30°C for 2 h, proteins were separated by SDS/PAGE and analyzed by PhosphorImaging (Upper) or immunoblotting (Lower). The cleavage product of MAVS is indicated as MAVS*. (B) HEK293 cells were transfected with expression vectors for Flag-NS3/4A and HA-MAVS as indicated. Cell lysates were immunoprecipitated with the MAVS Ab (lanes 3, 6, 9, and 12) or control IgG (lanes 2, 5, 8, and 11). The precipitated proteins were analyzed by immunoblotting with an Ab against HA (Upper) or Flag (Lower). (C) Expression vectors for Flag-NS3/4A and HA-MAVS or C508R mutant were transfected into HeLa cells, and the localization of these proteins was stained by the indicated Abs and visualized by confocal microscopy.
The cleavage of MAVS by NS3/4A suggests that these two proteins may interact directly. To investigate this possibility, we cotransfected MAVS and NS3/4A or their mutants into HEK293 cells, immunoprecipitated MAVS with the MAVS Ab, and then determined whether NS3/4A was coprecipitated with MAVS by immunoblotting (Fig. 3B). Interestingly, only the S139A mutant, but not the WT, NS3/4A coprecipitated with MAVS (Fig. 1D Bottom, compare lanes 3 and 9), suggesting that the cleaved MAVS dissociated from the WT protease, whereas the mutant protease was able to “trap” its substrate. Consistent with this interpretation, C508R also bound to either WT or S139A mutant of NS3/4A, but this binding was weaker than that observed between MAVS and the S139A mutant of NS3/4A (compare lanes 6 and 12 with lane 9), suggesting that C508R mutation may partially impair its binding to the protease.
NS3/4A Colocalizes with MAVS in the Mitochondria. It has been shown that HCV core protein and NS3/4A are localized to a mitochondrion-associated membrane structure in both cultured hepatocytes (24, 25) and liver biopsies of chronic hepatitis C patients (26). Because MAVS is a mitochondrial membrane protein, we examined whether MAVS and NS3/4A colocalize in the same membrane compartment. Expression vectors encoding Myc-NS3/4A and/or HA-MAVS were transfected into HeLa cells, which then were stained with the corresponding Abs followed by imaging with a laser scanning confocal microscope. To stain the mitochondria, cells were incubated with Mito Tracker, a fluorescent dye taken up by the mitochondria of living cells. The staining patterns of the WT NS3/4A overlapped with that of Mito Tracker, but not the endoplasmic reticulum resident protein calnexin (Fig. 6B), suggesting that NS3/4A is localized to the mitochondria or in a mitochondrion-associated membrane. When WT NS3/4A and MAVS were coexpressed, the majority of MAVS became cytosolic (presumably due to cleavage), as revealed by an Ab that detects the N terminus of MAVS (Fig. 3C). In sharp contrast, when the WT NS3/4A was coexpressed with MAVS-C508R, both NS3/4A and MAVS-C508R proteins colocalized in the mitochondrial membrane and showed an extensive overlapping staining pattern. These results indicate that MAVS and NS3/4A are colocalized in the mitochondrial membrane or are positioned in very close proximity, thus rendering MAVS vulnerable to cleavage by NS3/4A.
MAVS Is Cleaved in a HCV Replicon Cell Line. To determine whether MAVS is a target of HCV during viral replication, we used the HCV replicon cell culture system in which the subgenomic RNA of HCV replicates autonomously in the human hepatoma cell line Huh7 (27). The HCV subgenomic RNA encodes all of the NS proteins, including the NS3/4A protease and the NS5B RNA polymerase. It has been shown that the HCV replicon cell lines that replicate the viral RNA efficiently can suppress the host IFN induction through the proteolytic activity of NS3/4A (3). To determine whether the endogenous MAVS is cleaved in the replicon cells, we used subcellular fractionation and immunoblotting to analyze the MAVS protein from the WT Huh7 and a replicon cell line K2040 (19). As shown in Fig. 4A, the MAVS protein is present predominately in the mitochondrial membrane pellet isolated from Huh7 cells. In contrast, in the HCV replicon cells, a cleaved form of MAVS was found mostly in the soluble cytosolic fraction, whereas NS3 was detected mainly in the membrane pellets.
Fig. 4.

