

Abstract
The human base excision repair machinery must locate and repair DNA base damage present in chromatin, of which the nucleosome core particle is the basic repeating unit. Here, we have utilized fragments of the Lytechinus variegatus 5S rRNA gene containing site-specific U:A base pairs to investigate the base excision repair pathway in reconstituted nucleosome core particles in vitro. The human uracil-DNA glycosylases, UNG2 and SMUG1, were able to remove uracil from nucleosomes. Efficiency of uracil excision from nucleosomes was reduced 3- to 9-fold when compared with naked DNA, and was essentially uniform along the length of the DNA substrate irrespective of rotational position on the core particle. Furthermore, we demonstrate that the excision repair pathway of an abasic site can be reconstituted on core particles using the known repair enzymes, AP-endonuclease 1, DNA polymerase β and DNA ligase III. Thus, base excision repair can proceed in nucleosome core particles in vitro, but the repair efficiency is limited by the reduced activity of the uracil-DNA glycosylases and DNA polymerase β on nucleosome cores.
Keywords: DNA repair/nucleosomes/uracil-DNA glycosylase
Introduction
The organization of DNA into chromatin in eukaryotic cells is likely to afford little protection against formation of DNA base damage generated spontaneously through hydrolysis, since DNA remains hydrated in chromatin (Wolffe, 1998). Two frequently occurring hydrolytic reactions are depurination to produce non-coding abasic sites, and deamination of cytosine to uracil (U) (Lindahl, 1993). Uracil can also occur in DNA through misincorporation of dUMP opposite adenine (A) residues during replication to generate U:A base pairs, the rate of misincorporation being proportional to the size of the dUTP pool (Goulian et al., 1980). Uracil residues in DNA are removed rapidly by base excision repair (BER), initiated by one of the two major uracil-DNA glycosylase activities in mammalian cells, UNG2 and SMUG1 (Nilsen et al., 1997; Haushalter et al., 1999; Nilsen et al., 2001). Mice deficient in the conserved UNG2 uracil-DNA glycosylase have increased levels of dUMP in their genome without a corresponding increase in spontaneous mutagenesis (Nilsen et al., 2000), indicating that UNG2 primarily removes uracil misincorporated during replication (Otterlei et al., 1999). Both UNG2 and SMUG1 are able to remove uracil from U:A as well as U:G base pairs, and the resulting AP-sites are repaired by the short-patch BER pathway. Briefly, AP-endonuclease 1 (APE1) incises the damaged strand 5′ to the AP-site generating a 3′-hydroxyl terminus and a 5′-deoxyribose phosphate (dRP) moiety. DNA polymerase β (Polβ) fills the one-nucleotide gap and excises the dRP moiety, and the remaining strand interruption is sealed by DNA ligase III (LigIII). XRCC1 also participates in this pathway as a scaffold protein interacting with Polβ and LigIII. The short-patch BER pathway has been reconstituted with recombinant human proteins and uracil-containing oligonucleotide substrates (Kubota et al., 1996).
Chromatin is composed of repeating units known as nucleosome core particles, which are comprised of 147 bp of DNA wrapped around a histone octamer (Wolffe, 1998). The assembly of DNA into nucleosomes restricts the access of several trans-acting factors (Luger et al., 1997; Wolffe, 1998), and different strategies have been adopted to overcome the steric and structural constraints imposed. During replication, the nucleosomes are destabilized in front of the fork and fully reassembled ∼250 bp behind the fork (Sogo et al., 1986; Gasser et al., 1996). Active transcription is associated with regions of altered chromatin structure in vivo, and inhibition of transcription from chromatin substrates in vitro can be relieved by chromatin-remodelling factors in concert with post-translational modification of core histones (Orphanides and Reinberg, 2000).
Efficient DNA repair requires identification and removal of DNA damage throughout the genome (Green and Almouzni, 2002). The efficiency of nucleotide excision repair (NER) of UV-induced DNA lesions is reduced in chromatin substrates (Smerdon and Conconi, 1999; Green and Almouzni, 2002). This effect is observed even with nucleosome core particles (Hara et al., 2000; Liu and Smerdon, 2000; Kosmoski et al., 2001). The chromatin-remodelling factor ACF has been shown to stimulate NER of UV lesions in dinucleosomes ∼2-fold in vitro, but only when the lesion was in the linker DNA. ACF had no effect when the lesions were on the surface of the nucleosome core (Ura et al., 2001). As NER involves ∼30 polypeptides and an extended stretch of repair synthesis (de Laat et al., 1999), it is not surprising that some inhibition of repair is observed with nucleosome cores where the accessibility of DNA is restricted. Mechanistically, BER is a simpler process than NER, and it seemed possible that the BER pathway might function efficiently in nucleosomes. We chose to address this question by measuring the rates of uracil excision by UNG2 and SMUG1 from U:A base pairs in mononucleosome substrates reconstituted with the Lytechinus variegatus 5S rRNA gene.
Results
Preparation of nucleosome core particles
Chicken erythrocyte histone octamers (Figure 1A) were reconstituted by stepwise salt dilution (Steger and Workman, 1999) onto a 146 bp DNA fragment containing the strong nucleosome positioning sequence from the L.variegatus 5S rRNA gene. Uracil-containing DNA substrates were prepared by PCR using 5′ 32P-labelled oligonucleotide primers to introduce a single dUMP in place of a dTMP residue at defined positions along a 146 bp 5S rDNA fragment (positions U19, U22 or U51 of the sense strand). Packaging of these DNA fragments into nucleosome core particles resulted in a band shift after separation in a non-denaturing polyacrylamide gel (Figure 1B). Reconstitution efficiencies of ∼90% were routinely obtained using DNA containing a site-specific uracil residue. Neither reconstitution efficiency nor nucleosome stability were influenced by the presence of uracil in position 19, 22 or 51 (data not shown), indicating that the presence of a single uracil residue is tolerated in the core particle. The reconstitution efficiency was consistently much lower (∼60%) with DNA substrates containing U residues at multiple positions. These core particles were purified through 5–25% sucrose gradients (data not shown). Footprinting experiments showed that naked DNA was cleaved randomly by DNase I, albeit with some sequence specificity (Figure 1C). Core particle DNA was cleaved preferentially at sites where the minor groove faces the solvent (Wolffe, 1998), resulting in a characteristic 10 bp ladder upon DNase I digestion (Figure 1C). These results are in good agreement with DNase I footprinting of 146 bp 5S rDNA core particles described previously (Richmond et al., 1988). Core particle DNA was protected against restriction enzyme digestion (see Figure 2A for positioning of restriction sites) by FokI, AluI and MboI (Figure 4B, lanes 5–7; data not shown), confirming that the DNA is firmly attached to the octamers near the ends of the DNA fragment.

Fig. 1. Reconstitution of nucleosome core particles. (A) Histone octamers isolated from chicken erythrocytes, consisting of equimolar amounts of the four core histones, shown after 18% acrylamide/SDS–gel electrophoresis and staining with Coomassie Brilliant Blue R. (B) The 146 bp fragment of the L.variegatus 5S rRNA gene containing a single uracil residue in position 19 (lane 1) was reconstituted into nucleosome core particles upon serial salt dilution in the presence of histone octamers, producing a characteristic band shift in a native 5% polyacrylamide gel (lane 2). (C) Naked DNA (DNA) and nucleosome core particles (NCP) containing a single uracil residue (U19) were subjected to DNase I footprinting using 0.2 (DNA) or 4 U of DNase I (NCP). Aliquots were removed after 0, 0.5, 1, 3, 6 and 15 (NCP only) min, denatured, and analysed in 8% polyacrylamide/7 M urea/20% formamide gels.
