

Abstract
We present an expanded data set of 50 unlinked autosomal noncoding regions, resequenced in samples of Hausa from Cameroon, Italians, and Chinese. We use these data to make inferences about human demographic history by using a technique that combines multiple aspects of genetic data, including levels of polymorphism, the allele frequency spectrum, and linkage disequilibrium. We explore an extensive range of demographic parameters and demonstrate that our method of combining multiple aspects of the data results in a significant reduction of the compatible parameter space. In agreement with previous reports, we find that the Hausa data are compatible with demographic equilibrium as well as a set of recent population expansion models. In contrast to the Hausa, when multiple aspects of the data are considered jointly, the non-Africans depart from an equilibrium model of constant population size and are compatible with a range of simple bottleneck models, including a 50–90% reduction in effective population size occurring some time after the appearance of modern humans in Africa 160,000–120,000 years ago.
Keywords: bottlenecks, combining P values, human demographic inference, population growth
Elucidating how and when populations change in size is an important element in reconstructing evolutionary history because these changes often reflect crucial events in the history of a species, such as range expansions, environmental changes, and mixture between groups (1). In addition, making inferences based on population variation data typically requires the specification of a demographic model. Such applications include detecting the signature of natural selection or estimating recombination rates from patterns of linkage disequilibrium (LD) (2–5). Finally, better knowledge of demographic histories in human populations is particularly important for whole-genome, LD-based association studies (6, 7).
Motivated by the excess of rare variants observed in mitochondrial DNA data, attention initially focused on models of ancient population growth and on the idea that population expansions may have accompanied the dispersal out of Africa or the emergence of new tool technology in the Upper Paleolithic (8–13). However, the accumulation of nuclear sequence variation surveys showed that this simple growth model was consistent with the observed frequency spectrum only for a subset of the loci (14–16). Likewise, LD surveys revealed marked differences in the rate of LD decay in African populations compared with that in non-African populations (17–19). These results together with the higher levels of sequence variation in African populations compared with that in non-African populations led to the proposal that population size reduction, such as bottlenecks, account for patterns of variation and LD in non-African populations (15, 18, 19). This bottleneck was hypothesized to correspond with the dispersal of modern humans out of Africa (18).
However, the investigation of formal bottleneck models has typically used a single aspect of genetic variation data, either the allele frequency spectrum (15, 20) or patterns of LD (18, 21), raising the question of whether such models were indeed consistent with the data when multiple aspects of genetic variation were considered simultaneously (22–24). Specifically, it is not known whether simple bottleneck models can generate the marked differences in LD levels seen between Africans and non-Africans with only a limited reduction in polymorphism levels outside Africa. Although previous work suggested that variation in recombination rate may explain the decay in LD observed in a multiethnic sample (7), it is not obvious that it could also explain the differences between Africans and non-Africans.
Ideally, making inferences about population history should be based on data from a large number of unlinked and neutrally evolving loci and on statistical methodology that makes efficient use of all or most of the information in the data. Full resequencing studies, in which the sequence of the surveyed segments is determined for every individual in the sample, represent one scheme for generating data sets in which multiple aspects of sequence variation are characterized. With regard to data analysis, full likelihood methods have been successfully applied to nonrecombining data (Y chromosome or mitochondrial DNA) to reconstruct population histories (25–27). However, for regions with recombination, the currently available methods are computationally infeasible. As a result, a variety of statistics, each summarizing different aspects of genetic variation data, may be used (13, 15, 28), with the subsequent reduction in information content traded for computational tractability. It is still desirable to combine the results of tests based on individual statistics because the joint distributions of multiple summaries of the data should contain more information than the marginal distributions of multiple single summaries considered separately.
We previously developed a full resequencing scheme in which pairs of tightly but not completely linked segments, referred to as “locus pairs,” are surveyed (19). This study design aims to maximize the information content for a given amount of sequencing effort because, by skipping the intervening segment, many more independent loci can be surveyed. Using this scheme, we previously surveyed 10 noncoding regions in three human population samples: Hausa of Cameroon, Italians, and Chinese. Here, we survey an additional 40 locus pairs in the same samples. This data set allows the simultaneous characterization of polymorphism levels, allele frequency spectrum, and LD in each sample; in addition, it obviates the need to correct for ascertainment bias with its associated uncertainties and possible loss of information (29–31). In choosing only noncoding regions distant from genes, we limit the possibility that our analysis of demographic history will be confounded by the effects of natural selection. To analyze these data, we implement an approach to determine P values associated with several observed summaries of genetic data considered jointly over a grid of demographic parameter values. These summaries include the average Tajima's D (D̄) and the variance of Tajima's D across loci  (32), the average number of segregating sites across loci (S̄), and an overall composite likelihood estimator of the population cross-over rate parameter (
(32), the average number of segregating sites across loci (S̄), and an overall composite likelihood estimator of the population cross-over rate parameter ( ) (33). By combining P values obtained from these individual statistics into a single statistical test, we greatly improve the power to reject demographic scenarios incompatible with the data. Although it is well established that other demographic features apply to these populations (e.g., population subdivision and gene flow) (34, 35), we chose to focus solely on population size changes to reduce modeling complexity. We explore an extensive grid of the demographic parameter space that revealed a confidence set of relatively simple bottleneck models that explain the patterns of variation in the non-African samples. Our results combine aspects of genetic variation from allele frequency spectrum, LD, and polymorphism levels within noncoding autosomal regions to infer the history of human populations. Because our data set was collected without ascertainment, it may be useful for validating the results of SNP genotyping surveys.