MAVS is cleaved at Cys-508 in a HCV replicon cell line. (A) Cell lysates from Huh7 (lanes 1 and 2) or Replicon cells (K2040; lanes 3 and 4) were separated by centrifugation into the membrane pellet (P) and cytosolic supernatant (S), which then were analyzed by immunoblotting with an Ab against MAVS or NS3. The cleavage products of MAVS are indicated as MAVS*. MAVS-ΔN, N-terminal truncated fragment of MAVS (see ref. 10); MAVS-ΔN*, N-terminal truncated fragment of MAVS in which the TM domain is also cleaved by NS3/4A. (B and C) Expression vectors for MAVS, C508 mutant of MAVS, or RIG-I(N) were cotransfected with the IFN-β-Luc reporter into Huh7 (B) or Replicon (C) cell lines. Twenty-four hours after transfection, cells were infected with SeV or mock infected for another 24 h before harvesting for Luc assay.
Prevention of MAVS Cleavage Restores the IFN Response in HCV Replicon Cells. If the cleavage of MAVS is solely responsible for the failure to induce IFNs in HCV replicon cells, prevention of MAVS cleavage should restore IFN induction in these cells. To test this possibility, we introduced the C508R mutant of MAVS as well as the WT MAVS into the HCV replicon cells to examine the induction of IFN-β. As reported in ref. 3, SeV induced IFN-β in the Huh7 cells but failed to do so in the replicon cell line (Fig. 4 B and C). Even overexpression of RIG-I(N) failed to induce IFN-β in the replicon cells (Fig. 4C), presumably because the endogenous MAVS was cleaved into an inactive soluble fragment (Fig. 4A, lane 4). Overexpression of WT MAVS was able to induce partial IFN response in the replicon cells, and this response was further enhanced by SeV. This result may be due to the incomplete cleavage of the overexpressed MAVS by the NS3/4A protease in the replicon cells. Most significantly, expression of the protease-resistant mutant of MAVS (C508R) restored IFN-β induction in the replicon cells to the same level observed in Huh7 cells (Fig. 4 B and C). These results demonstrate that the evasion of innate immune response in the HCV replicon cells is due to the cleavage of MAVS by NS3/4A.
Discussion
We have previously shown that MAVS is an essential antiviral signaling protein and proposed that MAVS may be a prime target of viruses that have succeeded in evading the host immune system (10). In this work, we show that MAVS is indeed the proteolytic target of HCV NS3/4A protease. Specifically, we found that HCV cleaves MAVS at Cys-508, dislocating MAVS from the mitochondria, thus preventing the induction of IFN-β. NS3/4A binds to and colocalizes with MAVS at the mitochondrial membrane, and it can cleave MAVS at Cys-508 directly in vitro. We also provide direct evidence that the endogenous MAVS protein is cleaved in a HCV replicon cell line that failed to elicit IFN response. Importantly, a point mutation of MAVS at Cys-508 that prevents its cleavage restores IFN induction in the HCV replicon cell line. These results provide compelling evidence that MAVS is the prime target of the HCV protease. Our present work not only provides the direct biochemical evidence that MAVS is the proteolytic target of NS3/4A (Fig. 3) but also shows that the cleavage of endogenous MAVS at Cys-508 in the HCV replicon cell line is solely responsible for the suppression of IFN induction in these cells (Fig. 4). Furthermore, our finding that NS3/4A inactivates MAVS by cleaving and dislodging it from the mitochondria underscores the importance of mitochondrial localization of MAVS in antiviral immunity.
Infectious diseases are a manifestation of constant battles between the host and pathogenic microbes. This host–pathogen antagonism is now vividly demonstrated from the study of the interaction between MAVS and HCV. MAVS can orchestrate strong immune defense against HCV; however, HCV counterattacks by using the NS3/4A protease to cleave MAVS, thus crippling the immune response. Because a point mutation at Cys-508 of MAVS completely prevents its cleavage by NS3/4A and preserves its antiviral activity, it would be interesting to determine whether there are sequence variations of MAVS in human population that confer differential sensitivity to NS3/4A and, hence, differential immunity to HCV. However, the prevalence of persistent HCV infection in humans suggests that HCV might have won the battle between the virus and the host. In the next battle of “our wits versus their genes,” it may be possible to exploit the knowledge of MAVS–HCV interaction to fight back against HCV. For example, the cleavage of MAVS from the mitochondrial membrane may be a diagnostic marker for an established HCV infection. Furthermore, blocking the cleavage of MAVS by NS3/4A may be effective in the prevention and treatment of HCV.
