

Abstract
A semilinear in-slide model is introduced to remove the intensity effect in the scanning process. It is demonstrated that the intensity effect can be estimated accurately and removed effectively. This normalization step is vital for Affymetrix arrays to reveal relevant biological results when comparing gene expression in multiple arrays. The normalized expression ratios are analyzed further by a modified two-sample t test along with a sieved permutation scheme for computing P values. The improved specificity and sensitivity are demonstrated by using a study on the impact of macrophage migration inhibitory factor (MIF) reduction in neuroblastoma cells. With semilinear in-slide model analysis, expression of 166 genes was altered with a P value no greater than 0.001. Among those genes, 44 were altered >2-fold. MIF-regulated genes associated with tumor development including IL-8 and C-met, which are overexpressed in many tumors, were down-regulated in MIF-reduced cells. On the other hand, some tumor-suppressor genes such as EPHB6, visinin-like protein 1 (VSNL-1), and BLU were up-regulated in MIF-reduced cells. In addition, we demonstrated that down-regulation of MIF expression could result in a reduction in cell proliferation and tumor growth in vitro and in vivo. Our data not only demonstrate that targeting MIF expression is a promising therapeutic strategy in human neuroblastoma therapy but also indicate the MIF target genes for additional study.
Keywords: MIF, normalization, signficance analysis, SLIM
Affymetrix GeneChip arrays (1, 2) have been widely used for monitoring mRNA expression in many areas of biomedical research. The high-density oligonucleotide array technology allows researchers to monitor tens of thousands of genes in a single hybridization experiment as they are expressed differently in tissues and cells. The expression profile of an mRNA molecule of a gene is obtained by the combined intensity information from probes in a probe set, which consists of 11-20 probe pairs of oligonucleotides of 25 bp in length, interrogating a different part of the sequence of a gene. Several techniques have been proposed for extracting expression profiles from the information at probe level. These techniques include the detection signals that are prominently featured in Affymetrix Micorarray Suite 5.0 (mas 5.0) software, the model-based expression index (3), and the robust multichip average (4, 5). A comprehensive review and comparisons of various expression measures are given in refs. 6 and 7.
To account for the overall brightness of scanned images and other experimental variations such as differences in sample preparation and array production, normalization is needed for direct array-to-array comparisons. Several methods (7-10) have been proposed for normalizing data at probe level, and their impacts on the analysis of gene expressions have been examined. However, most researchers use detection signals from mas 5.0 as starting points of their investigation. Yet, as reported below, the techniques for probe-level normalization are not effective at the detection-signal level. Indubitably, there are strong demands for normalization methods based on the output of detection signals. Indeed, we are not aware of any methods for normalization of Affymetrix arrays at the detection-signal level.
Proper normalization is critical for revealing relevant biological results. Although the detection algorithm for mas 5.0 was designed carefully, systematic biases such as the intensity effect still exist, and they should be removed before making multiple-array comparisons. One aim of this article is to develop a normalization and analysis system based on sound statistical principles. The observed detection signals are decomposed into three components: the treatment effect, intensity effect, and random noise. With very mild statistical assumptions, the intensity effect can be estimated accurately and removed effectively as long as there are replications in treatment arrays. The normalized log ratios are then analyzed by a modified two-sample t test. The P values are estimated by a permutation technique that aims at reducing biases of the estimates and effective use of data. The resulting comprehensive system will be called the semilinear in-slide model (SLIM) system.
The sensitivity and specificity of the SLIM system are demonstrated by our application to microarray analysis of neuroblastoma cells with macrophage migration inhibitory factor (MIF) being reduced. Three treatment and three control arrays were analyzed by the SLIM. Among ≈200 genes that are identified significantly differently expressed, 12 important ones are selected for additional biological confirmation by using real-time quantitative RT-PCR, Western blot analysis, and ELISA. All of them were biologically confirmed. On the other hand, without normalization, two important genes were missed, giving a missed-discovery rate of 17%. By using the mas 5.0 system, the missed-discovery rate was 29% (see Discussion for additional details).
Neuroblastoma is a malignant tumor of neural crest origin that may arise anywhere along the sympathetic ganglia or within the adrenal medulla. Neuroblastoma is the most frequent solid extracranial neoplasia in children and is responsible for ≈15% of all pediatric cancer deaths (11). Spontaneous regressions and differentiation are common in infants and in early-stage tumors, whereas neuroblastoma is extremely aggressive in older children with late-stage tumors (12). MIF has begun to be recognized recently as a protumorigenic factor (13) in addition to its effects on proinflammatory and immune responses. Although a growing body of data have been gathered on the expression of MIF in several tumors, the exact mechanism of its function is unknown. Our previous results revealed that MIF was highly expressed in neuroblastoma and MIF could stimulate oncogene N-myc expression and up-regulate the expression of angiogenic factors (14). Furthermore, MIF is able to regulate the expression of genes that are related to tumor cell proliferation, migration, and antiapoptosis (15). These results suggested that MIF may play an important role in the development of neuroblastoma.