) (33). By combining P values obtained from these individual statistics into a single statistical test, we greatly improve the power to reject demographic scenarios incompatible with the data. Although it is well established that other demographic features apply to these populations (e.g., population subdivision and gene flow) (34, 35), we chose to focus solely on population size changes to reduce modeling complexity. We explore an extensive grid of the demographic parameter space that revealed a confidence set of relatively simple bottleneck models that explain the patterns of variation in the non-African samples. Our results combine aspects of genetic variation from allele frequency spectrum, LD, and polymorphism levels within noncoding autosomal regions to infer the history of human populations. Because our data set was collected without ascertainment, it may be useful for validating the results of SNP genotyping surveys.
Materials and Methods
DNA Samples. Sequence variation was surveyed in DNA samples from the same three human populations investigated in Frisse et al. (2001): 15 Hausa samples from Yaounde, Cameroon; 15 individuals from central Italy; and 15 Han Chinese from Taiwan. In addition, one common chimpanzee DNA sample was also sequenced at each region. This study was approved by the Institutional Review Board of the University of Chicago.
Resequencing Data Collection. We selected 40 unlinked genomic regions for resequencing using the locus pair approach (19): For each unlinked region, we sequenced two segments of ≈1 kb separated by ≈8 kb. The selection of genomic targets was aimed at regions that did not contain nor were tightly linked to known or strongly predicted coding regions. Most surveyed segments also did not contain and were not tightly linked to noncoding regions strongly conserved between human and mouse (as determined by inspection of the vista genome browser). These regions were selected as described in ref. 19 except that here we deliberately included regions with a broader range of cross-over rates and %G+C content. The local cM:Mb (Mb-megabase) ratio was obtained based on the interval defined by the two closest flanking markers on the DeCode Genetics (Reykjavik, Iceland) genetic map (36). The average and variance of the cM:Mb ratio across the 50 segments (i.e., 40 locus pairs from this study and the 10 given in ref. 19) are 1.31 and 0.83, respectively. The average and variance of %G+C across the 50 locus pairs are 38.3 and 46.6, respectively. Detailed information on each surveyed segment is provided in Table 2, which is published as supporting information on the PNAS web site. PCR and sequencing was performed as described in refs. 19 and 37. All sequencing reactions were run on automated capillary sequencers (ABI3100 and ABI3700). Sequence reads were scored by using polyphred (38); all putative polymorphisms and software-derived genotype calls were visually inspected and individually confirmed.
Testing Demographic Models. For each demographic model of interest, we performed a separate test for each summary statistic of genetic variation. In addition, for some of the models (equilibrium and bottleneck), we also calculated a test statistic, C, which combines the P values of multiple summary statistics as follows:
 |
[1] |
where Pi is the estimated P value of the ith summary statistic of k summary statistics.
For models defined by more than one demographic parameter (i.e., simple growth and bottleneck models), these tests were performed over a grid of parameter values. The combinations of parameter values that are compatible with the observed values of the test statistic(s) constitute the accepted portion of the parameter space for each model. For simple growth models, the test was based on Fu and Li's D* (39), whereas for bottleneck models, the test was based on combining P values from multiple summary statistics, as discussed below. The P values, Pi, for each individual summary statistic were estimated from Monte Carlo simulations using a modification of the program ms (40), as follows. We used coalescent simulations to generate 50,000 replicates, each consisting of 50 independent locus pairs, for each combination of parameter values; mutation and recombination rates were allowed to vary across locus pairs as described below. Samples of sequences 10 kb in length were generated in which the intervening 8 kb were ignored to mimic the locus pair data. The probability, P, of observing a value greater than that found in the data were estimated by simulations and converted to a two-tailed P value by applying the formula 1 - 2 · |0.5 - P|.
The P values for the combined test statistic C were estimated by using the empirical distribution of the statistic from simulations. For each combination of parameter values, we recorded the values of each summary statistic in each replicate and generate the distribution of these simulated values. For each replicate, we treated the value of each summary statistic as the “observed” value and determined its P value relative to the empirical distribution from the remaining 49,999 replicates. For each replicate, we combined these P values to calculate a value of C. By following this procedure for each of the 50,000 replicates (for a single demographic scenario of interest), we obtained a distribution of the combined statistic. This distribution can be used to estimate a one-tailed P value for the observed value of C.
Mutation Rate Model. We assumed an infinite sites model, where we modeled the variation in mutation rate across locus pairs by using a gamma(12.46, 2.11 × 10-9) distribution. The mean and variance for this distribution matched the observed mean and variance for the mutation rates estimated based on human–chimpanzee sequence divergence in our locus pair data (assuming 6 million years since divergence and a generation time of 25 years). The 90% central interval of this distribution is (1.54 × 10-8,3.96 × 10-8) with Eμ = 2.63 × 10-8.
Recombination Rate Model. We modeled the variation in the crossing-over rate, c, across locus pairs using a lognormal [-18.148, (0.5802)2] distribution; cross-over rate was assumed to be homogeneous within each locus pair. The 90% central interval of this distribution is (0.51 × 10-8, 3.41 × 10-8). The median of this distribution matched the overall recombination rate for the Hausa data (1.31 × 10-8) based on the composite likelihood estimator,  , of Hudson (33). Because we cannot accurately estimate the variance in recombination rate across surveyed segments as short as 10 kb, we matched the variance of the lognormal distribution to the variance of cM:Mb values estimated from the Marshfield genetic map for the interval containing each locus pair (41). We acknowledge that this model may capture some but not all of the recombination rate variation estimated across the human genome (42).
