

Abstract
Despite the presence of neural stem cells and ongoing neurogenesis in some regions of the adult mammalian brain, neurons are not replaced in most brain regions after injury. With the aim to unravel factors contributing to the failure of neurogenesis in the injured cerebral cortex, we examined the expression of cell fate determinants after acute brain injuries, such as stab wound or focal ischemia, and in a model of chronic amyloid deposition. Although none of the neurogenic factors, such as Pax6, Mash1, Ngn2, was detected in the injured parenchyma, we observed a strong up-regulation of the bHLH transcription factor Olig2, but not Olig1, upon acute and chronic injury. To examine the function of Olig2 in brain lesion, we injected retroviral vectors containing a dominant negative form of Olig2 into the lesioned cortex 2 days after a stab wound. Antagonizing Olig2 function resulted in a significant number of infected cells generating immature neurons that were not observed after injection of the control virus. These data, therefore, imply Olig2 as a repressor of neurogenesis in cells reacting to brain injury and open innovative perspectives toward evoking endogenous neuronal repair.
Keywords: amyloid, gliosis, mouse neocortex, pax6, stab wound
The discovery that adult neural stem cells in the mammalian brain continue to generate neurons throughout life has shed new light on the possibility to repair degenerated neurons by endogenous sources. The similarity of adult neural stem cells to astroglial cells (1, 2) prompts the question whether glial cells reacting to injury also may have a broader potential. Astrocytes mediate the wound healing reaction of the central nervous system (CNS) upon injury (3) and reexpress molecules [vimentin, nestin, tenascin-C, and glia-fibrillary acidic protein (GFAP); see refs. 3 and 4] that are absent in mature astrocytes but present during development in radial glial cells (5–7) and adult neural stem cells (8–13). Whereas during development adult neural stem cells and radial glia generate neurons (1, 2, 6, 14–16), reactive astrocytes do not, despite their partial dedifferentiation response. However, immature astrocytes can generate neurons in vitro upon stimulation with intrinsic or extrinsic factors (17, 18). This neurogenic potential is not restricted to astrocytes, because NG2-positive glial precursors also can generate neurons upon exposure to certain culture conditions (19) or transplantation into neurogenic zones in vivo (20) but fail to regenerate neurons upon injury in vivo (21). Thus, endogenous precursor and glial cell populations have the potential to generate neurons but fail to do so upon injury in vivo.
Here, we set out to elucidate the molecular cues involved in the glial fate restriction after injury. We examined transcription factors, such as Pax6, Ngn2, Mash1, Gsh2, Olig1, and Olig2, that regulate neuron-glia fate decisions during development and are present in precursors in the subependymal zone (SEZ) of the adult mouse forebrain (22–24). Our results identify Olig2 as a key factor in reaction of glial cells to brain injury and show that, by interfering with Olig2 function, some degree of endogenous neurogenesis can be evoked. Thus, these results show proof of principle evidence that neurogenesis can occur in the mammalian neocortex, even in severe injury conditions, such as acute stab-wound lesion.
Materials and Methods
Animals and Surgical Procedures. Forty-three CD1 mice (2–6 months, Charles River Laboratories, Sulzfeld, Germany) were anesthetized (ketamine, 100 mg/kg, Ketavet; Amersham Pharmacia, Erlangen, Germany, and xylazine, 5 mg/kg, Rompun; Bayer, Leverkusen, Germany) and underwent a stab wound in the right cerebral neocortex (Bregma from -0.9 mm to -2.7 mm, latero-lateral 1.5–2.5 mm) or hindbrain (lateral reticular nucleus, Bregma from -8 to -8.12 mm, 1.1 mm latero-lateral). Mice were killed at 24 h (n = 3) and 3 (n = 6), 7 (n = 9), and 30 days after lesion (n = 3; intact, n = 2). In some experiments, 1 μl of retroviral suspension containing green fluorescent protein (GFP), Olig2-VP16-IRES-GFP (Olig2VP16), or Pax6-IRES-GFP (titers: 106 to 107, produced as described in ref. 22) was injected 2 days after injury within 100–150 μm from the lesion track at 0.5- to 1-mm depth from the dura (n = 22, survival times: 2, 7, 14, and 30 days after injection).
To examine focal ischemia, seven C57BL/6 male mice (2 months old) underwent occlusion of the middle cerebral artery (MCAO) by the intraluminal filament model (25), according to ref. 26, for 2 h. Reperfusion was induced by thread withdrawal under short inhalation anesthesia. The animals were killed after 24 h (n = 3) or 7 days (n = 4).