The biological aim of this study was to investigate whether MIF would be a target for limiting neuroblastoma development, that is, whether reduction of MIF expression could control tumor proliferation and tumorigenicity in neuroblastoma. A decrease in MIF expression in neuroblastoma cells was observed after transfection with MIF antisense expression vector. We demonstrated that down-regulation of MIF expression could result in a reduction in cell proliferation and tumor growth in vitro and in vivo. To better understand the MIF molecular mechanism in neuroblastoma, we used Affymetrix GeneChip to identify genes in MIF-reduced neuroblastoma cells. As reported below, gene-expression profiling analyzed by SLIM, validated at mRNA and protein levels for selected gene products, provides a comprehensive view of molecular changes in MIF-reduced tumor cells and reveals possible underlying molecular unidentified pathogenesis.
Materials and Methods
Cells. Human neuroblastoma cell line SK-N-DZ was obtained from the American Type Culture Collection. Cells were maintained in DMEM supplemented with 10% FBS (GIBCO/BRL-Life Technologies, Grand Island, NY) and 1% penicillin-streptomycin in a humidified incubator (95% air/5% CO2) at 37°C.
Construction of Expression Vectors and Transfection of Neuroblastoma Cells. An MIF antisense expression plasmid was constructed by cloning human MIF cDNA that corresponds to nucleotides 98-445 (GenBank accession no. NM_002415) with HindIII and XhoI adaptors in 3′-5′ orientation into pCMV-containing expression vector pSecTag2/Hygro (Invitrogen). The pSec/antisense MIF expression vector or pSec vector alone was transfected into neuroblastoma cell line SK-N-DZ with Lipofectamine 2000 (Invitrogen), and stable clones were selected by resistance to hygromycin (300 μg/ml; Geneticin-Life Technologies, Gaithersburg, MD) and expanded into cell lines. Three clones (3, 5, and 24) transfected with MIF antisense-expressing vectors were analyzed for MIF expression by Western blot. All three clones showed low levels of MIF expression (Fig. 1A). The cells transfected with vector alone were used as a control.
Fig. 1.

Normalization of detection signals based on mas 5.0.(A) Clone 3. Log ratios were plotted against log intensities for a treatment array along with the lowess fit for the conditional mean function (middle curves) and the conditional SD function. (B) The conditional mean and SD functions for all three treatment arrays are depicted for comparison. The bottom three curves are the mean curves, and the top three curves are their associated SD curves.
RNA Isolation and Application. Total RNA was prepared from cell culture with TRIzol reagent (Invitrogen) according to manufacturer directions. Probe synthesis and hybridization of Affymetrix GeneChip HG-U133A 2.0 microarrays (Affymetrix, Santa Clara, CA) were performed by following manufacturer instructions. Gene-expression data (CHP file of mas 5.0 software) were normalized to a global target intensity of 500.
Preprocessing of Detection Signals. The detection signals of 22,283 probe sets were extracted from the Affymetrix mas 5.0 software, which resulted in three expression profiles, respectively, from the control and treatment groups (three cell lines 3, 5, and 24). The repeatability, assessed by the coefficient of variation, is low for genes with small detection signals. Thus, only genes with all detection signals >50 were considered, which filtered 37.3% of genes, with 13,980 genes remaining for additional investigation. The percentage is actually smaller than those called “absent” by the Affymetrix mas 5.0 software (somewhat >50%). The remaining genes had much smaller coefficients of variations (better repeatability). The repeatability in the control group is somewhat better but is in the same bulk.
SLIM Normalization. Let xgi and ygi be the log-detection signal of the gth probe set in the ith control and treatment array, respectively. Denote by x̄g the average of the log-detection signals over the control arrays for the g probe set. Similar to that in ref. 16, we first computed the log intensities and log ratios, respectively, as follows:
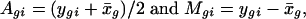 |
[1] |
for i = 1,..., n and g = 1,..., G. In our particular application, n = 3 and G = 13,980. Fig. 1A depicts the log ratios versus log intensities for a treatment array, along with the lowess fit for the conditional mean and conditional SD curves (17, 18) for the array.
The SLIM in ref. 15 and two-way semilinear model in refs. 19 and 20 were extended to remove the intensity effect. Let μg be the treatment effect on gene g and fi(·) represent the array-dependent intensity effect. The model
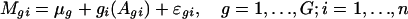 |
[2] |
was used to assess the intensity effect fi(Agi) and treatment effect μg on genes. Once the intensity effect is estimated, the normalized log ratios are given by
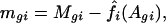 |
[3] |
for an estimated intensity effect f̂i(·).
The parameters in SLIM can be estimated by the profile least-squares method. Let Mi, μ, and fi denote, respectively, the vector of length G for the log ratios, treatment effect, and intensity effect. For the given estimated treatment effect 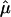 , the intensity effect is estimated by smoothing {
, the intensity effect is estimated by smoothing {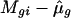 } on the intensities {Agi} separately for each array, resulting in
} on the intensities {Agi} separately for each array, resulting in
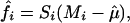 |
[4] |
for a smoothing operator Si (see, e.g., ref. 21), whereas for given estimated intensity effect functions f̂i,
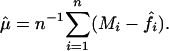 |
[5] |
Solving Eqs. 3 and 4 yields the explicit solution
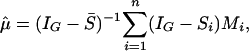 |
[6] |
and f̂i in Eq. 3, where IG is the G × G identity matrix and 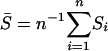 . Inverting the matrix in the order of tens of thousands is not practically feasible, and the solution can be computed by the G-Seidel or back-fitting algorithm (21, 22), which iteratively computes Eqs. 3 and 4.