, of Hudson (33). Because we cannot accurately estimate the variance in recombination rate across surveyed segments as short as 10 kb, we matched the variance of the lognormal distribution to the variance of cM:Mb values estimated from the Marshfield genetic map for the interval containing each locus pair (41). We acknowledge that this model may capture some but not all of the recombination rate variation estimated across the human genome (42).
Summary Statistics. We summarize the locus pair data in terms of the average Tajima's D (D̄), the variance of Tajima's D  ), the average Fu and Li's D* (D̄*), the average number of segregating sites (S̄), and the average nucleotide diversity across the 50 locus pairs
), the average Fu and Li's D* (D̄*), the average number of segregating sites (S̄), and the average nucleotide diversity across the 50 locus pairs  , as well as
, as well as  , an overall estimate of the population crossing-over parameter (4Nc) as obtained by composite likelihood (33). Because there is not enough information in our data to accurately estimate
, an overall estimate of the population crossing-over parameter (4Nc) as obtained by composite likelihood (33). Because there is not enough information in our data to accurately estimate  and the gene conversion parameters (43), we assumed a model of gene conversion with rate (f) twice that of cross-over and tract lengths exponentially distributed with mean (L) 500 bp and estimate
and the gene conversion parameters (43), we assumed a model of gene conversion with rate (f) twice that of cross-over and tract lengths exponentially distributed with mean (L) 500 bp and estimate  . Alternative models of gene conversion (f = 10, L = 55 bp) based on sperm-typing data (44) yielded qualitatively similar results (data not shown).
. Alternative models of gene conversion (f = 10, L = 55 bp) based on sperm-typing data (44) yielded qualitatively similar results (data not shown).
Results
Summary of Sequence Variation and Tests of the Equilibrium Model. We resequenced 40 unlinked locus pairs in 15 individuals from each of three population samples: Hausa, Italians, and Chinese. The results of this survey are analyzed together with data for an additional 10 unlinked locus pairs previously resequenced in the same population samples (19), for a total of 50 unlinked locus pairs. The average surveyed length per locus pair was 2,365 bp (for a total of 118,259 bp surveyed in each individual), and the average unsurveyed intervening segment was 7,921 bp long.
The values of summary statistics used for demographic testing are shown in Table 1, with a synopsis of the summary statistics for the 40 new locus pairs presented in Table 3, which is published as supporting information on the PNAS web site. The allele frequency spectrum was summarized by the average and variance of Tajima's D and Fu and Li's D* across loci, polymorphism levels are summarized by the average number of polymorphic sites (S̄) across loci, and LD decay was summarized in terms of an overall composite likelihood estimator of the population cross-over rate parameter 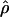 (33). The results of this expanded data set are in qualitative agreement with those from our previous survey (19) and with other similar data sets (2, 5, 15, 16). With regard to the allele frequency spectrum, the Hausa show a skew toward rare variants and a low variance across loci, whereas both non-African samples have an excess of intermediate frequency variants and high variance across loci. In addition, polymorphism levels and LD decay are higher in the Hausa compared with both non-African samples, but this difference is greater for LD decay (1.9- to 3.2-fold) than polymorphism levels (1.6-fold).
(33). The results of this expanded data set are in qualitative agreement with those from our previous survey (19) and with other similar data sets (2, 5, 15, 16). With regard to the allele frequency spectrum, the Hausa show a skew toward rare variants and a low variance across loci, whereas both non-African samples have an excess of intermediate frequency variants and high variance across loci. In addition, polymorphism levels and LD decay are higher in the Hausa compared with both non-African samples, but this difference is greater for LD decay (1.9- to 3.2-fold) than polymorphism levels (1.6-fold).
Table 1. Observed summary statistics.
| Population | D̄ | 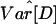 |
D̄* | S̄ |
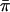 , % , % |
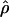 |
|---|---|---|---|---|---|---|
| Hausa | –0.20 | 0.55 | –0.17 | 11.1 | 0.110 | 0.0006 |
| Italian | 0.28* | 1.19** | 0.18 | 7.1 | 0.085 | 0.0003 |
| Chinese | 0.18 | 1.08* | 0.05 | 6.9 | 0.079 | 0.0002* |
Observed summary statistics of polymorphism data for 50 locus pairs. *, P < 0.05; **, P < 0.01; under an equilibrium model.
To determine whether the levels of LD decay and the frequency spectrum were consistent with a model of constant population size, we conducted coalescent simulations under equilibrium to determine the P values of the observed summary statistics. We obtained the effective population size, denoted NA, for each population by using an estimator of the population mutation rate parameter (4NAμ) based on the number of polymorphic sites and sample size (45), and an estimate of μ based on sequence divergence between human and chimpanzee for the 50 locus pairs. Each summary statistic for the Hausa data are consistent with the equilibrium model (Table 1). However, for the non-African populations, the skew toward intermediate frequency variants, and the elevated LD are incompatible with a simple equilibrium model; a combined statistic based on D̄, 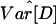 , and
, and 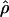 , obtained by using Eq. 1, is significant for the Italian (P ≤ 0.0148) and Chinese data (P ≤ 0.0052).