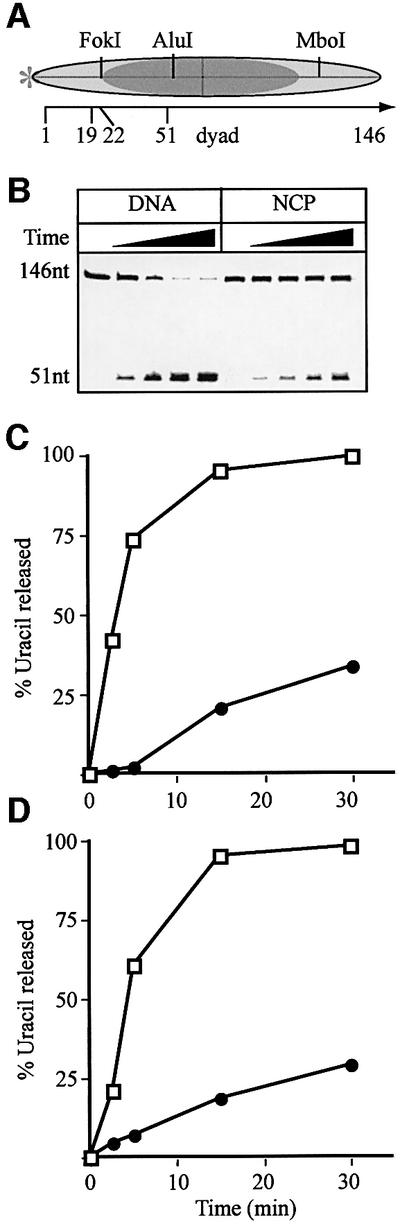
Fig. 2. Excision of uracil by the UNG2 DNA glycosylase. (A) Cartoon showing the histone octamer (shaded grey oval) positioned on the 5′-end-labelled (asterisk) 146 bp 5S rDNA fragment with the central 80 bp region (dark grey) tightly bound by the octamer. The positions of uracil residues, numbered in the 5′ to 3′ direction of the sense strand, as well as recognition sites for restriction enzymes, are indicated. (B) An 8% polyacrylamide/7 M urea/20% formamide gel showing the time course of excision of an internal uracil residue (U51) from naked DNA (DNA) and nucleosome core particles (NCP). (C) Average rate of uracil excision of U51 from naked DNA (open squares) and NCP (closed circles) from three independent experiments. Rates of uracil removal from core particles are corrected for uracil removal from naked DNA present in the core particle preparation. (D) Rates of excision from naked DNA (open squares) and NCP (closed circles) of a uracil residue situated at position 19 (U19; average of three independent experiments) after correcting for the presence of naked DNA. The SEM of uracil removal from U51 or U19 was <20%.
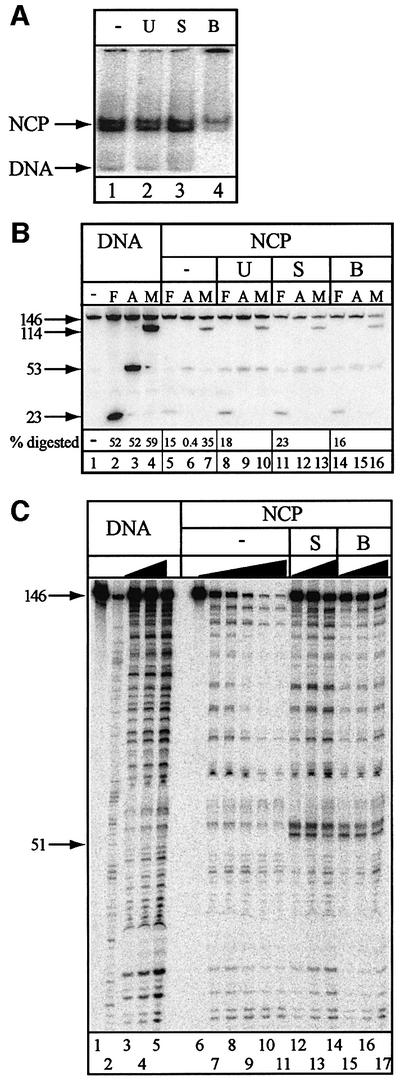
Fig. 4. Stability of 146 bp nucleosome core particles. (A) The stability of core particles was analysed by incubating uracil-containing (U51) core particles alone (–), with UNG2 (U), SMUG1 and APE1 (S) or with BER enzymes (UNGΔ84 in combination with APE1, Polβ and LigIII; B) for 30 min at 37°C and loaded directly on a native 5% polyacrylamide gel. (B) An 8% polyacrylamide/7 M urea/20% formamide gel showing restriction enzyme digestion of naked 146 bp DNA (lanes 1–4), with FokI (F), AluI (A) or MboI (M). Core particles prior to uracil removal and repair (–), and after incubation with UNG2 (U), SMUG1/APE1 (S) or BER enzymes (B) were digested with FokI (F), AluI (A) or MboI (M). (C) Naked DNA (DNA) and nucleosome core particles (NCP) were subjected to DNase I footprinting prior to uracil removal and repair. Aliquots were removed from naked DNA after 0, 0.5, 1 and 3 min (lanes 1 and 3–5) and from core particles after 0, 0.5, 1, 3, 6 and 15 min (lanes 6–11). A Maxam–Gilbert T-reaction was included (lane 2). Core particles incubated in the presence of SMUG1/APE1 (S) or BER enzymes (B) were subjected to DNase I digestion. Aliquots were taken after 0.5, 1 and 3 min, denatured and analysed in 8% polyacrylamide/7 M urea/20% formamide gels.
Removal of uracil by UNG2 from core particles containing a single U:A base pair
The effect of the nucleosome core particle on the initial step of BER was addressed by studying uracil removal by the UNG2 DNA glycosylase from substrates containing a single dUMP residue at a defined position along the 5S rDNA (positions U19, U22 or U51) (Figure 2A). The U:A-containing naked DNA or core particles were treated with equal amounts of UNG2 DNA glycosylase under standard enzyme assay conditions. Aliquots taken at various time points were heated in piperidine in order to cleave abasic sites. Uracil release was measured as the appearance of a shorter product band after denaturing polyacrylamide gel electrophoresis; representative results are shown for the U51 substrate (Figure 2B). After correcting for uracil removal from naked DNA present in the nucleosome substrate, a time course of uracil removal from U51 was generated (Figure 2C). The initial rate of uracil removal from the core particles (filled circles) was ∼30-fold slower than with naked DNA (open squares). However, after a 30 min incubation, only a 3-fold reduction in uracil removal was observed. A straightforward comparison of initial uracil excision rates in free DNA and nucleosome core particles is difficult because reconstituted core particles inevitably contain a small fraction of naked DNA (5–10%), which is repaired more rapidly than the core particles. This naked DNA competitor exacerbates the delay in core particle repair. A similar situation has been described previously with mixed sequence substrates (Nilsen et al., 1995).
Histone–DNA interactions are stronger in the central 80 bp of the nucleosome where the H3–H4 tetramer contacts the DNA (Figure 2A, dark oval) than in flanking sequences where the H2A–H2B dimers contact the DNA (Figure 2A, shaded oval) (Wolffe, 1998). However, uracil removal from U19 after 15 and 30 min incubations, when most of the uracil was removed from naked DNA, was impaired as strongly as from the more internal U51 residue (Figure 2D). Similar inhibition was observed with a U22 substrate (data not shown). The addition of purified replication protein A (RPA) or proliferating cell nuclear antigen (PCNA), which bind to the N-terminus of UNG2 (Otterlei et al., 1999), or APE1 did not facilitate uracil release from nucleosome cores (data not shown). Whereas uracil removal by UNG2 from naked DNA was essentially complete after 15 min, only ∼60% of the uracil residues were removed from core particles, even after prolonged incubation (up to 240 min) (Figure 2C and D; data not shown).