As a model for amyloid plaque deposition, we examined four pairs of 6- to 9-month-old C57BL/6-TgN (Thy1-APPKM670/671NL; Thy1-PS1L166P) mice (Thy1-APPPS) and wild-type littermates. The transgenic mice were generated by coinjection of Thy1-APPKM670/671NL and Thy1-PS1L166P constructs into B6 oocytes (line 21; R. Radde and M.J., unpublished observations, but see refs. 27 and 28). The Thy1 promoter restricted transgene expression to CNS neurons. For the present study, line 21 was used. Cerebral amyloidosis (Fig. 5, which is published as supporting information on the PNAS web site) developed at the age of 6–8 weeks.
Proliferation Analysis and Histological Procedures. The DNA base analog 5-bromodeoxyuridine (BrdUrd, Sigma) was injected i.p. (100 mg/kg body weight) 2 h before perfusion to label fast proliferating cells (short pulse). Alternatively, animals received BrdUrd in the drinking water (long pulse, 1 mg/ml) from the first postsurgery day for either 2 (3 days survival) or 6 days (7 days survival) or 2 weeks (Thy1-APPPS mice). The mice were transcardially perfused with 4% paraformaldehyde in phosphate buffer, and sections were cut after cryoprotection. In MCAO-treated brains, volumetric analysis (25) showed infarction in the basal ganglia and cortex, corresponding to the territory supplied by the middle cerebral artery. Immunostainings and in situ hybridization were performed according to standard protocols, and the antibodies used are listed in the Supporting Materials, which is published as supporting information on the PNAS web site).
Quantitative Analysis. Quantifications (cell densities, marker coexpression and nuclear size; three sections per animal, three animals for each time point or lesion model) were performed by means of neurolucida and neuroexplorer (Microbright-field, Colchester, VT), connected to an Axiophot Zeiss microscope (×40 objective). The analysis was performed on 200,000 μm2 areas within the cortical gray matter (GM) and localized 100 μm away from the stab-lesion track (both laterally and medially) or the edge of the ischemic core, and in corresponding areas of the intact hemisphere, wild-type, and Thy1-APPPS cortices. All results are presented as averages and standard deviations, unless indicated differently. Statistical analysis was performed by the Student or one-sample t test.
Results
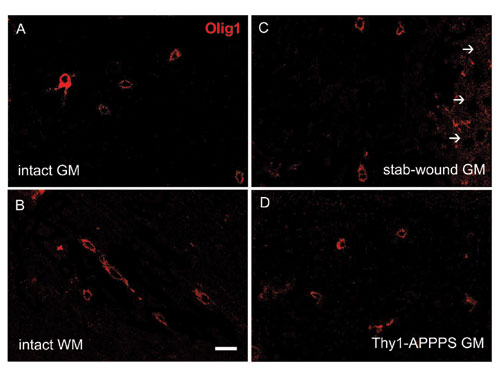
Increase in Olig2-Immunoreactive Cells in the Cortex After Stab-Wound Injury. After stab-wound injury of the adult cerebral cortex, microglia is the first cell type to react (29), followed by an increase in NG2-positive cells, astroglia reaction (increase in number and hypertrophy), and proliferation of various cell types (Fig. 6, which is published as supporting information on the PNAS web site; see also Fig. 2I). In this context, we examined the transcription factors Pax6, Mash1, Gsh2, Ngn2, Olig1, and Olig2 1–30 days after the stab wound by immunochemistry. No Pax6-, Mash1-, Gsh2- or Ngn2-immunopositive cells were detectable in the lesioned cortex despite positive signals in the adult subependymal zone or the embryonic telencephalon (data not shown), consistent with the absence of even a low degree of neurogenesis around the lesion as monitored by doublecortin (dcx) staining (data not shown) or retroviral infection (see below). In contrast, 3–7 days after the stab wound, we observed a prominent increase in the number of Olig2-immunoreactive cells (spread in the cortex up to 400–500 μm from the lesion) in comparison with the intact contralateral cortex, where Olig2-positive cells were much more rare (Fig. 1 A–C). The increase in Olig2 was also detected at the mRNA level, as revealed by in situ hybridization (Fig. 1 D and E), and this reaction returned to control levels 30 days after lesion (dpl, Fig. 1C). Notably, the increase in Olig2-expressing cells was specific because even the close family member Olig1 was not affected in its expression levels upon stab-wound injury (Fig. 7, which is published as supporting information on the PNAS web site).