, obtained by using Eq. 1, is significant for the Italian (P ≤ 0.0148) and Chinese data (P ≤ 0.0052).
Estimating the Ancestral Population Size Under a Growth Model. Even though a model of constant population size could not be rejected for the Hausa, human populations certainly experienced rapid growth recently and, perhaps, in more ancient times. Thus, the negative but nonsignificant values of Tajima's D and Fu and Li's D* in the Hausa may simply reflect limited power and suggest that some expansion models are appropriate for this population. By following the approach in ref. 28, we considered a model in which an ancestral population at equilibrium size NA grows exponentially beginning tonset generations in the past at rate α, such that the present population size is NAeαtonset (8). To test this model, we fixed the ancestral population size for each combination of demographic parameter values, such that the expected number of segregating sites matched the average number observed in the Hausa sample (28).
Unlike in ref. 28, we estimated the best-fit growth parameters for the Hausa data, α and tonset, along with the associated point estimate of NA, via approximate maximum likelihood (ML) based on the summary statistic, Fu and Li's D*. We focused on the average D* across the 50 locus pairs, denoted 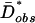 , because it was previously shown to be the most informative for discriminating between equilibrium and growth models (28). For each demographic growth model, we obtained distributions of D̄* by simulation and estimated the probability that
, because it was previously shown to be the most informative for discriminating between equilibrium and growth models (28). For each demographic growth model, we obtained distributions of D̄* by simulation and estimated the probability that 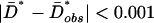 and then chose the model for which this probability was highest. This procedure returns the approximate ML estimate of the growth parameters, α and tonset, compatible with the Hausa data based on D̄*obs. Note that we refer to this as approximate ML on a summary statistic because we do not use the full data and because we approximate rather than obtain the probabilities exactly. We found that the model with the highest overall probability was at an α of 0.75 × 10-3 and tonset of 1,000 generations, which corresponds to a model with ≈2–fold growth starting 25,000 years ago, assuming a generation time of 25 years, from an ancestral population size of 10,659. We present confidence sets of α and tonset for which
and then chose the model for which this probability was highest. This procedure returns the approximate ML estimate of the growth parameters, α and tonset, compatible with the Hausa data based on D̄*obs. Note that we refer to this as approximate ML on a summary statistic because we do not use the full data and because we approximate rather than obtain the probabilities exactly. We found that the model with the highest overall probability was at an α of 0.75 × 10-3 and tonset of 1,000 generations, which corresponds to a model with ≈2–fold growth starting 25,000 years ago, assuming a generation time of 25 years, from an ancestral population size of 10,659. We present confidence sets of α and tonset for which 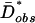 are consistent with the observed Hausa data in Fig. 3, which is published as supporting information on the PNAS web site. The span of acceptable models is consistent with previous reports (28), with a slight reduction in confidence set due to the inclusion of additional data.
are consistent with the observed Hausa data in Fig. 3, which is published as supporting information on the PNAS web site. The span of acceptable models is consistent with previous reports (28), with a slight reduction in confidence set due to the inclusion of additional data.
To asses the uncertainty in NA, we obtain a range of NA consistent with the ML estimate of 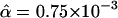 and t̂onset = 1,000 as follows. We performed additional coalescent simulations as described earlier, where we used the ML parameters for the demographic history and gradually lowered or raised the value of NA until S̄ was incompatible with the observed data at the 5% level. We found these high and low values of NA to be 9,450 and 12,300, respectively. Later, we will use this information to assess the effect of our choice of NA in testing bottleneck models.
and t̂onset = 1,000 as follows. We performed additional coalescent simulations as described earlier, where we used the ML parameters for the demographic history and gradually lowered or raised the value of NA until S̄ was incompatible with the observed data at the 5% level. We found these high and low values of NA to be 9,450 and 12,300, respectively. Later, we will use this information to assess the effect of our choice of NA in testing bottleneck models.
Testing Bottleneck Models in the Non-African Data. The positive D̄ values and large 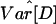 along with the low polymorphism and high LD levels observed in the non-African populations (Table 1) suggest that models including a reduction in population size may be compatible with the data. We considered one family of bottleneck models for these data, where a population of constant size NA instantaneously shrinks in size to b · NA at time tstart generations before the present. The population remains at that size for tdur generations and then instantaneously recovers to its original size (Fig. 4, which is published as supporting information on the PNAS web site).
along with the low polymorphism and high LD levels observed in the non-African populations (Table 1) suggest that models including a reduction in population size may be compatible with the data. We considered one family of bottleneck models for these data, where a population of constant size NA instantaneously shrinks in size to b · NA at time tstart generations before the present. The population remains at that size for tdur generations and then instantaneously recovers to its original size (Fig. 4, which is published as supporting information on the PNAS web site).
Under the assumption that non-African populations originated from an ancestral population in sub-Saharan Africa, we set the ancestral population size in the bottleneck simulations to the values of NA obtained by ML based on the Hausa data and the simple growth model (NA = 10,659). This assumption has important implications for our subsequent inferences about compatible bottleneck scenarios. We then used coalescent simulations to estimate the P values for each summary statistic, point on a grid of bottleneck severities (b), bottleneck duration (tdur), and time since the beginning of the bottleneck (tstart). This procedure allows defining the portion of the multidimensional parameter space that is compatible with the data.