Removal of uracil by SMUG1 from core particles containing a single U:A base pair
The role of human UNG2 DNA glycosylase in removing uracil residues at DNA replication forks (Otterlei et al., 1999; Nilsen et al., 2000) suggests that this enzyme may not act on mature nucleosomes in vivo. The reduced uracil excision from core particles could therefore be a consequence of UNG2 having evolved to act on naked DNA or immature nucleosome structures immediately in front of or behind the replication fork (Krude, 1999). The SMUG1 DNA glycosylase, on the other hand, presumably functions throughout the genome independently of DNA replication (Nilsen et al., 2001). We therefore asked whether SMUG1 was better suited to remove uracil from nucleosome core particles. However, just as observed with UNG2, there was a 9-fold reduction of uracil excision from U51 (Figure 3), U19 and U22 (data not shown) when nucleosome core particles (filled circles) were compared with naked DNA (open squares) after correcting for the presence of free DNA in the nucleosome preparation. Thus, contrary to expectation, SMUG1 does not appear to have evolved specialized properties that facilitate efficient removal of uracil from nucleosomal DNA.
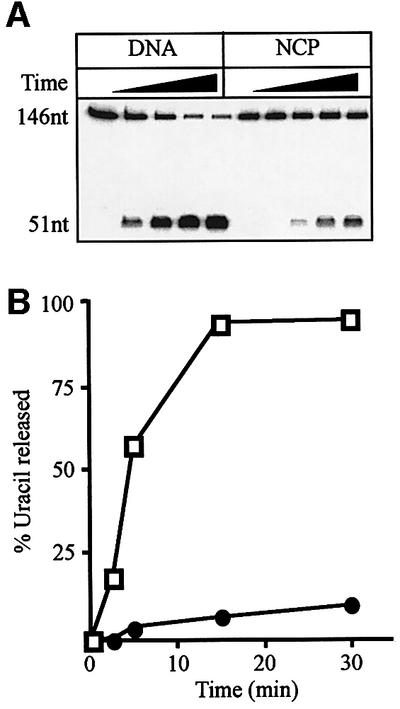
Fig. 3. Excision of uracil by the SMUG1 DNA glycosylase. (A) An 8% polyacrylamide/7 M urea/20% formamide gel showing the time course of excision of an internal uracil residue (U51) from naked 146 bp DNA (DNA) and nucleosome core particles (NCP) by the SMUG1 glycosylase in the presence of APE1. (B) Average rate of uracil excision of U51 from naked DNA (open squares) and NCP (closed circles) after correcting for the presence of free DNA from two independent experiments. The SEM of uracil removal was <15%.
Stability of nucleosomes during uracil removal and repair
Nucleosomes reconstituted on the 5S rDNA can occupy more than one translational position depending on reaction and temperature conditions (Flaus et al., 1996). Further more, the stability of nucleosomes may be compromised by small changes in reaction conditions (Godde and Wolffe, 1995). It was therefore important to determine the stability of the core particles under the reaction conditions employed. No disruption of the core particles was observed. The fraction of naked DNA remained unchanged after native polyacrylamide gel electrophoresis prior to (Figure 4A, lane 1) or after 30 min incubation with UNG2 (Figure 4A, lane 2), SMUG1 and APE1 (Figure 4A, lane 3) and after BER (Figure 4A, lane 4). However, incomplete repair of the uracil-containing substrates led to the formation of stable repair intermediates persisting on naked DNA as well as core particles. This was apparent from the presence of high molecular weight intermediates that did not enter the gel (Figure 4A, lane 4). Finally, the relative amounts of two distinctly migrating nucleosome bands, which represent different translational positions of the octamer along the DNA, were preserved during uracil removal (Figure 4A). Restriction enzyme mapping was employed to analyse whether histone octamer position along the DNA changed as a consequence of repair (see Figure 2A for positions of restriction sites). Whereas naked DNA was digested readily by FokI, AluI and MboI (Figure 4B, lanes 2–4), histone octamers protected nucleosomal DNA from digestion. The highest degree of protection was seen for digestion by the centrally located AluI (Figure 4B, lane 6), but FokI and MboI were also inhibited on nucleosomes (Figure 4B, lanes 5 and 7). Incubations with UNG2 alone (Figure 4B, lanes 8–10), SMUG1/APE1 (Figure 4B, lanes 11–13) or the full complement of BER proteins (Figure 4B, lanes 14–16) did not relieve inhibition. This was consistent with a lack of severe perturbations or even short-range sliding of the octamer during the course of the reaction. Bands co-migrating with the 53 nucleotide AluI product (Figure 4B, lanes 8–16) resulted from removal of uracil from position 51 and subsequent cleavage of AP-sites by APE1 or during denaturation and gel electrophoresis. As these bands might arise from undigested material as well as products digested with AluI and MboI, the calculation of digestion efficiency for these enzymes is not possible.
Despite the presence of more than one translational position, one rotational setting was found to be preferred on the 146 bp L.variegatus 5S rDNA (Flaus et al., 1996), and the DNase I footprinting patterns observed here confirmed this finding (Figures 1C, and 4C, lanes 7–11). To test whether the rotational positioning of the histone octamers changed as a consequence of uracil removal and repair, DNase I footprinting was performed after 30 min incubation with repair enzymes. The DNase I footprint after uracil removal by UNG2 (data not shown) was identical to the footprint observed from digestion of nucleosomal DNA alone (Figure 4C, lanes 7–11). This pattern was also largely unchanged after incubation with SMUG1/APE1 (Figure 4C, lanes 12–14) or after BER (Figure 4C, lanes 15–17), apart from the appearance of strong DNase I-sensitive sites 3′ to the damaged site (Figure 4C) where the repair proteins remained bound either to the substrate uracil or to repair intermediates. The absence of this hypersensitive site after incubation with UNG2 was probably due to the low affinity of this enzyme for AP-sites compared with SMUG1 and APE1. Importantly, since there was no overall change in the DNase I footprint, we conclude that uracil removal and repair of the resulting AP-sites does not severely affect rotational positioning.
Uracil removal from a 202 bp nucleosome substrate
In a recent study, DNA ligase I was found to be more strongly inhibited when nucleosome cores contained shorter DNA fragments (154 bp as opposed to 218 bp), as a consequence of inappropriately localized histone tails interfering with ligation when nucleosome cores contained the shorter DNA fragments (Chafin et al., 2000). To investigate this point, we reconstituted nucleosome cores on a 202 bp fragment of the L.variegatus 5S rRNA gene. Two histone octamer positions along the 202 bp DNA were detected in nucleoprotein gels (Figure 5E). Restriction enzyme mapping revealed that the uracil residue at position 57 was protected by the octamer in both positions, as illustrated in Figure 5A. The DNase I protection pattern of these 202 bp nucleosome core particles showed that the histone octamers predominantly occupied one rotational setting (Figure 5B, lanes 1 and 2). DNase I footprinting confirmed that the uracil residue at position 57 was situated in the region protected by the histone octamer (Figure 5B, lane 3). Nucleosomes or naked DNA were incubated with UNG2 (Figure 5C) or SMUG1 (data not shown), and uracil removal was measured as before. A 3-fold inhibition of uracil removal was observed when the 202 bp nucleosome cores (Figure 5D, closed circles) were compared with naked DNA (Figure 5D, open squares). A very similar inhibition was observed when the core particles were reconstituted using hyperacetylated histone octamers isolated from HeLa cells treated with sodium butyrate (data not shown), arguing that inhibition of UNG2 or SMUG1 DNA glycosylases was not mediated primarily by the core histone tails. Upon prolonged incubation, maximum uracil removal from the 202 bp nucleosome cores was slightly higher (∼75%, data not shown) than with 146 bp nucleosome cores (∼60%). Nucleoprotein gels revealed that the 202 bp nucleosome cores remained intact during uracil removal and repair (Figure 5E). Furthermore, changes in octamer position did not occur, as was evident from unchanged protection from FokI and ScaI digestion (Figure 5F). As was observed for the 146 bp core particles (Figure 4C), the rotational positioning of the DNA was also conserved throughout the course of the reaction, except for an additional DNase I-sensitive site 3′ to the damaged site after incubation with SMUG1 and BER factors (data not shown).