Fig. 2.

Identification of Olig2-immunoreactive cells after stab-wound and proliferation analysis. (A–E) Frontal sections of the intact neocortical GM immunostained as indicated. Olig2 immunoractivity was detected in CC1+ oligodendrocytes (A), NG2+ glial precursors (B), and S100β+ astrocytes (C). Arrows point to colabeled cells, and arrowheads point to cells devoid of Olig2. (D) Olig2 and Sox10 coexpression (arrows) in the cortical GM are shown. (E) The micrograph depicts Olig2+ nuclei triple labeled for BrdUrd (red) and a mixture of antibodies (in blue: NG2, CC1, and S100β) close to the stab-wound track: Several proliferating Brdu+/Olig2+ cells are lineage marker-negative (arrow). Histograms in F depict the proportions of cell types in the Olig2+ population in the intact cortex and after a stab wound, whereas in G, the percentage of Olig2+ cells amongst different glial populations is shown. The fractions of Olig2+ astrocytes increased at 7 dpl (P = 0.012) compared to the intact situation. The opposite was observed for the NG2+ cells in comparison with the intact cortex (P = 0.003 at 3 days; P = 0.042 at 7 days) and the proportion of Olig2+/CC1+ oligodendrocytes rose 7 dpl (P = 0.044). (H) Olig2/BrdUrd colabeled cells (indicated also as percentages) are shown over the density of BrdUrd-positive cells in the intact and stab-lesioned cortex (2-h BrdUrd pulse). (I) The composition of BrdUrd-positive cells is depicted after 2 h (Left) or several days of BrdUrd pulse (Right). (Scale bars: A–C and E, 20 μm; D, 100 μm.)
Fig. 1.

Increase in the number of Olig2-immunoreactive cells after a stab wound. (A and B) Frontal sections of the gray matter (GM) in the adult mouse neocortex stained for Olig2. Note the increase in the number of Olig2+ cells after a stab wound (s-w, B; yellow line indicates the lesion site, dashed contours outline some unspecific/autofluorescent staining, and arrowheads point to some Olig2+ cells) compared to the intact cortex (A, arrowheads indicate Olig2+ cells) as quantified in the histogram (C). *, statistically significant differences; hs, hours; dpl, days after lesion. In situ hybridization reveals Olig2 mRNA up-regulation after a stab wound (E) compared to the intact cortex (D). (Scale bars: 100 μm.)
Identity of Olig2-Immunopositive (Olig2+) Cells in the Intact and Stab-Wound Injured Cortex. Next, we characterized the identity of cells expressing Olig2 (see Table 1, which is published as supporting information on the PNAS web site, for cell type identification criteria). In the intact adult CNS, Olig2 is expressed in mature oligodendrocytes and NG2-positive glial precursors (30–32). Consistent with these data, the majority of NG2-positive (NG2+) cells in the intact cortical GM (≈70%) contained Olig2, but they only accounted for 26% of the Olig2+ cell pool (Fig. 2 B, F, and G). Mature CC1-expressing (CC1+) oligodendrocytes and S100β-positive (S100β+/NG2-/CNPase-, see Table 1) astrocytes also comprised a small proportion of Olig2+ cells (19% and 11%, respectively; Fig. 2 A, C, F, and G, see also ref. 33). The considerable proportion of remaining Olig2+ cells was actually negative for all the tested lineage markers (referred to as LM-, Fig. 2F) for all tests, including neuronal, microglial, leukocyte, endothelial markers, nestin, platelet-derived growth factor receptor-α, and Lewis-X (see also Table 1). However, virtually all (91 ± 3%) Olig2+ nuclei were Sox10-immunopositive (Sox10+; Fig. 2D) (34), including the LM- cells, that thus appear to belong to the oligodendrocyte lineage. Because the mean nuclear size of Olig2+/Sox+/LM- cells (52 ± 6 μm2) resembles the one of NG2+ cells but is notably distinct from that of mature oligodendrocytes (20 ± 6 μm2), these cells seem to represent a precursor stage in the oligodendrocyte lineage.