By combining P values of different summaries as described by Eq. 1, we can make of multiple aspects of the data to narrow the confidence region of compatible parameter values. The value of such an approach is depicted in Fig. 1. We found that, for all possible combinations of two or more summary statistics, the combination of 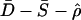 was the most powerful to discriminate between bottlenecks and a constant size model over the parameter range depicted in Fig. 1. Therefore, we use the combination of
was the most powerful to discriminate between bottlenecks and a constant size model over the parameter range depicted in Fig. 1. Therefore, we use the combination of 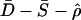 in our subsequent analyses of bottleneck models.
in our subsequent analyses of bottleneck models.
Fig. 1.

Power of combining multiple summary statistics. The power to reject a constant size demographic model for combined summaries with an NA of 10,659 under bottleneck models with a 70% reduction in NA and a total time of 40,000 years for various bottleneck durations (tdur) (a) and a bottleneck duration of 20,000 years for a total demographic epoch of 40,000 years for various bottleneck severities (b). The type-I error rate was held at 5%.
The confidence sets for the Italian and Chinese data for a tstart value of 40,000 years and NA = 10,659 are shown in Fig. 2 b and e; in all cases, the accepted portion of the parameter space tends to lie on the diagonal of the plots, indicating that bottleneck severity and duration have inversely related effects on patterns of variation. The Italian data are compatible with a range of bottleneck models that include shorter and more severe bottlenecks (e.g., b = 0.1, tdur = 400 generations) at one end and longer and milder bottlenecks (e.g., b = 0.4, tdur = 1,600 generations) at the other. If tstart = 80,000 years ago, this range is slightly shifted to the right, including longer and less severe bottlenecks (Fig. 5, which is published as supporting information on the PNAS web site). For the Chinese data, if tstart is 40,000 years, the compatible parameter space is similar to that of the Italian data, except that it includes slightly more severe bottleneck scenarios (Fig. 2 b and e). The most severe and longest bottleneck occurs where b = 0.005 and tdur = 300–600 generations, but fewer combinations of parameter values corresponding to mild bottlenecks are accepted. If tstart = 80,000 years, milder bottlenecks cannot be rejected, and even a long-lasting and mild bottleneck with b = 0.4 cannot be rejected (Fig. 5). For NA = 10,659 and for any value of tstart, no bottleneck of <100 generations is accepted in either population.
Fig. 2.

Confidence sets for a bottleneck with tstart of 40,000 years. Results are shown for the Italian (a–c) and Chinese (d–f) data sets for NA values of 9,450 (a and d), 10,659 (b and e), and 12,300 (c and f). The combined statistics are 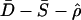 . The contours represent the confidence region of parameter space with P values of 0.1, 0.05, 0.02, and 0.01 from innermost to outermost, with darker shading indicating lower P values.
. The contours represent the confidence region of parameter space with P values of 0.1, 0.05, 0.02, and 0.01 from innermost to outermost, with darker shading indicating lower P values.
We also considered values for tstart of 20,000 and 120,000 years (Figs. 6 and 7, which are published as supporting information on the PNAS web site). In all cases, the lower tstart values showed a confidence set that was shifted toward scenarios of longer and more severe bottlenecks. Conversely, at higher tstart values, more severe bottlenecks were rejected in favor of milder bottleneck scenarios.
To assess the effect of the uncertainty associated with the estimates of NA, we repeated the above analyses by using different values of NA that were obtained from estimating the uncertainty around NA from the Hausa ML growth models described above. As shown in Fig. 2, the effect of NA on the accepted parameter space is substantial. As expected, for the larger value of NA, the accepted portion of the parameter space is reduced such that only relatively severe and long bottlenecks are compatible with the data, whereas a larger range of less severe bottlenecks are compatible with the smaller value of NA.
Discussion
By resequencing unlinked noncoding regions, we assessed patterns of polymorphism levels, frequency spectrum and LD for the same set of genomic segments and population samples. To achieve greater resolution of different demographic scenarios, we use an analytical approach that combines information from individual summary statistics of sequence variation; computer simulations showed that combinations of summaries allow for more powerful tests of each demographic scenario. Rather than focusing on a single best-fitting demographic model, we construct an acceptance region of the parameter space that is compatible with the demographic model of interest (in this case population growth or bottleneck), thus providing an inclusive picture of the uncertainty in inferences of human demography. A major conclusion of this analysis is that the non-African population samples are compatible with simple bottleneck models even when multiple aspects of sequence variation are considered simultaneously. Consistent with our previous analysis (28), the Hausa sample from sub-Saharan Africa is compatible with the equilibrium model and with relatively recent population growth.
Modeling human population history is central to a variety of questions in human biology, but most recently the search for signatures of natural selection has given new importance to this line of inquiry (2, 5, 46, 47). The impact of natural selection on the human genome can be detected by contrasting patterns of neutral variation, i.e., those shaped solely by demography, to those observed at test loci that may be shaped by natural selection in addition to demography. Traditionally, this contrast used the theoretical predictions of the standard neutral model in which the population was assumed to be constant in size and randomly mating. However, studies of human variation have shown genome-wide departures from this model, suggesting that human demography is complex (7, 15, 21, 48). Thus, the development of a more realistic null model of evolutionary neutrality is necessary for improving inferences about natural selection (2, 5).