Fig. 5. Removal of uracil from nucleosome core particles reconstituted on a 202 bp DNA fragment. An extended fragment of the L.variegatus 5S rRNA gene (202 bp) containing a uracil residue at position 57 (numbered from the 5′ end of the 202 bp fragment) was constructed. (A) Cartoon showing the major and minor (dashed line) translational positions of the histone octamers based on digestion with the indicated restriction enzymes. (B) DNase I footprinting of core particles (lanes 1 and 2) shown next to the cleaved DNA substrate following UNG2 and piperidine treatment (lane 3) to verify that the uracil residue is positioned within DNA protected by octamers. The 202 bp DNA substrate (lane 4) and DNase I footprinting of the naked DNA (lanes 5 and 6) are shown for comparison. (C) An 8% polyacrylamide/7 M urea/20% formamide gel showing the time course of uracil excision by the UNG2 glycosylase from naked DNA (DNA) and nucleosome core particles (NCP). (D) Rates of uracil excision in naked DNA (open squares) and NCP (closed circles) after correction for uracil removal from naked DNA present in the core particle preparation (average of three independent experiments). The SEM of uracil removal was <20%. (E) Native 5% polyacrylamide gel after a 30 min incubation, showing naked DNA (D), and core particles incubated alone (–) or with UNG2 (U), SMUG1/APE1 (S) or BER proteins (B). (F) An 8% polyacrylamide/7 M urea/20% formamide gel showing restriction enzyme digestion of 202 bp naked DNA (lanes 1–3) with FokI (F) or ScaI (Sc) and, similarly, core particles alone (–) or after uracil removal by UNG2 (U), SMUG1/APE1 (S) or BER enzymes (B) after a 30 min incubation.
Removal of uracil incorporated along the length of the 5S rDNA
The previous experiments showed that the rates of uracil excision from three nucleotide positions in the core particle (U19, U22 and U51) were remarkably similar. In order to extend this approach to other U:A positions along the length of the 5S rDNA, including residues closer to the dyad axis (Figure 2A), DNA substrates containing U:A base pairs partially substituting T:A pairs in all possible positions along the 146 bp 5S rDNA were produced by primer extension in the presence of low amounts of dUTP (10% of the dTTP concentration). Treating these substrates with a uracil-DNA glycosylase allowed inspection of uracil removal from several positions along the core particle simultaneously. DNase I footprinting with core particles purified by sucrose gradient centrifugation showed nuclease protection by the core particles (Figure 6, lane 12), that was absent in naked DNA (Figure 6, lane 1). With nucleosome core particles, the major nuclease-accessible sites were around positions (numbered from the 5′-labelled primer) 35, 45, 57, 65 and 75. A time course of uracil removal by the UNG2 DNA glycosylase is shown for naked DNA (lanes 2–6) and core particles (lanes 7–11). Consistent with the observations from core particles containing site-specific U:A base pairs, the overall level of uracil excision from these substrates was reduced when compared with naked DNA (∼36% uracil removal in lane 3 compared with ∼11% in lane 8). Although subtle variations in repair efficiency along the 5S rDNA were observed, the pattern of uracil removal from core particles followed the pattern in naked DNA, and was largely uniform along the core particles (compare lanes 2–6 and 7–11). Surprisingly, uracil residues in the central core (U35–U78) were removed as readily as residues near the site of DNA entry (U22 and U27) into the core particle (Figure 2A). Thus, the weaker histone–DNA interactions in this region do not make it a better substrate for UNG2 (Figure 6) or SMUG1 (data not shown). Importantly, rotational setting did not appear to influence the rate of uracil excision; there was no simple correlation between the rate of uracil removal and whether uracil residues were facing towards or away from the histone octamer.
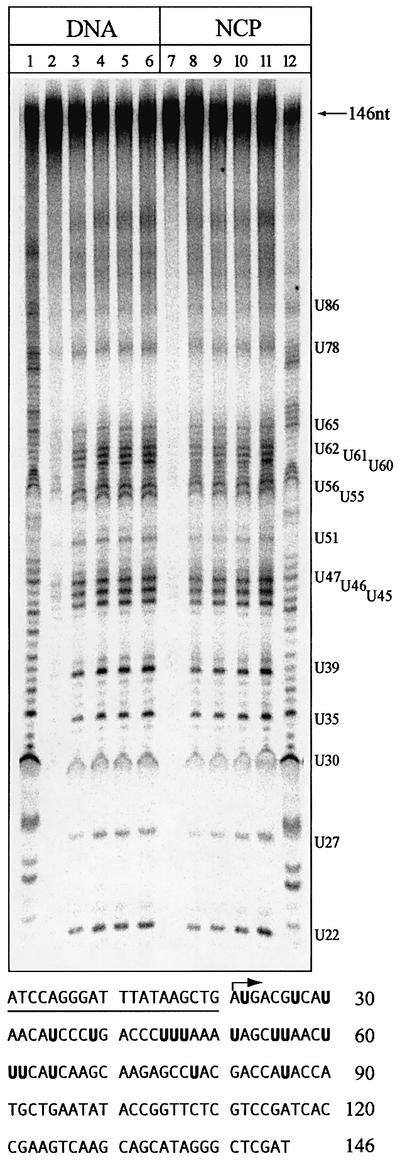
Fig. 6. UNG2 excises uracil essentially uniformly along the nucleosome core particle. A 146 bp substrate was prepared by incorporation of dUMP residues along the length of the 5S rDNA by primer extension (lanes 2 and 7). Reconstituted nucleosome core particles (NCP) show the expected DNase I protection pattern (lane 12), which is not observed in naked DNA (lane 1). DNA (lanes 3–6) and NCP (lanes 8–11) were treated with UNG2. Aliquots were removed after 5, 15, 30 and 60 min, treated with piperidine to cleave abasic sites and analysed in 8% polyacrylamide/7 M urea/20% formamide gels. Positions where dTMP residues have been partly substituted with dUMP, with numbers corresponding to distance from the 5′ terminus, are shown on the right. The sequence of the L.variegatus 5S rRNA gene (sequence given for the sense strand in the 5′ to 3′ direction), showing the substituted uracil residues in bold, is illustrated in the bottom panel. The primer used for primer extension is underlined, and the arrow indicates the start of DNA synthesis.