. Inverting the matrix in the order of tens of thousands is not practically feasible, and the solution can be computed by the G-Seidel or back-fitting algorithm (21, 22), which iteratively computes Eqs. 3 and 4.
A Theoretical Analysis of SLIM. For the normalization purpose, the number of nuisance parameters {μg} is large, consisting of 1/n of the total sample size nG. Can the functions fj be estimated accurately? In a different context and somewhat different model, it was demonstrated (20, 22) that the rate of convergence for estimating fi is G-2/5. Because G is large, the intensity functions can be estimated accurately.
To appreciate the estimability of the intensity effect in Eq. 1, consider the case that n = 2. Let zg = Mg1 - Mg2. Then,
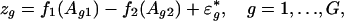 |
[7] |
where 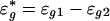 is the new random noise. Eq. 6 is an additive model (21), and its components f1 and f2 can be estimated as though one of them were known (23-26). The cost for estimating parameters {μg} reflects in the increase of the variability of the noise {
is the new random noise. Eq. 6 is an additive model (21), and its components f1 and f2 can be estimated as though one of them were known (23-26). The cost for estimating parameters {μg} reflects in the increase of the variability of the noise {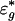 }. High correlation between the intensities of arrays {Ag1} and {Ag2}, however, reduces the accuracy of the estimated intensity effect.
}. High correlation between the intensities of arrays {Ag1} and {Ag2}, however, reduces the accuracy of the estimated intensity effect.
To gain an idea of the degree of accuracy in the estimation of the intensity effect, we simulated 10 data sets from the model in Eq. 1, with true parameters and functions taken from those estimated from our data. The new random noises in Eq. 1 were generated from the normal distribution with a mean of 0 and SD of 0.12. The mean absolute deviation error for the estimation of the intensity effect is ≈0.0134 ± 0.0003, which is negligible for our application. See ref. 22 for more intensive simulation studies.
Analysis of Treatment Effect. After obtaining the normalized ratio mgi, significant genes were obtained by testing the following hypothesis separately:
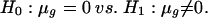 |
[8] |
As to be demonstrated, it is reasonable to assume that the errors in Eq. 1 are homoscedastic across arrays, because the intensity Agi does not vary much across arrays. Indeed, we adapted the weighted t statistic in ref. 15 to the current problem and did not find significantly different results.
Let 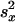 and
and 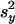 represent the sample variance of {xgj} and {ygj}, which are of sample sizes n1 and n2, respectively. Regarding the log intensities {xgj} and {ygj} as two random samples from two populations with the same variance, a test statistic (15, 27) for Eq. 7 is
represent the sample variance of {xgj} and {ygj}, which are of sample sizes n1 and n2, respectively. Regarding the log intensities {xgj} and {ygj} as two random samples from two populations with the same variance, a test statistic (15, 27) for Eq. 7 is
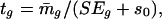 |
[9] |
where m̄g is the average of the normalized log ratios in Eq. 2, and 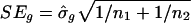 with
with
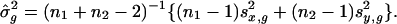 |
[10] |
The constant s0 is used here to guard against the zero denominator in Eq. 8. Note that m̄g is the difference between averages of treatment and control arrays with the intensity effect removed.
The constant s0 plays two important roles in identifying significant genes. First, it reduces the false-discovery rate (FDR) (28, 29) by an appropriate choice of s0. Because tens of thousands of genes are involved, there is a high chance that some of the SDs 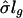 will be extremely small, which leads to a large test statistic and a possible falsely discovered gene. Second, a gene needs to satisfy a minimum fold-change requirement to be identified as a statistically significant gene. For example, if a gene is considered significant when |tg| > 3, then the fold change |m̄g| > 3s0. Thus, with the tuning constant s0, the P values and the fold changes are both taken into consideration in one single statistic. In our implementation, s0 = 0.07 was used.
will be extremely small, which leads to a large test statistic and a possible falsely discovered gene. Second, a gene needs to satisfy a minimum fold-change requirement to be identified as a statistically significant gene. For example, if a gene is considered significant when |tg| > 3, then the fold change |m̄g| > 3s0. Thus, with the tuning constant s0, the P values and the fold changes are both taken into consideration in one single statistic. In our implementation, s0 = 0.07 was used.
Results
MIF Expression in Cells Transfected with MIF Antisense Expression Vector. MIF expression in control (vector alone) and MIF antisense expression vector-transfected cells (clones 3, 5, and 24) was detected by Western blot analysis. The result showed that the amount of MIF was substantially reduced in three clones with MIF antisense vector-transfected cells compared to control cells (empty vector transfection; Fig. 2A). The protein expression levels of β-actin (used as internal control to confirm equivalent protein loading) in control and MIF antisense vector-transfected cells were nearly identical.
Fig. 2.