HCV may not be the only pathogen that targets MAVS to evade the host immune system. Although the mitochondrial localization of MAVS may allow the host immune system to detect many viruses that replicate in intracellular membrane locations proximal to the mitochondria, it also may render MAVS vulnerable to viral attack. For example, it has recently been shown that the hepatitis A virus (HAV) inhibits IFN response at a step downstream of RIG-I but upstream of TBK1/IKKε (28), raising the possibility that MAVS may be a target of a HAV protein. Future studies should uncover more examples of host–pathogen interaction that revolve around the battle for MAVS to gain control of the host immune system.
Supplementary Material
Acknowledgments
We thank Dr. Michael Gale for providing the HCV replicon cell line K2040 (19). This work was supported by National Institutes of Health Grant R01-A I60919 and American Cancer Society Grant RSG0219501TBE. Confocal microscopy was supported by funding from a National Institutes of Health shared instrumentation grant 1-S10-RR19406. Z.J.C. is a Burroughs Wellcome Fund Investigator in Pathogenesis of Infectious Diseases.
Author contributions: X.-D.L., L.S., R.B.S., G.P., and Z.C. designed research; X.-D.L., L.S., R.B.S., and G.P. performed research; X.-D.L., L.S., R.B.S., G.P., and Z.C. analyzed data; and Z.C. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: CARD, caspase recruitment domain; HA, hemagglutinin; HCV, hepatitis C virus; IRF3, IFN regulatory factor 3; Luc, luciferase; MAVS, mitochondrial antiviral signaling protein; NS, nonstructural; RIG-I, retinoic acid-induced gene I; SeV, Sendai virus; TM, transmembrane; miniMAVS, truncated MAVS protein containing only the CARD and TM domains.
See Commentary on page 17539.
Note. While this manuscript was in preparation, Meylan et al. (12) also reported the identification of MAVS/CARDIF as the target of NS3/4A.
References
- 1.Lindenbach, B. D. & Rice, C. M. (2005) Nature 436, 933–938. [DOI] [PubMed] [Google Scholar]
- 2.Chisari, F. V. (2005) Nature 436, 930–932. [DOI] [PubMed] [Google Scholar]
- 3.Foy, E., Li, K., Wang, C., Sumpter, R., Jr., Ikeda, M., Lemon, S. M. & Gale, M., Jr. (2003) Science 300, 1145–1148. [DOI] [PubMed] [Google Scholar]
- 4.Maniatis, T., Falvo, J. V., Kim, T. H., Kim, T. K., Lin, C. H., Parekh, B. S. & Wathelet, M. G. (1998) Cold Spring Harb. Symp. Quant. Biol. 63, 609–620. [DOI] [PubMed] [Google Scholar]
- 5.McWhirter, S. M., Tenoever, B. R. & Maniatis, T. (2005) Cell 122, 645–647. [DOI] [PubMed] [Google Scholar]
- 6.Silverman, N. & Maniatis, T. (2001) Genes Dev. 15, 2321–2342. [DOI] [PubMed] [Google Scholar]
- 7.Fitzgerald, K. A., McWhirter, S. M., Faia, K. L., Rowe, D. C., Latz, E., Golenbock, D. T., Coyle, A. J., Liao, S. M. & Maniatis, T. (2003) Nat. Immunol. 4, 491–496. [DOI] [PubMed] [Google Scholar]
- 8.Sharma, S., tenOever, B. R., Grandvaux, N., Zhou, G. P., Lin, R. & Hiscott, J. (2003) Science 300, 1148–1151. [DOI] [PubMed] [Google Scholar]
- 9.Yoneyama, M., Kikuchi, M., Natsukawa, T., Shinobu, N., Imaizumi, T., Miyagishi, M., Taira, K., Akira, S. & Fujita, T. (2004) Nat. Immunol. 5, 730–737. [DOI] [PubMed] [Google Scholar]