Several conditions must be satisfied to achieve these goals. One is the availability of sequence variation data for many unlinked and neutrally evolving regions. Although several whole-genome variation data sets are available, they consist mainly of genotyping data for ascertained polymorphisms (49, 50). Resequencing data are also available, but they tend to focus on gene regions that may have been targets of selection and, hence, are less suitable for demographic inference (2, 3, 5). An additional challenge derives from the complexity of human demography and the fact that realistic models are defined by multiple unknown demographic parameters, which implies that, for any given value of one parameter (e.g., bottleneck severity), there may be a range of values for the other parameters (e.g., time of onset and duration of bottleneck) that are equally consistent with the data. It is particularly important in this context to make efficient use of the information in the data. Although it may be useful to generate point estimates of the demographic parameters, it is even more important to obtain the multidimensional confidence set if specific hypotheses about human evolution are to be tested.
The present study represents an important step toward improving our inferences about human demography. Although the present data set is not as large as other resequencing surveys (2, 5), it was specifically designed for demographic inference and will provide a useful reference for analyses of gene regions, because, in an attempt to select neutrally evolving regions, we focused on segments that neither contain nor are tightly linked to coding regions. In addition, most of these segments neither contain nor are tightly linked to noncoding sequences conserved between human and mouse. Our scheme for data collection aimed at maximizing the information content of the data so that multiple aspects of genetic variation could be analyzed for the same set of independent loci. Owing to the use of ethnically identified samples, we could provide evidence for different demographic histories in different populations.
Our analytical approach also improves on previous studies of human demography. First, it provides a full characterization of the uncertainty around the best-fitting model by identifying the portion of the multidimensional parameter space that is consistent with genetic variation data in each population. The inclusion of multiple aspects of genetic variation by combining the P values for different summary statistics provides greater power than any single summary alone, allowing us to reduce substantially the accepted space for each model. Our study is based on an extensive exploration of the demographic parameter space including onset, duration, and severity of the bottleneck. It is important to note that the reduction in bottleneck parameter space was greatly aided by our inference about NA based on the Hausa data. Because the NA is restricted, the range of compatible values for summary statistics that depend on NA (i.e., 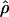 and S̄) is also constrained.
and S̄) is also constrained.
An important limitation of our analysis is that we considered only models of randomly mating populations. Although this is a common assumption in modeling studies of population size change, it is unlikely to be satisfied by human populations, even if geographically defined (34, 51). In fact, it is possible that population structure alone could account for the observed patterns of human variation (2, 5, 15, 35). Interestingly, the addition of 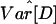 into the bottleneck analysis results in a further reduction of the accepted parameter space (Figs. 8–11, which are published as supporting information on the PNAS web site), although combining this statistic with D̄, S̄, and
into the bottleneck analysis results in a further reduction of the accepted parameter space (Figs. 8–11, which are published as supporting information on the PNAS web site), although combining this statistic with D̄, S̄, and 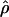 reduces the power to reject the constant size model (Fig. 1). This observation suggests that additional features, such as population structure, are required to produce
reduces the power to reject the constant size model (Fig. 1). This observation suggests that additional features, such as population structure, are required to produce 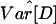 values that are more consistent with our data. Although it is desirable and certainly more realistic to include elements of population structure in models of human demography (52), there is insufficient data to indicate the most plausible family of such models. For these reasons, testing simple growth and bottleneck models is a reasonable first step toward developing more complex and realistic models. Obviously, if changes in population size and population structure were considered jointly rather than separately, the accepted range of values for the growth and bottleneck parameters is likely to be different.
values that are more consistent with our data. Although it is desirable and certainly more realistic to include elements of population structure in models of human demography (52), there is insufficient data to indicate the most plausible family of such models. For these reasons, testing simple growth and bottleneck models is a reasonable first step toward developing more complex and realistic models. Obviously, if changes in population size and population structure were considered jointly rather than separately, the accepted range of values for the growth and bottleneck parameters is likely to be different.
A main conclusion of this study is that simple bottleneck models can explain the non-African data even when multiple aspects of genetic variation are considered simultaneously. Several previous studies of human sequence variation had modeled specific bottleneck scenarios on the basis of either frequency spectrum information (2, 5, 15, 48), LD decay (18), or polymorphism levels (21). Wall and Przeworski (15) analyzed full resequencing data and proposed that a bottleneck and selective sweeps at some loci could explain the frequency spectrum observed in non-Africans but did not provide information regarding the likely parameter values. The frequency spectrum was used also by Marth et al. (20) to estimate a best-fit bottleneck model for Europeans and East Asians. We used our simulation scheme to estimate the probability of the Italian and Chinese data for the corresponding best-fit models of Marth et al. (20). In our parameterization, the best fit model for the Asian sample in Marth et al. corresponds to an NA of 10,000, b of 0.3, tdur of 400 generations, a tstart of 90,000 years; note that this model includes growth after the bottleneck to a size of 25,000. The best-fit model for the European sample in Marth et al. corresponds to an NA of 10,000, b of 0.2, tdur of 500 generations, and a tstart of 87,500 years, with growth after the bottleneck to a size of 20,000. Using our simulation scheme, our data turned out to be incompatible with these models (P < 0.0001). It should be noted, however, that Marth et al. (20) analyzed a data set of ascertained SNPs and attempted to correct for the resulting bias. Hence, the discrepancy between the two studies may be due to incomplete ascertainment correction and highlights the value of resequencing data.