Notably, a similar composition of the Olig2+ population was observed 7 days after a stab wound (Fig. 2F). GFAP is a hallmark of reactive astrocytes expressed in a subset of the total astrocyte S100β+ population after injury. Olig2 was detected only in a fraction of the GFAP-positive astrocytes in the injured cortical GM (10.2 ± 1.3% at 3 dpl and 14.65 ± 4.6% at 7 dpl) and the GFAP-positive cells constituted only 7–9% of the Olig2+ cells (7.1 ± 1.4 at 3 dpl and 8.9 ± 4.6 at 7 dpl). Thus, Olig2 expression was not restricted to the GFAP-expressing reactive astrocytes, but occurred more widespread in astrocytes and NG2+ cells. Because the proportion of these glial populations amongst the Olig2+ cells remained relatively constant after the 3-fold increase in the Olig2+ cells after injury, this evidence suggests that all of these diverse cell types contribute to the increase in the number of Olig2+ cells. As the total number of S100β+ astrocytes and NG2+ cells increased after a stab wound (Fig. 6 A–C), the increase in the number of Olig2+ cells may be partially due to cell proliferation.
To characterize the proliferating cell types after a stab wound, we labeled the fast proliferating cells by injecting the DNA base analog BrdUrd 2 h before analysis or we supplied BrdUrd for several days in the drinking water to label all dividing cells, including those proliferating very slowly (see Materials and Methods). Both paradigms revealed a massive increase in the density of BrdUrd-positive cells 3 dpl (12-fold increase, 2-h pulse) that decreased later and reached control levels 4 weeks after injury (Figs. 2H and 6A). Olig2+ cells, which constituted about half of all of the fast cycling cells in the intact parenchyma, also comprised up to 60% of the expanded fast dividing cell pool around the lesion (Fig. 2H), suggesting that they are highly proliferative and that this proliferation is responsible, at least partially, of the increase in Olig2+ cells after a stab wound. Notably, hardly any LM- cells were labeled with a short BrdUrd pulse, whereas they did incorporate BrdUrd supplied for several days (Fig. 2I), indicating that these cells are slowly proliferating. Consistent with the data presented above, several of the slowly dividing LM- cells were Olig2+ (Fig. 2E). In summary, the transcription factor Olig2 is prominent in diverse sets of fast proliferating glia and slow dividing LM- cells in reaction to injury.
Lineage Analysis of Proliferating Cells After a Stab Wound. To assess the cell fate and lineage of the proliferating cells described above, we used replication-incompetent retroviral vectors that incorporate stably into the genome of proliferating cells and inherit their genome encoding, e.g., for the GFP, to the progeny generated from the infected precursors (see, e.g., ref. 35). When we injected a retroviral vector containing only GFP (22) in the cortical parenchyma close to the lesion site 2 days after a stab wound (Fig. 3A), infected cells were detected in the gray or white matter at all examined times after injection (dpi, Fig. 3B). As expected, the composition of infected cells analyzed at 2 dpi (Fig. 3J) closely resembled the composition of the fast proliferating cells as determined by BrdUrd-pulse labeling around the time of viral injection (Fig. 2I). Notably, 54 ± 15% of the infected cells were Olig2+ (n = 120) and they largely consisted of NG2+ precursors (Fig. 3J), whereas few LM- cells were labeled, in line with the failure of MLV-based retroviral genomes to incorporate into the genome of slow proliferating cells (36). However, 1 week after virus injection, almost 50% of all GFP-labeled (GFP+) cells were LM- (Fig. 3 F and K), suggesting that these cells derive from some of the infected glial cell types, most likely the NG2+ cells because they decrease in number when LM- cells increase. When the progeny of infected cells was analyzed at 14 or 30 dpi, the proportion of the LM- cells had decreased again, but now mostly S100β+ astroglial cells had increased in number (Fig. 3K). These data suggest that the LM- cells increase as reaction to injury, originating from NG2+ cells and later differentiating into astrocytes, consistent with a previous analysis with adenoviral vectors (21). Although these data would be consistent with some transition between glial lineages, no neurons were generated because none of the infected GFP+ cells contained any neuronal markers (dcx, HuC/D, NeuN) at all of the examined time points (Fig. 3K), reconfirming the absence of neurogenesis described above.
Fig. 3.

Repression of Olig2 function after a stab wound. (A) The viral injection experimental design is illustrated: Animals were injected with either the GFP or the Olig2VP16 virus 2 days after a stab wound and killed later. The micrograph (B) illustrates the distribution of GFP+-infected cells in the cortex 7 days after control virus injection (GM, gray matter; WM, white matter). (C–I) GFP+ cells 7 days after viral transduction (as specified in the images) stained for the antigens indicated. (F) Arrow points to a marker-negative infected cell. The micrographs (I–I′′′) show examples of Pax6 and dcx coexpression in Olig2VP16-infected cells. The graphs in J–L illustrate the phenotypes of virus-infected cells as indicated (three mice analyzed for cell type identification at each time point; 100–200 analyzed cells unless differently indicated in the text; dpi, days after injection). (Scale bars: B, 200 μm; C–G,20 μm; H,40 μm; I–I′′′,10 μm.)