Effect of MIF suppression in vitro and in animal model. (A) Expression of MIF in control and MIF-suppressed neuroblastoma cells assessed by Western blot. (Upper) MIF has successfully been reduced compared with control. (Lower) Confirmation of the equivalent protein loading by using the house-keeping gene “β-actin” as internal control. (B) The average tumor growth curves for the MIF-reduced and control groups. The black curve indicates the control group (n = 10), and the three colored curves indicate the MIF-reduced group (total sample size, n = 30).
Tumor Growth in Nude Mice. The effect of suppressing MIF expression on tumor growth was further investigated in athymic nude mice. Six-week-old BALB/c nude mice were injected s.c. with 2 × 106 SN-K-DZ cells transfected with vector alone (n = 10, control group) or MIF-reduced cells (i.e., clones 3, 5, and 24; n = 10 for each clone). The mice were monitored for tumor growth after 20 days of inoculation for 10 days. Tumor growth in two groups was monitored everyday by two-dimensional measurements of individual tumors from each mouse. Mice in the treatment group showed an obvious reduction in tumor growth compared to control mice (Fig. 2B).
Estimated Intensity Effect. Fig. 3A depicts the estimated intensity effects for three treatment arrays that are contrasted with the average of the control arrays. The intensity effect tends to be smaller in the middle than at tails. When the algorithm converges, the residuals {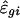 } can be obtained. To examine the degree of heteroscedasticity for the significant analysis of the treatment effect, the squared partial residuals {
} can be obtained. To examine the degree of heteroscedasticity for the significant analysis of the treatment effect, the squared partial residuals {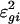 } were smoothed against the log intensity {Agi} for each treatment array, which results in three SD curves (Fig. 3B).
} were smoothed against the log intensity {Agi} for each treatment array, which results in three SD curves (Fig. 3B).
Fig. 3.

Effect of normalization of detection signals from mas5.0 and summary of treatment effects on gene expressions. (A) Estimated intensity effects for three treatment arrays. They were removed from the log ratios for significant analysis. (B) Estimated SD curves for the residuals in the model in Eq. 1. They were used to judge whether weighted t statistics are needed. (C) Summary of fold changes {μg} caused by the treatment. A reduced range is used to get a better view of the histogram. (D) The average log intensities for the treatment and control arrays along with genes (magenta) being selected at a level of α = 0.001.
P Value and FDRs. The P value was computed by using the permutation test (30, 31). To reduce possible biases in the permutation, only genes with P values, computed under the t distribution with degree of freedom m + n - 2, of >5% were considered. This criterion filtered some genes that are likely significant and left us with 13,897 genes. This step reduces the biases in the permutation test. There are 10 possible permutations among three treatment and three control arrays, resulting in 138,970 test statistics. By using these statistics as an estimate of the null distribution of tg, the P values were obtained. We called such a technique the sieved permutation. The basic assumption in the approach is that the null distribution of tg is the same for all insignificantly differently expressed genes, and filtering those that might be significant reduces the bias of the estimated null distribution. Another possible method for reducing the bias in permutation is the balanced permutation (15, 27), in which half of the treatment arrays swap with half of the control arrays in each permutation. In contrast with the balanced permutation, the sieved permutation uses effectively more sample (permuting all permutations on the sieved genes) and can be applicable to the case in which the number of treatment or control arrays is odd. The technique can be combined with the balanced permutation method to reduce further the biases in the permutation test when the numbers of treatment and control arrays are even and large.
Genes with P values no greater than α = 0.001 were selected as statistically significant genes. The FDR among the selected genes was estimated by p̂ = (Gα)/N, where N is the number of genes “discovered,” having P values no greater than α, and Gα is the expected number of falsely discovered genes. For example, at α = 0.001, 166 genes were discovered, and false discovery of 0.001 × 13,980 = 13.98 genes is expected, giving an FDR of 8.4%. This simple method is somewhat more conservative but is almost equivalent to the Benjamini-Hochberg method (28) for controlling FDR at the data-dependent level p̂. In other words, if the Benjamini-Hochberg method is used with the FDR controlled at p̂, the same set of genes will be selected frequently. Our method provides an intuitive understanding to the Benjamini-Hochberg method and also gives statistical justification (28, 32) for our method.
To appreciate this connection, recall that to control FDR at level p, the Benjamini-Hochberg selects
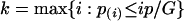 |
[11] |
genes with the k smallest P values, where p(1) ≤ p(2) ≤... ≤ p(G) are the order statistic of the P values. Now, if we take p = p̂ = Gα/N, we would select
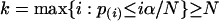 |
[12] |
genes with the smallest P values. If p(n+1) > α(n + 1)/N, then k = N; namely, we have selected the same set of genes as the Benjamini-Hochberg method with FDR controlled at p̂. This simple connection can also provide deeper understanding to the probabilistic properties of the Benjamini-Hochberg method. For example,
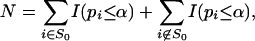 |
[13] |
where S0 = {g: μg = 0} is the set of genes that do not express differently under treatment. The probabilistic aspect N can be understood easily. Assume ideally that there are G1 very differently expressed genes. Under very mild conditions, N ≈ Gα + G1, where G1 is the number of significantly differently expressed genes. Hence, p̂ ≈ Gα/(Gα + G1). Hence, the smaller the α, the lower the FDR.