Reconstitution of short-patch base excision repair in nucleosome core particles
Having established that uracil excision was suppressed in nucleosome core particles, we studied the entire short-patch BER reaction in nucleosome cores. In order to maximize the efficiency of uracil excision in nucleosomal DNA substrates to allow subsequent repair of AP-sites to be monitored, naked DNA and core particles were treated with the catalytic domain of UNG2 (UNGΔ84), which was able to remove 90% of the uracil residues (Figure 7A, lanes 2 and 8), presumably due to the higher specific activity of UNGΔ84. The core particles were stable during this prolonged incubation at 37°C (Figure 4A, lane 4). Efficient strand incision 5′ to the AP-site by APE1 was observed on naked DNA (Figure 7A, lane 2) as well as on the core particle (Figure 7A, lane 8). Polβ was able to extend the resulting 3′-OH terminus by one nucleotide in both naked DNA and core particles (Figure 7A, lanes 3 and 9), although with lower efficiency in the latter. Strand displacement synthesis was observed with both substrates (Figure 7A, lanes 3 and 9, +2 nucleotide insertions), but synthesis beyond two nucleotides normally was not observed at position U22. XRCC1 did not significantly facilitate Polβ activity on either substrate when included in the reaction (Figure 7A, lanes 4 and 10). The reappearance of the full-length 146mer showed that BER could be reconstituted upon addition of LigIII to naked DNA (Figure 7A, lane 5) as well as to the core particle (Figure 7A, lane 11). In addition to sealing +1 nucleotide extended products, LigIII reduced strand displacement synthesis by Polβ (Figure 7A, lanes 5 and 11). XRCC1 did not facilitate ligation of either substrate (Figure 7A, lane 6 and 12). Similar results were obtained with the U51 substrate (Figure 7B). In this position, however, Polβ strongly favoured +1 over +2 nucleotide extension in naked DNA (Figure 7B, lane 3), and strand displacement synthesis was reduced further by including XRCC1 (Figure 7B, lane 4). In contrast, the ability of Polβ to extend from this position in core particles was poor in both the absence (Figure 7B, lane 7) and presence of XRCC1 (Figure 7B, lane 8). Interestingly, the preference for a +1 over a +2 nucleotide extension, seen with naked DNA, was lost in nucleosome core particles (Figure 7B, compare lanes 3 and 7 or lanes 4 and 8). The +1 nucleotide extended products were religated readily by LigIII in both substrates (Figure 7B, lanes 5 and 9). Thus, BER of nucleosome core particles was reconstituted without including putative accessory factors to increase damage accessibility, but the activity of Polβ appeared to be impaired at some positions along the 5S rDNA.
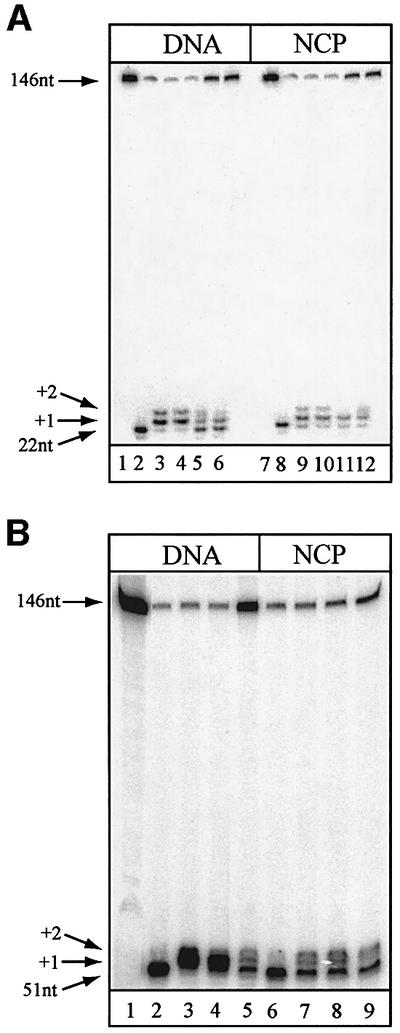
Fig. 7. Reconstitution of BER in 146 bp nucleosome core particles. Substrates containing abasic sites were produced in naked DNA (DNA) or nucleosome core particles (NCP) with UNGΔ84. (A) Substrate containing uracil in position 22 (lanes 1 and 7) is incised by treatment with UNGΔ84 and APE1 (lanes 2 and 8). The incised 3′ terminus is extended when Polβ is included (lanes 3 and 9). Addition of XRCC1 did not facilitate extension by the polymerase detectably (lanes 4 and 10). The nicked substrate is religated when DNA ligase III is included in the reaction (lanes 5 and 11). The addition of XRCC1 did not improve ligation (lanes 6 and 12). (B) Substrate containing uracil in position 51 (lane 1) is incised by treatment with UNGΔ84 and APE1 (lanes 2 and 6). Polβ extends the incised 3′ terminus equally well in the absence (lanes 3 and 7) or presence (lanes 4 and 8) of XRCC1, but strand displacement synthesis (+2 nucleotides) was reduced by XRCC1 in naked DNA (lane 4) but not on core particles (lane 8). The addition of DNA ligase III seals the resulting nick to complete repair (lanes 5 and 9).
Discussion
We have investigated BER in vitro using reconstituted nucleosome core particles and recombinant human repair enzymes. Uracil-containing substrates were assembled into mononucleosomes by stepwise salt dilution. The following conclusions can be drawn: (i) the two major human uracil-DNA glycosylases, UNG2 and SMUG1, can excise uracil from nucleosome core particles; (ii) uracil excision is impaired by ∼3- to 9-fold for UNG2 and SMUG1, respectively, when nucleosome core particles are compared with naked DNA; (iii) uracil is excised essentially uniformly along the length of the core particle without detectable displacement or sliding of the octamer; (iv) repair of nucleosome core particles containing abasic sites can be reconstituted using only the known repair proteins, APE1, Polβ and LigIII; and (v) the two limiting steps for repair of core particles are the initial excision of uracil and the activity of Polβ.
Removal of uracil from core particles
DNA repair in nucleosomes has been studied previously for cis-syn cyclobutyl pyrimidine dimers (CPDs) and (6–4) photoproducts, lesions repaired by the NER pathway (Smerdon and Conconi, 1999; Hara et al., 2000; Liu and Smerdon, 2000; Kosmoski et al., 2001). These studies showed that NER can take place in nucleosomes, but the repair efficiency was reduced by ∼2- (Kosmoski et al., 2001) to 10-fold (Hara et al., 2000). In line with these results, we show that uracil excision from a site-specific U:A base pair by UNG2 and SMUG1 was reduced ∼3- to 9-fold, respectively, on reconstituted nucleosomes core particles (Figures 2 and 3). Neither increased DNA substrate length nor hyperacetylation of histone tails relieved this inhibition (Figure 5; data not shown), showing that inappropriate binding of histone tails to nucleosome core DNA cannot explain our observations. DNase I is very strongly inhibited at sites facing the histone octamer surface, giving rise to a characteristic 10 bp DNA ladder upon digestion (Wolffe, 1998). In contrast, the lack of a clear correlation between rotational positioning and rate of repair of CPDs by direct reversal (Schieferstein and Thoma, 1998) or NER (Liu and Smerdon, 2000) challenges the intuitive assumption that DNA damage facing the histone octamer surface, rather than the solvent, may be less accessible for repair. Using purified core particles containing randomly incorporated uracil residues, we have shown that both UNG2 (Figure 6) and SMUG1 (data not shown) remove uracil from several positions at remarkably uniform rates, refuting a simple correlation with rotational positioning. Although reconstituted nucleosome core particles occupy more than one position along the 146 bp 5S rDNA (Figure 4A), there is clearly one predominant rotational setting (Figures 1C and 4C). It is therefore unlikely that variations in rates of uracil excision resulting from preferential repair of accessible residues along the 5S rDNA are masked by subtle variations in translational positioning of the octamer along the DNA.
A possible model to explain the lack of correlation between rotational setting and excision rates would involve the DNA glycosylase binding the core particles where DNA is exposed to the solvent. Upon binding, histone–DNA interactions may be destabilized locally, thereby allowing the DNA glycosylase to traverse the core particle in a processive manner until a uracil residue is encountered. Access to nearby buried residues would then be increased in a co-operative manner without completely releasing the DNA from the octamer, akin to RNA polymerase III transcription through a single nucleosome (Studitsky et al., 1997). There is evidence for a partially processive action of the Escherichia coli Ung protein from kinetic studies of uracil removal (Higley and Lloyd, 1993; Bennett et al., 1995).