- 10.Seth, R. B., Sun, L., Ea, C. K. & Chen, Z. J. (2005) Cell 122, 669–682. [DOI] [PubMed] [Google Scholar]
- 11.Kawai, T., Takahashi, K., Sato, S., Coban, C., Kumar, H., Kato, H., Ishii, K. J., Takeuchi, O. & Akira, S. (2005) Nat. Immunol. 6, 981–988. [DOI] [PubMed] [Google Scholar]
- 12.Meylan, E., Curran, J., Hofmann, K., Moradpour, D., Binder, M., Bartenschlager, R. & Tschopp, J. (2005) Nature. [DOI] [PubMed]
- 13.Xu, L. G., Wang, Y. Y., Han, K. J., Li, L. Y., Zhai, Z. & Shu, H. B. (2005) Mol. Cell 19, 727–740. [DOI] [PubMed] [Google Scholar]
- 14.Breiman, A., Grandvaux, N., Lin, R., Ottone, C., Akira, S., Yoneyama, M., Fujita, T., Hiscott, J. & Meurs, E. F. (2005) J. Virol. 79, 3969–3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foy, E., Li, K., Sumpter, R., Jr., Loo, Y. M., Johnson, C. L., Wang, C., Fish, P. M., Yoneyama, M., Fujita, T., Lemon, S. M. & Gale, M., Jr. (2005) Proc. Natl. Acad. Sci. USA 102, 2986–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li, K., Foy, E., Ferreon, J. C., Nakamura, M., Ferreon, A. C., Ikeda, M., Ray, S. C., Gale, M., Jr., & Lemon, S. M. (2005) Proc. Natl. Acad. Sci. USA 102, 2992–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akira, S. & Takeda, K. (2004) Nat. Rev. Immunol. 4, 499–511. [DOI] [PubMed] [Google Scholar]
- 18.Kato, H., Sato, S., Yoneyama, M., Yamamoto, M., Uematsu, S., Matsui, K., Tsujimura, T., Takeda, K., Fujita, T., Takeuchi, O. & Akira, S. (2005) Immunity 23, 19–28. [DOI] [PubMed] [Google Scholar]
- 19.Sumpter, R., Jr., Wang, C., Foy, E., Loo, Y. M. & Gale, M., Jr. (2004) J. Virol. 78, 11591–11604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim, J. L., Morgenstern, K. A., Lin, C., Fox, T., Dwyer, M. D., Landro, J. A., Chambers, S. P., Markland, W., Lepre, C. A., O'Malley, E. T., et al. (1996) Cell 87, 343–355. [DOI] [PubMed] [Google Scholar]
- 21.Love, R. A., Parge, H. E., Wickersham, J. A., Hostomsky, Z., Habuka, N., Moomaw, E. W., Adachi, T. & Hostomska, Z. (1996) Cell 87, 331–342. [DOI] [PubMed] [Google Scholar]
- 22.Grakoui, A., McCourt, D. W., Wychowski, C., Feinstone, S. M. & Rice, C. M. (1993) J. Virol. 67, 2832–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kolykhalov, A. A., Agapov, E. V. & Rice, C. M. (1994) J. Virol. 68, 7525–7533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwer, B., Ren, S., Pietschmann, T., Kartenbeck, J., Kaehlcke, K., Bartenschlager, R., Yen, T. S. & Ott, M. (2004) J. Virol. 78, 7958–7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mottola, G., Cardinali, G., Ceccacci, A., Trozzi, C., Bartholomew, L., Torrisi, M. R., Pedrazzini, E., Bonatti, S. & Migliaccio, G. (2002) Virology 293, 31–43. [DOI] [PubMed] [Google Scholar]
- 26.Kasprzak, A., Seidel, J., Biczysko, W., Wysocki, J., Spachacz, R. & Zabel, M. (2005) Liver Int. 25, 896–903. [DOI] [PubMed] [Google Scholar]
- 27.Lohmann, V., Korner, F., Koch, J., Herian, U., Theilmann, L. & Bartenschlager, R. (1999) Science 285, 110–113. [DOI] [PubMed] [Google Scholar]
- 28.Fensterl, V., Grotheer, D., Berk, I., Schlemminger, S., Vallbracht, A. & Dotzauer, A. (2005) J. Virol. 79, 10968–10977. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}