Table 1 summarizes the results by using the SLIM, the quantile normalization in refs. 5 and 7, and no normalization.
Table 1. Number of genes that are up- or down-regulated after MIF being inhibited.
| α = 5%
|
α = 1%
|
α = 0.5%
|
α = 0.1%
|
α = 0.05%
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Methods | Up | Down | FDR, % | Up | Down | FDR, % | Up | Down | FDR, % | Up | Down | FDR, % | Up | Down | FDR, % |
| SLIM | 1452 | 1754 | 21.8 | 540 | 483 | 13.6 | 321 | 263 | 12.0 | 101 | 65 | 8.4 | 59 | 43 | 6.9 |
| Q-norm | 1037 | 1046 | 33.6 | 339 | 318 | 21.3 | 212 | 176 | 18.0 | 73 | 54 | 11.0 | 42 | 38 | 8.7 |
| No-norm | 944 | 1149 | 33.4 | 303 | 345 | 21.6 | 190 | 199 | 18.0 | 62 | 60 | 11.5 | 43 | 39 | 8.5 |
| CommGene† | 718 | 894 | 217 | 265 | 135 | 145 | 46 | 26 | 29 | 55 | |||||
| CommGene‡ | 914 | 1017 | 290 | 300 | 182 | 170 | 62 | 51 | 38 | 37 | |||||
| CommGene§ | 732 | 1006 | 219 | 298 | 137 | 170 | 45 | 50 | 29 | 37 | |||||
Number of common genes (CommGene) selected by the following combinations: SLIM normalization and the quantile normalization (Q-norm) (†), quantile normalization and no normalization (No-norm) (‡), and SLIM normalization and no normalization (§).
Gene-Expression Profiling in MIF-Reduced Cells. Of a total of ≈22,283 probe sets, we discovered 166 genes showing significantly (P ≤ 0.001) changed expression in MIF-reduced cells compared to control cells, 44 of which were altered >2-fold. The genes that were found to be up- or down-regulated are shown in Table 2.
Table 2. Affymetrix analysis of genes regulated by antisense MIF transfection.
| Affymetrix ID no. | GenBank accession no. | Description | Fold change | P value† |
|---|---|---|---|---|
| 205943_at‡ | NM_005651 | Tryptophan 2,3-dioxygenase (TDO2) | −3.15 ± 0.16 | 0.0000 |
| 202859_at‡ | NM_000584 | IL-8 | −13.22 ± 0.40 | 0.0000 |
| 202023_at | NM_004428 | Ephrin-A1 (EFNA1) | −2.66 ± 0.15 | 0.0000 |
| 213624_at | AA873600 | Acid sphingomyelinase-like phosphodiesterase | −2.38 ± 0.14 | 0.0000 |
| 202644_at‡ | NM_006290 | Tumor necrosis factor, α-induced protein 3 (TNFAIP3) | −3.48 ± 0.18 | 0.0000 |
| 211343_at‡ | M33653 | Type IV collagen (COL4A2) | 4.27 ± 0.28 | 0.0000 |
| 203797_at‡ | AF039555 | VSNL1 | 1.58 ± 0.08 | 0.0007 |
| 204718_at‡ | NM_004445 | EphB6 | 2.64 ± 0.22 | 0.0022 |
| 207196_at | NM_006058 | Nef-associated factor 1 (NAF1) | −2.79 ± 0.23 | 0.0036 |
| 205205_at‡ | NM_006509 | Reticuloendotheliosis viral oncogene homolog B | −1.91 ± 0.16 | 0.0043 |
| 215223_at | W46388 | Mitochondrial superoxide dismutase 2 | −4.8 ± 0.40 | 0.0094 |
| 205067_at | NM_000576 | IL-1 | −3.92 ± 0.35 | 0.0108 |
| 201438_at‡ | NM_004369 | Collagen type VI, 3 (COL6A3) | 2.37 ± 0.23 | 0.0160 |
| 207096_at | NM_006512 | Serum amyloid A4 (SAA4) | −1.86 ± 0.17 | 0.0209 |
| 208378_at‡ | NM_004464 | Fibroblast growth factor 5 (FGF5) | −1.46 ± 0.11 | 0.0259 |
| 203381_at | N33009 | Apolipoprotein E | 3.12 ± 0.37 | 0.0532 |
| 213136_at | AI828880 | Protein tyrosine phosphatase, type 2 | −2.39 ± 0.28 | 0.0532 |
| 213807_at‡ | BE870509 | C-met protooncogene (hepatocyte growth factor receptor) | −1.58 ± 0.14 | 0.0338 |
| 213416_at | BG532690 | Integrin 4 (antigen CD49D) | 2.62 ± 0.31 | 0.0669 |
| 216663_at‡ | AC002481 | BLU | 1.79 ± 0.20 | 0.0799 |
| 208820_at | AL037339 | Protein tyrosine kinase 2 (PTK2) | −1.54 ± 0.14 | 0.0835 |
| 200636_at | NM_002840 | Protein tyrosine phosphatase, receptor type F (PTPRF) | −1.43 ± 0.14 | 0.0741 |
| 205534_at | NM_002589 | BH-protocadherin (brain-heart) (PCDH7) | 2.49 ± 0.29 | 0.0446 |
| 203582_at‡ | NM_004578 | RAS oncogene family (RAB4) | −1.36 ± 0.12 | 0.1710 |
Multiplied by 100.