Based on the frequency spectrum observed in a large resequencing study of genes involved in inflammation, Akey et al. (2) concluded that the European data were consistent with a bottleneck starting 40,000 years and a bottleneck intensity, as measured by the inbreeding coefficient (F) of 0.175. This best-fit model can be translated to a range of models in our notation by using
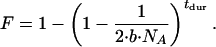 |
[2] |
This bottleneck model corresponds to a number of points that are well within the accepted portion of the parameter space for our non-African data (for example, b = 0.2 and tdur = 820 generations assuming our best-fit NA of 10,659). Because only the best-fit model is reported by Akey et al. (2), the overall agreement between these two data sets cannot be assessed.
Similar conclusions were obtained through an analysis of pairwise LD data of ascertained SNPs in a European population sample (18); however, a narrow portion of the parameter space was investigated. We determined that there are points in our accepted parameter space that correspond to the estimated time of onset and F reported by Reich et al. (18), indicating agreement between the two methods and data sets. Finally, a recent analysis of resequencing data from a pool of ethnically diverse samples detected evidence for very recent population growth (3). Although this model is compatible with our Hausa data, it does not provide a good explanation for the Italian and Chinese data, hence, pointing to the need for population-specific demographic inferences.
Supplementary Material
Acknowledgments
We thank W.-H. Li (University of Chicago, Chicago), G. Galluzzi (Catholic University, Rome), and J. Donfack (Center for Genomic Sciences, Pittsburgh) for DNA samples; M. Przeworski for helpful comments on an earlier version of the manuscript; and D. S. Angulo (DePaul University, Chicago) and J. K. Pritchard (University of Chicago, Chicago) for use of computational resources. This work was supported by National Institutes of Health Grant R01 HG02098 (to A.D.R.). L.A.F. was supported by National Research Service Award F32 HG00219. A.M.A. was supported by U.S. Department of Education Grant P200A030043. B.F.V. was supported by National Institutes of Health Grants R01 DK55889, R01 HG02772, and T32 GM07197.
Author contributions: R.R.H. and A.D.R. designed research; B.F.V., A.M.A., L.A.F., and Y.Q. performed research; R.R.H. contributed new reagents/analytic tools; B.F.V., A.M.A., and L.A.F. analyzed data; and B.F.V., A.M.A., and A.D.R. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: LD, linkage disequilibrium; ML, maximum likelihood.
References
- 1.Lahr, M. M. & Foley, R. A. (1998) Year Book Phys. Anthropol. 41, 137-176. [DOI] [PubMed] [Google Scholar]
- 2.Akey, J. M., Eberle, M. A., Rieder, M. J., Carlson, C. S., Shriver, M. D., Nickerson, D. A. & Kruglyak, L. (2004) PLoS Biol. 2, e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williamson, S. H., Hernandez, R., Fledel-Alon, A., Nielsen, R. & Bustamante, C. D. (2005) Proc. Natl. Acad. Sci. USA 102, 7882-7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith, C. N. & Fearnhead, P. (2005) Genetics, 10.1534/genetics.104.036293. [DOI] [PMC free article] [PubMed]
- 5.Stajich, J. E. & Hahn, M. W. (2005) Mol. Biol. Evol. 22, 63-73. [DOI] [PubMed] [Google Scholar]
- 6.Goldstein, D. B. & Chikhi, L. (2002) Annu. Rev. Genomics Hum. Genet. 3, 129-152. [DOI] [PubMed] [Google Scholar]
- 7.Reich, D. E., Schaffner, S. F., Daly, M. J., McVean, G., Mullikin, J. C., Higgins, J. M., Richter, D. J., Lander, E. S. & Altshuler, D. (2002) Nat. Genet. 32, 135-142. [DOI] [PubMed] [Google Scholar]
- 8.Slatkin, M. & Hudson, R. R. (1991) Genetics 129, 555-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Di Rienzo, A. & Wilson, A. C. (1991) Proc. Natl. Acad. Sci. USA 88, 1597-1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rogers, A. R. & Harpending, H. (1992) Mol. Biol. Evol. 9, 552-569. [DOI] [PubMed] [Google Scholar]
- 11.Sherry, S. T., Rogers, A. R., Harpending, H., Soodyall, H., Jenkins, T. & Stoneking, M. (1994) Hum. Biol. 66, 761-775. [PubMed] [Google Scholar]
- 12.Rogers, A. R. & Jorde, L. B. (1995) Hum. Biol. 1, 1-36. [PubMed] [Google Scholar]
- 13.Weiss, G. & von Haeseler, A. (1998) Genetics 149, 1539-1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hey, J. (1997) Mol. Biol. Evol. 14, 166-172. [DOI] [PubMed] [Google Scholar]
- 15.Wall, J. & Przeworski, M. (2000) Genetics 155, 1865-1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Przeworski, M., Hudson, R. R. & Di Rienzo, A. (2000) Trends Genet. 16, 296-302. [DOI] [PubMed] [Google Scholar]