Base excision repair in nucleosome core particles
Short-patch BER was reconstituted in vitro, using recombinant human repair proteins and core particles containing site-specific abasic sites. As APE1 was able to introduce nicks efficiently in nucleosomal DNA, the reduced activity of Polβ in core particles cannot be ascribed to differences in effective substrate concentration, but reflects properties of the enzyme itself (Figure 7A and B; data not shown). The marked variation in the activity of Polβ at different positions along the 5S rDNA might be explained by structural requirements for interaction between Polβ and the DNA substrate. Polβ contacts both the 3′-OH primer terminus and the 5′ dRP moiety and, upon productively binding the substrate, induces a 90° kink in DNA (Sawaya et al., 1997). This structural distortion might be either discouraged or facilitated depending on the local conformation of the DNA on the surface of the nucleosome (Luger et al., 1997). It seems likely that Polβ may have reduced ability to extend primer termini where the energetic cost of introducing a kink in the DNA is too large, for example at the internal residue U51.
The lack of modulation of BER by XRCC1 in core particles is surprising in light of the postulated trimeric complex of nicked DNA–Polβ–XRCC1 (Marintchev et al., 1999) and recent studies describing a direct stimulation of BER by XRCC1 (Vidal et al., 2001; Whitehouse et al., 2001). The need for XRCC1 in in vitro repair assays may be mitigated when starting from an AP-site-containing, rather than a nick-containing substrate. However, we did observe the expected suppression of strand displacement synthesis by XRCC1 (Kubota et al., 1996) during repair of U51 in naked DNA (Figure 7B).
A recent study demonstrated that DNA ligase I was able to seal DNA nicks in core particles, the rate of ligation being reduced 4- to 6-fold on nucleosomes containing a 218 bp DNA fragment (Chafin et al., 2000). Here we obtained similar results with DNA ligase III. However, a direct comparison of ligation kinetics between naked DNA and nucleosome cores was not possible because the effective substrate concentration was low in the core particle substrates as a result of diminished Polβ activity.
A possible explanation of the lack of effect of XRCC1 and the poor activity of Polβ observed might be that the formation of productive BER complexes on the core particles was impaired due to steric constraints. It appears that BER proteins can remain bound to the core particle (Figures 4A, lane 4, and 5E, lane 5), resulting in a local structural distortion that generates a DNase I-hypersensitive site 3′ to the lesion (Figure 4C). It has been suggested that poly(ADP-ribose) polymerase 1 (PARP-1) may function as a surveillance protein for a stalled BER intermediate (Lavrik et al., 2001), and that PARP-1, in concert with Flap endonuclease 1 (FEN1), could stimulate long-patch synthesis by Polβ in the absence of NAD (Prasad et al., 2001). The inhibition of Polβ in core particles, however, was not relieved by PARP-1 alone nor PARP-1 in combination with FEN1, in the presence or absence of NAD (data not shown). Therefore, we favour the view that PARP-1 might increase the access to DNA damage in higher order chromatin structure (d’Erme et al., 2001), rather than in nucleosome core particles.
It seems possible that accessory factors might be required to assist later steps of BER and to restore the nucleosome structure after repair. An indication that chromatin alterations take place during BER comes from studies of bleomycin-induced strand breaks in cell lines (Sidik and Smerdon, 1990, 1992). We did not, however, observe increased repair efficiency when using nucleosomes containing hyperacetylated histone octamers.
In conclusion, we have shown that human BER enzymes are able to act on nucleosome core particles, but the activities of the major human uracil-DNA glycosylases, as well as Polβ, are impaired. Nucleosome core particles appear to modulate BER as they do NER, suggesting that they present a similar obstacle to both repair pathways.
Materials and methods
DNA substrates
Lytechinus variegatus 5S rRNA gene fragments were isolated from plasmid pTJR2 (Richmond et al., 1988) following EcoRV digestion (146 bp) and purified from 1.9% NuSieve GTG agarose gels (FMC) using the QIAex gel extraction kit (Qiagen). The following oligonucleotide primers were used in the study (all listed in the 5′ to 3′ direction of the sense strand where position 1 in the 146 bp fragment corresponds to position –74 relative to the transcription start of the 5S rRNA gene): U19, ATCCAGGGATTTATAAGCUGATGACGTCATAAC; U22, ATCCAGGGATTTATAAGCTGAUGACGTCATAAC; U51, ATCCAGGGATTTATAAGCTGATGACGTCATAACATCCCTGACCCTTTAAAUAGCTTAACTTTCATCAAGC; 146-Start, ATCCAGGGATTT ATAAGCTG; and 146-End, ATCGAGCCCTATGCTGC. A synthetic, 146 nucleotide oligonucleotide (146-Temp) with sequence complementary to the 146 bp 5S rRNA gene fragment was used as a template in primer extension reactions. The longer (202 bp) template was prepared similarly from pBUless (Jesper Svejstrup) by EcoRI digestion. This fragment comprises a longer stretch of the 5S rRNA gene, containing 17 and 39 nucleotide 5′- and 3′-extensions, respectively, compared with the 146 bp fragment. Primers used in PCRs to generate a 202 bp uracil-containing substrate were 202-Start, AATTAAAACGAATAACTTCCAGGGATTTATAAGCCGATGACGTCATAACATCCCTGUCCCTTTAAATAG; and 202-End, AATTCGGTATTCCCAGGCGGT CTCCCATCCAAG.
Substrate preparation
Uracil-containing oligonucleotides were 5′ end-labelled with T4 polynucleotide kinase in the presence of [32γ-P]ATP and purified through Microspin™ G25 columns (Amersham Pharmacia). For preparation of substrates containing site-specific uracil residues, 5′ end-labelled dUMP-containing primers were used in combination with the appropriate reverse primers in 50 µl PCRs containing 100 pmol of each primer, 10 ng of DNA template, 400 µM each dNTP, 1 U of Taq polymerase, 50 mM KCl, 3.3 mM MgCl2, 10 mM Tris–HCl pH 8 for 30 cycles at 96°C for 30 s, 68°C for 15 s and 72°C for 30 s. The mixed damaged substrate was generated by primer extension from a labelled undamaged primer (146-Start) and 146-Temp using Taq polymerase as described above, in the presence of dUTP (dUTP:dTTP ratio of 1:10) in the nucleotide mix. The full-length product was purified from 1.9% NuSieve GTG agarose gels using the QIAex gel extraction kit. Maxam–Gilbert thymine mapping was performed as described (Sambrook and Russell, 2001).
Reconstitution of nucleosome core particles
Histone octamers were purified from chicken erythrocytes as described previously (Simon and Felsenfeld, 1979). Reconstitution of nucleosome core particles was performed according to the serial salt dilution method developed by Steger and Workman (1999), with minor modifications introduced in order to stabilize the nucleosomes (Godde and Wolffe, 1995). Briefly, 500 000 c.p.m. (∼20 pmol) of 5′-end-labelled substrate DNA was co-precipitated with 5 µg of sheared salmon sperm DNA. The washed and dried pellet was resuspended in 2 M NaCl, 100 µg/ml bovine serum albumin (BSA) and chicken erythrocyte octamers in a volume of 10 µl. After incubation at 37°C for 15 min, the sample was transferred to 30°C and subjected to step-wise dilution in 50 mM Na-HEPES pH 7.5, 1 mM EDTA, 5 mM dithiothreitol (DTT), 1× Complete™ proteinase inhibitor cocktail (Roche) down to 200 mM NaCl. Finally, 100 µl of storage buffer (10 mM Tris–HCl pH 7.5, 1 mM EDTA, 5 mM DTT, 0.1% NP-40, 20% glycerol, 100 µg/ml BSA, 1× Complete™) was added to bring the NaCl concentration down to 100 mM. The reconstitution efficiency was analysed on 5% polyacrylamide/20% glycerol gels run at 100 V for 2.5 h in 20 mM Na-HEPES/0.1 mM EDTA. Reconstitution reactions resulting in >85% of the labelled DNA present as nucleosome core particles were used directly in experiments. Otherwise, the core particles were purified through 5–25% sucrose gradients (10 mM HEPES–KOH pH 7.9, 1 mM EDTA, 0.1% NP-40) (Hara et al., 2000) by centrifugation at 25 000 r.p.m. for 18 h in an SW41T rotor (Beckman). Nucleosome cores recovered from the gradient were subjected to buffer exchange using Microcon™ YM100 filter units (Millipore).