The gene has been confirmed by real-time RT-PCR, ELISA, or Western blot.
Validation of a Subset of Differentially Expressed Genes. To examine the reliability of the microarray data and identify molecules important in the pathogenesis of MIF, 12 sequences (six up-regulated and six down-regulated) were chosen from the lists of up- or down-regulated genes for additional validation. These 12 genes were chosen on the basis of their association with tumor growth, angiogenesis, and metastasis. Each gene or its protein products were validated by real-time quantitative RT-PCR, Western blot, or ELISA. All validation results for these 12 genes on changes in gene expression or protein products are consistent with Affymetrix microarray data. The most interesting genes confirmed were c-met, visinin-like protein 1 (VSNL-1), EPHB6, BLU, and interleukin 8 (IL-8).
Discussion
Comparison with Other Methods. The effectiveness of SLIM was compared with no normalization and the quantile normalization (although it was proposed for normalization at probe level). The distributions for the differences between the log ratios after and before normalizations are summarized in Fig. 4. It is clear that the quantile normalization does not much alter the log ratios of detection signals; they are approximately the same as those without normalization. From Table 1, the quantile normalization and no normalization result in higher FDRs. They also discovered some different genes from those with normalization. For example, among 12 confirmed ones, two important genes (BLU and RAS) were accorded statistical significance if the SLIM normalization was used, but not under the quantile normalization or no normalization, which provides additional evidence to support the SLIM-normalization method.
Fig. 4.

Histograms for the differences between the log ratios after and before the SLIM normalization (A), the quantile normalization for the treatment group (B), and the quantile normalization for the pooled sample (C). The pooled sample consists of both treatment and control arrays. Reduced ranges are used to get a better view of the histograms.
The pairwise comparisons were also conducted by using the Affymetrix mas 5.0 software. A total of nine possible pairwise comparisons were conducted. Among 12 confirmed ones, three important genes (VSNL-1, BLU, and C-met) were called “NC,” indicating no change consistently across all nine comparisons. Among 9 × 12 = 108 comparisons, 29 were falsely called “NC,” which gives a missed-discovery rate of 27%. The lesson is very clear: even with the most advanced Affymetrix microarrays to date, normalization is required, and replications are needed for the discovery of slightly and moderately differently expressed genes. Replications are needed for both treatment and control arrays to reduce sampling variability.
Biological Implications. C-met was identified as an activated oncogene (33, 34). A large number of studies reveal that C-met is frequently expressed in carcinomas, in other types of human solid tumors, and in their metastases. Overexpression of C-met is often associated with poor prognosis (35). Our unpublished data showed that MIF expression significantly correlated with C-met protein expression in human neuroblastoma specimens (r = 0.635; P = 0.002; n = 32), which suggests that C-met and MIF may be closely related. In the present study, C-met was decreased in MIF-reduced cells, which strongly indicates that MIF may have a function in the up-regulation of C-met expression.
VSNL-1 is expressed in the central nervous system, where it plays a crucial role in regulating cAMP levels, cell signaling, and differentiation. High-level VSNL-1 expression has been found in less aggressive and high-grade squamous cell carcinoma (36). These results indicate that VSNL-1 plays an important role in regulating tumor cell invasiveness and that its loss could help to enhance the advanced malignant phenotype.
EPHB6 is a favorable neuroblastoma gene. EPHB6 is down-regulated in the most aggressive neuroblastoma cell lines, and a prognostic indicator in neuroblastoma and other tumors such as melanoma and breast cancer (37-40). High-level expression of EPHB6 predicts favorable neuroblastoma outcome, and the expression of this gene inhibits growth of unfavorable neuroblastoma cells (38). However, it is not clear how EPHB6 is regulated. It was very interesting to find that EPHB6 was increased in MIF-reduced neuroblastoma cells. This result suggests a valuable line of advance for additional study of the relationship between MIF and EPHB6.
BLU is abundantly expressed in normal lung tissue. However, its expression is markedly reduced in a subset of lung tumor cell lines (41). BLU is also inactivated in neuroblastoma. Methylation of the BLU promoter region (inactivation of BLU) in neuroblastoma was inversely correlated with tumor stage (41). These data suggest that BLU is one of the candidate tumor-suppressor genes.
IL-8 is a multifunctional CXC chemokine. It was identified as an angiogenesis-regulating molecule that induced angiogenesis (42, 43). The expression of IL-8 has been found in various human cancers (44). Recent studies have demonstrated that IL-8 regulates tumor cell growth and metastasis in many tumors such as melanoma and carcinoma of the breast, stomach, pancreas, and liver. Our previous results indicate that serum IL-8 from patients with hepatocellular carcinoma significantly correlates with tumor size, tumor stage, and venous invasion, which suggests that IL-8 may be involved directly or indirectly in the progression of hepatocellular carcinoma (45). We also demonstrated that MIF was able to stimulate neuroblastoma cells to express IL-8 (45). The present results revealed that IL-8 gene expression was markedly decreased in MIF-reduced cells (>13-fold), and this was confirmed further by the ELISA technique, which showed that IL-8 protein produced by MIF-reduced cells was 10 times less than that of control.