- 17.Tishkoff, S. A., Dietzsch, E., Speed, W., Pakstis, A. J., Kidd, J. R., Cheung, K., Bonne-Tamir, B., Santachiara-Benerecetti, A. S., Moral, P. & Krings, M. (1996) Science 271, 1380-1387. [DOI] [PubMed] [Google Scholar]
- 18.Reich, D. E., Cargill, M., Bolk, S., Ireland, J., Sabeti, P. C., Richter, D. J., Lavery, T., Kouyoumjian, R., Farhadian, S. F., Ward, R., et al. (2001) Nature 411, 199-203. [DOI] [PubMed] [Google Scholar]
- 19.Frisse, L., Hudson, R. R., Bartoszewicz, A., Wall, J., Donfack, J. & Di Rienzo, A. (2001) Am. J. Hum. Genet. 69, 831-843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marth, G. T., Czabarka, E., Murvai, J. & Sherry, S. T. (2004) Genetics 166, 351-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marth, G., Schuler, G., Yeh, R., Davenport, R., Agarwala, R., Church, D., Wheelan, S., Baker, J., Ward, M., Kholodov, M., et al. (2003) Proc. Natl. Acad. Sci. USA 100, 376-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ardlie, K. G., Krugliak, L. & Seilstad, M. (2002) Nat. Rev. Genet. 3, 299-309. [DOI] [PubMed] [Google Scholar]
- 23.Ardlie, K. G., Krugliak, L. & Seilstad, M. (2002) Nat. Rev. Genet. 3, 566. [DOI] [PubMed] [Google Scholar]
- 24.Eswaran, V., Harpending, H. & Rogers, A. R. (2005) J. Hum. Evol. 49, 1-18. [DOI] [PubMed] [Google Scholar]
- 25.Kuhner, M. K., Yamato, J. & Felsenstein, J. (1998) Genetics 149, 429-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nielsen, R. (1999) Genetics 154, 931-942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beerli, P. & Felsenstein, J. (2001) Proc. Natl. Acad. Sci. USA 98, 4563-4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pluzhinikov, A., Di Rienzo, A. & Hudson, R. R. (2002) Genetics 161, 1209-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nielsen, R. (2004) Hum. Genomics 1, 218-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kreitman, M. & Di Rienzo, A. (2004) Trends Genet. 20, 300-304. [DOI] [PubMed] [Google Scholar]
- 31.Soldevila, M., Calafell, F., Helgason, A., Stefansson, K. & Bertranpetit, J. (2005) Trends Genet. 21, 389-391. [DOI] [PubMed] [Google Scholar]
- 32.Tajima, F. (1989) Genetics 123, 585-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hudson, R. R. (2001) Genetics 159, 1805-1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosenberg, N. A., Pritchard, J. K., Weber, J. L., Cann, H. M., Kidd, K. K., Zhivotovsky, L. A. & Feldman, M. W. (2002) Science 298, 2381-2385. [DOI] [PubMed] [Google Scholar]
- 35.Wakely, J. & Lessard, S. (2003) Genetics 164, 1043-1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kong, A., Gudbjartsson, D. F., Sainz, J., Jonsdottir, G. M., Gudjonsson, S. A., Richardsson, B., Sigurdardottir, S., Barnard, J., Hallbeck, B., Masson, G., et al. (2002) Nat. Genet. 31, 241-247. [DOI] [PubMed] [Google Scholar]
- 37.Wall, J. D., Frisse, L. A., Hudson, R. R. & Di Rienzo, A. (2003) Am. J. Hum. Genet. 74, 1330-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nickerson, D. A., Tobe, V. O. & Taylor, S. L. (1997) Nucleic Acids Res. 25, 2745-2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fu, Y.-X. & Li, W.-H. (1993) Genetics 133, 693-709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hudson, R. R. (2002) Bioinformatics 18, 337-338. [DOI] [PubMed] [Google Scholar]
- 41.Yu, A., Zhao, C., Fan, Y., Jang, W., Mungall, A. J., Deloukas, P., Olsen, A., Doggett, N. A., Ghebranious, N., Broman, K. W., et al. (2001) Nature 409, 951-953. [DOI] [PubMed] [Google Scholar]
- 42.McVean, G. A., Myers, S. R., Hunt, S., Deloukas, P., Bently, D. R. & Donnelly, P. J. (2004) Science 304, 581-584. [DOI] [PubMed] [Google Scholar]
- 43.Ptak, S. E., Voelpel, K. & Przeworski, M. (2004) Genetics 167, 387-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jeffreys, A. J. & May, C. A. (2004) Nat. Genet. 36, 151-156. [DOI] [PubMed] [Google Scholar]
- 45.Watterson, G. A. (1975) Theo. Popul. Biol. 7, 256-276. [DOI] [PubMed] [Google Scholar]
- 46.Jensen, J. D., Kim, Y., Dumont, V. B., Aquadro, C. F. & Bustamante, C. D. (2005) Genetics 170, 1401-1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu, L. & Bustamante, C. D. (2005) Genetics 170, 1411-1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adams, A. M. & Hudson, R. R. (2004) Genetics 168, 1699-1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.The International Hapmap Consortium (2003) Nature 426, 789-796. [DOI] [PubMed] [Google Scholar]
- 50.Hinds, D. A., Stuve, L. L., Nilsen, G. B., Halperin, E., Eskin, E., Ballinger, D. G., Frazer, K. A. & Cox, D. R. (2005) Science 307, 1072-1079. [DOI] [PubMed] [Google Scholar]
- 51.Harding, R. M. & McVean, G. (2004) Curr. Opin. Genet. Dev. 14, 667-674. [DOI] [PubMed] [Google Scholar]
- 52.Wakeley, J., Nielsen, R., Liu-Cordero, S. N. & Ardlie, K. (2001) Am. J. Hum. Genet. 69, 1332-1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.