Characterization of reconstituted nucleosomes
The rotational positioning of reconstituted core particles was determined by DNase I digestion essentially as described by Schieferstein and Thoma (1996). Briefly, 50 000 c.p.m. (∼2 pmol) of substrate was treated with 4 U (NCP) or 0.2 U (naked DNA) of DNase I (Roche) in 20 mM Tris–HCl pH 7.5, 5 mM MgCl2 at room temperature. Aliquots were removed after 0.5, 1, 3, 6 and 15 min, and the reactions stopped by the addition of 25 mM EDTA and 95% formamide loading buffer. The samples were separated in an 8% polyacrylamide/7 M urea/20% formamide/1× TBE gel. The gels were fixed in 10% acetic acid, dried and analysed by PhosphorImaging.
Translational positioning was determined by monitoring the degree of inhibition of restriction enzyme cleavage along the length of the nucleosome by treating ∼1 pmol of substrate with 5–10 U of restriction enzymes XmnI, FokI, DraI, AluI, MboI and ScaI (New England Biolabs) in buffers provided by the manufacturer. After incubation at 37°C for 30 min, the reactions were stopped by the addition of 95% formamide loading buffer before separation in an 8% polyacrylamide/7 M urea/ 20% formamide/1× TBE gel.
Assays for DNA glycosylase activity
To determine DNA glycosylase activity, 25 000 c.p.m. (∼1 pmol) of substrate was incubated with UNG2 (7.5 fmol) in 20 mM Tris–HCl pH 7.5, 20 mM NaCl, 5 mM MgCl2, 100 µM EDTA, 1 mM DTT, 100 µg/ml BSA, 2 mM ATP, 20 µM each all four dNTPs (1× repair buffer). SMUG1 (1.7 pmol) was assayed in the presence of APE1 (13 fmol) in order to increase the off-rate of the DNA glycosylase (Nilsen et al., 2001) in 20 mM Tris–HCl pH 8, 20 mM NaCl, 1 mM EDTA, 1 mM DTT. Enzyme concentrations were chosen that gave ∼50% uracil removal from naked DNA after 5 min incubation under standard reaction conditions for either UNG2 or SMUG1/APE1 to enable us to compare the rates of uracil removal from naked DNA and core particles. Aliquots were removed after 0, 2.5, 5, 15 and 30 min at 37°C, and abasic sites were cleaved by heating at 90°C in 1 M piperidine for 20 min. The samples were dried and resuspended in 95% formamide loading buffer before separation in an 8% polyacrylamide/7 M urea/20% formamide/1× TBE gel. The gels were fixed in 10% acetic acid, dried, and visualized by PhosphorImaging. Uracil removal was quantified using ImageQuant software (Molecular Dynamics) and corrected for the presence of free DNA in the nucleosome substrate. The SEM of uracil removal within the same nucleosome preparation was <20%.
Parallel samples were prepared as above in order to characterize the nucleosomes during the course of the UNG2 and SMUG1 treatment. After a 30 min incubation at 37°C, aliquots were removed and loaded directly on 5% polyacrylamide/20% glycerol gels as described above to determine stability. Aliquots were also subjected to DNase I digestion as described above to determine rotational positioning after repair.
Base excision repair assays
Repair assays were performed in two steps. First, 25 000 c.p.m. (∼1 pmol) of substrate was treated extensively with UNGΔ84 (0.56 pmol) at 37°C for 1 h in 1× repair buffer in order to convert uracil residues into abasic sites. This was necessary since the UNGΔ84 catalytic fragment (Slupphaug et al., 1995) was more efficient than full-length UNG2 for removal of uracil from nucleosomes. Then, aliquots (∼200 fmol of substrate per reaction) were subjected to repair for 30 min at 37°C by the addition of APE1 (13 fmol), Polβ (6.5 fmol), XRCC1 (13 fmol) and DNA ligase III (3 fmol) in 1× repair buffer. Reactions were stopped by the addition of 95% formamide loading buffer on ice, and processed as described above. Nucleosome stability and rotational positioning prior to and after BER were determined as described above.
Acknowledgments
Acknowledgements
We thank Drs Hans E.Krokan and Geir Slupphaug (Norwegian University of Science and Technology, Trondheim, Norway) for providing purified UNG2 and UNGΔ84, Jesper Svejstrup (Cancer Research UK) for plasmid pBUless, and Martin Singleton (Cancer Research UK) for discussions. This work was supported by Cancer Research UK (T.L. and A.V). H.N. was the recipient of an EMBO long-term Fellowship and a Marie Curie Fellowship.
References
- Bennett S.E., Sanderson,R.J. and Mosbaugh,D.W. (1995) Processivity of Escherichia coli and rat liver mitochondrial uracil-DNA glycosylase is affected by NaCl concentration. Biochemistry, 34, 6109–6119. [DOI] [PubMed] [Google Scholar]
- Chafin D.R., Vitolo,J.M., Henricksen,L.A., Bambara,R.A. and Hayes,J.J. (2000) Human DNA ligase I efficiently seals nicks in nucleosomes. EMBO J., 19, 5492–5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Erme M., Yang,G., Sheagly,E., Palitti,F. and Bustamante,C. (2001) Effect of poly(ADP-ribosyl)ation and Mg2+ ions on chromatin structure revealed by scanning force microscopy. Biochemistry, 40, 10947–10955. [DOI] [PubMed] [Google Scholar]
- de Laat W.L., Jaspers,N.G. and Hoeijmakers,J.H. (1999) Molecular mechanism of nucleotide excision repair. Genes Dev., 13, 768–785. [DOI] [PubMed] [Google Scholar]
- Flaus A., Luger,K., Tan,S. and Richmond,T.J. (1996) Mapping nucleosome position at single base-pair resolution by using site-directed hydroxyl radicals. Proc. Natl Acad. Sci. USA, 93, 1370–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser R., Koller,T. and Sogo,J.M. (1996) The stability of nucleosomes at the replication fork. J. Mol. Biol., 258, 224–239. [DOI] [PubMed] [Google Scholar]
- Godde J.S. and Wolffe,A.P. (1995) Disruption of reconstituted nucleosomes. J. Biol. Chem., 270, 27399–27402. [DOI] [PubMed] [Google Scholar]
- Goulian M., Bleile,B. and Tseng,B. (1980) Methotrexate-induced misincorporation of uracil into DNA. Proc. Natl Acad. Sci. USA, 77, 1956–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green C.M. and Almouzni,G. (2002) When repair meets chromatin. EMBO rep., 3, 28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara R., Mo,J. and Sancar,A. (2000) DNA damage in the nucleosome core is refractory to repair by human excision nuclease. Mol. Cell. Biol., 20, 9173–9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haushalter K.A., Stukenberg,P.T., Kirschner,M.W. and Verdine,G.L. (1999) Identification of a new uracil-DNA glycosylase family by expression cloning using synthetic inhibitors. Curr. Biol., 9, 174–185. [DOI] [PubMed] [Google Scholar]
- Higley M. and Lloyd,R.S. (1993) Processivity of uracil DNA glycosylase. Mutat. Res., 294, 109–116. [DOI] [PubMed] [Google Scholar]