In summary, we have demonstrated that the use of antisense transfection when coupled with microarray and SLIM analysis provides a very useful system for defining the role of specific genes that are dysregulated in cancer. With this system, we are able to show that reduction of MIF results in tumor growth inhibition, and this suppression may be achieved by inhibiting angiogenic-related genes such as IL-8, oncogenes (C-met), and protein kinase. On the other hand, increasing the expression of tumor-suppressor genes (BLU, VSNL-1) and neuroblastoma favorite gene (EPHB6) may also contribute to tumor reduction. Therefore, these genes could potentially be key players in the development of neuroblastoma. Our data not only demonstrate that targeting MIF expression is a promising therapeutic strategy in human neuroblastoma therapy but also indicate the MIF target genes for additional study.
Acknowledgments
This research was supported by National Science Foundation Grant DMS-0354223 and University of Hong Kong Grant CRCG 10204242/35644/21700/323/01.
Author contributions: J.F., P.K.H.T., and Y.R. designed research; J.F., Y.C., H.M.C., and Y.R. performed research; J.F., Y.C., H.M.C., and Y.R. contributed new reagents/analytic tools; J.F. and Y.R. analyzed data; and J.F. and Y.R. wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: mas 5.0, Affymetrix Micorarray Suite, version 5.0; SLIM, semilinear in-slide model; MIF, macrophage migration inhibitory factor; FDR, false-discovery rate; VSNL-1, visinin-like protein 1.
References
- 1.Lockhart, D. J., Dong, H., Byrne M., Follettie, M., Gallo, M., Chee, M., Mittmann, M., Wang, C., Kobayashi, M., Horton, H., et al. (1996) Nat. Biotech. 14, 1675-1680. [DOI] [PubMed] [Google Scholar]
- 2.Lipschutz, R. J., Fodor, S., Gingeras, T. & Lockhart, D. J. (1999) Nat. Genet. 21, 20-24. [DOI] [PubMed] [Google Scholar]
- 3.Li, C. & Wong, W. H. (2001) Proc. Natl. Acad. Sci. USA 98, 31-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Irizarry, R. A., Hobbs, B., Collin, F., Beazer-Barclay, Y. D., Antonellis, K. J., Scherf, U. & Speed, T. P. (2003) Biostatistics 4, 249-264. [DOI] [PubMed] [Google Scholar]
- 5.Irizarry, R. A., Bolstad B. M., Collin, F., Cope, L. M., Hobbs, B., Speed, T. P. (2003) Nucleic Acids Res. 31, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lemon, W., Palatini, J., Krahe, R. & Wright, F. (2002) Bioinformatics 18, 1470-1476. [DOI] [PubMed] [Google Scholar]
- 7.Bolstad, B. M., Irizarry, R. A., Astrand, M. & Speed, T. P. (2003) Bioinformatics 19, 185-193. [DOI] [PubMed] [Google Scholar]
- 8.Hill, A. A., Brown, E. L., Whitley, M. Z., Tucker-Kellogg, G., Hunter, G. P. & Slonim, D. K. (2001) Genome Biol. 2, Research0055.1-Research0055.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schadt, E. E., Li, C., Ellis, B. & Wong, W. H. (2001) J. Cell. Biochem. Suppl. 37, 120-125. [DOI] [PubMed] [Google Scholar]
- 10.Astrand, M. (2003) J. Comput. Biol. 10, 95-102. [DOI] [PubMed] [Google Scholar]
- 11.Maris, J. M. & Matthay, K. K. (1999) J. Clin. Oncol. 17, 2264-2279. [DOI] [PubMed] [Google Scholar]
- 12.Pritchard, J. & Hickman, J. A. (1994) Lancet 344, 869-870. [DOI] [PubMed] [Google Scholar]
- 13.Michell, R. A. & Bucala, R. (2000) Semin. Cancer Biol. 10, 359-366. [DOI] [PubMed] [Google Scholar]
- 14.Ren, Y., Chan, H. M., Li, Z., Lin, C. L., Nicholls, J., Chen, Z. F., Lee, P. Y., Lui, V., Bacher, M. & Tam P. (2004) Oncogene 23, 4146-4154. [DOI] [PubMed] [Google Scholar]
- 15.Fan, J., Tam, P., Vande Woude, G. & Ren, Y. (2004) Proc. Natl. Acad. Sci. USA 101, 1135-1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dudoit, Y., Yang, Y. H., Callow, M. J. & Speed, T. P. (2002) Stat. Sin. 12, 111-139. [Google Scholar]
- 17.Cleveland, W. S. (1979) J. Am. Stat. Assoc. 74, 829-836. [Google Scholar]
- 18.Fan, J. & Yao, Q. (1998) Biometrika 85, 645-660. [Google Scholar]
- 19.Huang, J., Kou, H. C., Koroleva, I., Zhang, C. H. & Soares, M. B. (2003) Technical Report 321 (Dept. of Statistics, University of Iowa, Iowa City).
- 20.Huang, J., Wang, D. & Zhang, C. H. (2005) J. Am. Stat. Assoc. 100, 814-829. [Google Scholar]
- 21.Hastie, T. J. & Tibshirani, R. (1990) Generalized Additive Models (Chapman and Hall, London).
- 22.Fan, J., Peng, H. & Huang, T. (2005) J. Am. Stat. Assoc. 100, 781-796. [Google Scholar]
- 23.Stone, C. J. (1986) Ann. Stat. 14, 590-606. [Google Scholar]
- 24.Stone, C. J. (1994) Ann. Stat. 22, 118-184. [Google Scholar]
- 25.Fan, J., Härdle, W. & Mammen, E. (1998) Ann. Stat. 26, 943-971. [Google Scholar]
- 26.Mammen, E., Linton, O. & Nielsen, J. (1999) Ann. Stat. 27, 1443-1490. [Google Scholar]
- 27.Tusher, V. G., Tibshirani, R. & Chu, G. (2001) Proc. Natl. Acad. Sci. USA 98, 5116-5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benjamini, Y. & Hochberg, Y. (1995) J. R. Stat. Soc. B 57, 289-300. [Google Scholar]
- 29.Reiner, A., Yekutieli, D. & Benjamini, Y. (2003) Bioinformatics 19, 368-375. [DOI] [PubMed] [Google Scholar]
- 30.Dudoit, S., Shaffer, J. P. & Boldrick, J. C. (2003) Stat. Sci. 18, 71-103. [Google Scholar]
- 31.Ge, Y., Dudoit, S. & Speed, T. (2003) TEST 12, 1-77. [Google Scholar]
- 32.Benjamini, Y. & Yekutieli, D. (2001) Ann. Stat. 29, 1165-1188. [Google Scholar]
- 33.Cooper, C. S., Park, M., Blair, D. G., Tainsky, M. A., Huebner, K., Croce, C. M. & Vande Woude, C. F. (1984) Nature 311, 29-33. [DOI] [PubMed] [Google Scholar]
- 34.Park, M., Dean, M., Cooper, C. S., Schmidt, M., O'Brien, S. J., Blair, D. G. & Vande Woude, G. F. (1986) Cell 45, 895-904. [DOI] [PubMed] [Google Scholar]
- 35.Birchmeier, C., Birchmeier, W., Gherardi, E. & Vande Woude, C. F. (2003) Nat. Rev. Mol. Cell. Biol. 4, 915-925. [DOI] [PubMed] [Google Scholar]
- 36.Mahloogi, H., Gonzalez-Guerrico, A. M., Lopez De Cicco, R., Bassi, D. E., Goodrow, T., Braunewell, K. H. & Klein-Szanto, A. J. (2003) Cancer Res. 63, 4997-5004. [PubMed] [Google Scholar]
- 37.Tang, X. X., Robinson, M. E., Riceberg, J. S., Kim, D. Y., Kung, B., Titus, T. B., Hayashi, S., Flake, A. W., Carpentieri, D. & Ikegaki, N. (2004) Clin. Cancer Res. 10, 5837-5844. [DOI] [PubMed] [Google Scholar]
- 38.Tang, X. X., Zhao, H., Robinson, M. E., Cohen, B., Cnaan, A., London, W., Cohn, S. L., Cheung, N. K., Brodeur, G. M., Evans, A. E., et al. (2000) Proc. Natl. Acad. Sci. USA 97, 10936-10941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fox, B. P. & Kandpal, R. P. (2004) Biochem. Biophys. Res. Commun. 318, 882-892. [DOI] [PubMed] [Google Scholar]
- 40.Hafner, C., Bataille, F., Meyer, S., Becker, B., Roesch, A., Landthaler, M. & Vogt, T. (2003) Int. J. Oncol. 23, 1553-1559. [PubMed] [Google Scholar]
- 41.Agathanggelou, A., Dallol, A., Zochbauer-Muller, S., Morrissey, C., Honorio, S., Hesson, L., Martinsson, T., Fong, K. M., Kuo, M. J., Yuen, P. W., et al. (2003) Oncogene 22, 1580-1588. [DOI] [PubMed] [Google Scholar]
- 42.Desbaillets, I., Diserens, A. C., Tribolet, N., Hamou, M. F. & Van Meir, E. G. (1997) J. Exp. Med. 186, 1201-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koch, A. E., Polverini, P. J., Kunkel, S. L., Harlow, L. A., DiPietro, L. A., Elner, V. M., Elner, S. G. & Strieter, R. M. (1992) Science 258, 1798-1801. [DOI] [PubMed] [Google Scholar]
- 44.Xie, K. (2001) Cytokine Growth Factor Rev. 12, 375-391. [DOI] [PubMed] [Google Scholar]
- 45.Ren, Y., Poon, R. T. P., Tsui, H. T., Li, Z., Lau, C., Yu, W. C. & Fan, S. T. (2003) Clin. Cancer Res. 9, 5996-6001. [PubMed] [Google Scholar]