- Kosmoski J.V., Ackerman,E.J. and Smerdon,M.J. (2001) DNA repair of a single UV photoproduct in a designed nucleosome. Proc. Natl Acad. Sci. USA, 98, 10113–10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krude T. (1999) Chromatin assembly during DNA replication in somatic cells. Eur. J. Biochem., 263, 1–5. [DOI] [PubMed] [Google Scholar]
- Kubota Y., Nash,R.A., Klungland,A., Schar,P., Barnes,D.E. and Lindahl,T. (1996) Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase β and the XRCC1 protein. EMBO J., 15, 6662–6670. [PMC free article] [PubMed] [Google Scholar]
- Lavrik O.I., Prasad,R., Sobol,R.W., Horton,J.K., Ackerman,E.J. and Wilson,S.H. (2001) Photoaffinity labeling of mouse fibroblast enzymes by a base excision repair intermediate. Evidence for the role of poly(ADP-ribose) polymerase-1 in DNA repair. J. Biol. Chem., 276, 25541–25548. [DOI] [PubMed] [Google Scholar]
- Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature, 362, 709–715. [DOI] [PubMed] [Google Scholar]
- Liu X. and Smerdon,M.J. (2000) Nucleotide excision repair of the 5S ribosomal RNA gene assembled into a nucleosome. J. Biol. Chem., 275, 23729–23735. [DOI] [PubMed] [Google Scholar]
- Luger K., Mader,A.W., Richmond,R.K., Sargent,D.F. and Richmond,T.J. (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature, 389, 251–260. [DOI] [PubMed] [Google Scholar]
- Marintchev A., Mullen,M., Maciejewski,M., Pan,B., Gryk,M. and Mullen,G. (1999) Solution structure of the single-strand break repair protein XRCC1 N-terminal domain. Nat. Struct. Biol., 6, 884–893. [DOI] [PubMed] [Google Scholar]
- Nilsen H., Yazdankhah,S.P., Eftedal,I. and Krokan,H.E. (1995) Sequence specificity for removal of uracil from U·A pairs and U·G mismatches by uracil-DNA glycosylase from Escherichia coli and correlation with mutational hotspots. FEBS Lett., 362, 205–209. [DOI] [PubMed] [Google Scholar]
- Nilsen H., Otterlei,M., Haug,T., Solum,K., Nagelhus,T.A., Skorpen,F. and Krokan,H.E. (1997) Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res., 25, 750–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen H. et al. (2000) Uracil-DNA glycosylase (UNG)-deficient mice reveal a primary role of the enzyme during DNA replication. Mol. Cell, 5, 1059–1065. [DOI] [PubMed] [Google Scholar]
- Nilsen H., Haushalter,K.A., Robins,P., Barnes,D.E., Verdine,G.L. and Lindahl,T. (2001) Excision of deaminated cytosine from the vertebrate genome: role of the SMUG1 uracil-DNA glycosylase. EMBO J., 20, 4278–4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orphanides G. and Reinberg,D. (2000) RNA polymerase II elongation through chromatin. Nature, 407, 471–475. [DOI] [PubMed] [Google Scholar]
- Otterlei M. et al. (1999) Post-replicative base excision repair in replication foci. EMBO J., 18, 3834–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad R., Lavrik,O.I., Kim,S.J., Kedar,P., Yang,X.P., Vande Berg,B.J. and Wilson,S.H. (2001) DNA polymerase β-mediated long patch base excision repair. Poly(ADP-ribose)polymerase-1 stimulates strand displacement DNA synthesis. J. Biol. Chem., 276, 32411–32414. [DOI] [PubMed] [Google Scholar]
- Richmond T.J., Searles,M.A. and Simpson,R.T. (1988) Crystals of a nucleosome core particle containing defined sequence DNA. J. Mol. Biol., 199, 161–170. [DOI] [PubMed] [Google Scholar]
- Sambrook J. and Russell,D. (2001) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sawaya M.R., Prasad,R., Wilson,S.H., Kraut,J. and Pelletier,H. (1997) Crystal structures of human DNA polymerase β complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry, 36, 11205–11215. [DOI] [PubMed] [Google Scholar]
- Schieferstein U. and Thoma,F. (1996) Modulation of cyclobutane pyrimidine dimer formation in a positioned nucleosome containing poly(dA–dT) tracts. Biochemistry, 35, 7705–7714. [DOI] [PubMed] [Google Scholar]
- Schieferstein U. and Thoma,F. (1998) Site-specific repair of cyclobutane pyrimidine dimers in a positioned nucleosome by photolyase and T4 endonuclease V in vitro. EMBO J., 17, 306–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidik K. and Smerdon,M.J. (1990) Nucleosome rearrangement in human cells following short patch repair of DNA damaged by bleomycin. Biochemistry, 29, 7501–7511. [DOI] [PubMed] [Google Scholar]
- Sidik K. and Smerdon,M.J. (1992) Correlation between repair patch ligation and nucleosome rearrangement in human cells treated with bleomycin, UV radiation or methyl methanesulfonate. Carcinogenesis, 13, 135–138. [DOI] [PubMed] [Google Scholar]
- Simon R.H. and Felsenfeld,G. (1979) A new procedure for purifying histone pairs H2A + H2B and H3 + H4 from chromatin using hydroxylapatite. Nucleic Acids Res., 6, 689–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slupphaug G., Eftedal,I., Kavli,B., Bharati,S., Helle,N.M., Haug,T., Levine,D.W. and Krokan,H.E. (1995) Properties of a recombinant human uracil-DNA glycosylase from the UNG gene and evidence that UNG encodes the major uracil-DNA glycosylase. Biochemistry, 34, 128–138. [DOI] [PubMed] [Google Scholar]
- Smerdon M.J. and Conconi,A. (1999) Modulation of DNA damage and DNA repair in chromatin. Prog. Nucleic Acid Res. Mol. Biol., 62, 227–255. [DOI] [PubMed] [Google Scholar]
- Sogo J.M., Stahl,H., Koller,T. and Knippers,R. (1986) Structure of replicating simian virus 40 minichromosomes. The replication fork, core histone segregation and terminal structures. J. Mol. Biol., 189, 189–204. [DOI] [PubMed] [Google Scholar]
- Steger D.J. and Workman,J.L. (1999) Transcriptional analysis of purified histone acetyltransferase complexes. Methods, 19, 410–416. [DOI] [PubMed] [Google Scholar]
- Studitsky V.M., Kassavetis,G.A., Geiduschek,E.P. and Felsenfeld,G. (1997) Mechanism of transcription through the nucleosome by eukaryotic RNA polymerase. Science, 278, 1960–1963. [DOI] [PubMed] [Google Scholar]
- Ura K., Araki,M., Saeki,H., Masutani,C., Ito,T., Iwai,S., Mizukoshi,T., Kaneda,Y. and Hanaoka,F. (2001) ATP-dependent chromatin remodeling facilitates nucleotide excision repair of UV-induced DNA lesions in synthetic dinucleosomes. EMBO J., 20, 2004–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal A.E., Boiteux,S., Hickson,I.D. and Radicella,J.P. (2001) XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein–protein interactions. EMBO J., 20, 6530–6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse C.J., Taylor,R.M., Thistlethwaite,A., Zhang,H., Karimi-Busheri,F., Lasko,D.D., Weinfeld,M. and Caldecott,K.W. (2001) XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell, 104, 107–117. [DOI] [PubMed] [Google Scholar]
- Wolffe A. (1998) Chromatin Structure and Function. Academic Press, London, UK.