Transduction of a Dominant-Negative Form of Olig2 Allows Neurogenesis. Next, we set out to determine the role of Olig2 in these proliferating cells after injury. Toward this end, we used retroviral vectors carrying Olig2 cDNA, where the Olig2 repressor domain was replaced by the herpes simplex VP16 protein and linked by an IRES domain to GFP (Olig2VP16) (22, 37–39). Previous studies have shown that the VP16 domain exerts effects specifically and exclusively in cells expressing Olig2, where it antagonizes the endogenous role of Olig2 (22, 37–39). Because half of all infected cells contained Olig2, this construct should affect Olig2 function in these cells. The phenotype of Olig2VP16-infected cells at 2 dpi was similar to that after control virus injections (data not shown), whereas we observed an obvious decrease in the proportion of the LM- cells 7 days after Olig2VP16 transduction compared to the control virus (Fig. 3 K and L). Conversely, the proportion of S100β+ cells amongst the GFP+ cells was increased upon Olig2VP16 infection compared to control virus injected cells (Fig. 3 K and L). Most excitingly, a significant proportion of Olig2VP16-infected cells (5 ± 2%, n = 228, three mice) acquired neuronal characteristics such as dcx immunoreactivity and an elongated morphology reminiscent of migrating neuroblasts 7 dpi (Fig. 3 G and L). Notably, this proportion further increased to 12 ± 3% at 14 dpi (P = 0.008, n = 320, Fig. 3L), indicating a considerable time required for respecification toward the neuronal lineage. Consistent with a neurogenic lineage redirection, we also observed Pax6 immunoreactivity in some Olig2VP16-infected cells (9 ± 3% at 7 dpi, n = 93; 20 ± 2% at 14 dpi, n = 103; Fig. 3 H, H′, and I′′), suggesting that Olig2 represses Pax6 also in this context, as in the developing spinal cord (37) and the adult subependymal zone (39). Because the majority of Pax6-positive cells (57 ± 14%) upon Olig2VP16 infection also expressed neuronal traits such as dcx immunoreactivity (Fig. 3 I–I′′′), the transcription factor Pax6 may act as an important intermediate for the lineage change toward a neuronal fate. In fact, transduction of cells with a Pax6-containing retrovirus (22, 39) led to an earlier and further increase in neurogenesis (19 ± 2%, P = 0.007, n = 228, three mice, 7 dpi). However, only some of the dcx-positive cells remained and differentiated into more mature neurons at later stages after Olig2VP16 infection (Fig. 3K). As for glial cells, no CC1+ oligodendrocytes differentiated amongst the Olig2VP16-infected cells compared to 10% of control virus injected cells that acquired this fate. Taken together, these data show that in an adult stab-lesioned cortex, endogenous neurogenesis can be evoked, and that a relief of the Olig2-mediated repression allows the induction of the neurogenic transcription factor Pax6, thereby instructing neurogenesis. Nevertheless, other strategies are required to promote neuronal survival and further differentiation.
The Increase in Olig2 as a Pan-Lesion Phenomenon. To understand to which extent the results described above have general implications for adult brain injury, we examined other lesion models, comprising a further acute injury, the MCAO as a model for stroke (25), and a chronic injury paradigm with amyloid plaque deposition in transgenic mice expressing the mutated forms of APP (27) and PS1 (28) under the Thy-1 promoter (Thy1-APPPS, see Materials and Methods). Despite the huge difference between these injury models, the number of Olig2+ cells increased in all of these injury paradigms (Fig. 4), 3-fold 7 days after MCAO (Fig. 4 A and C) and even 6-fold in the cortex of Thy1-APPPS transgenic mice compared to littermate controls (Fig. 4 B and D). Also, upon amyloid plaque deposition, the increase in Olig2+ cells was observed at the mRNA level (Fig. 4 E and F).
Fig. 4.

Increase in the number of Olig2-immunoreactive cells after focal ischemia (MCAO) and in the Thy1-APPPS cortex. (A and B) Anti-Olig2 immunostaining of frontal sections of the cortical GM shows an increased number of Olig2+ cells after MCAO (A) and after amyloid plaque formation (B) compared to the control cortex (see Fig. 1 A). Histograms in C and D display the corresponding quantifications. hs, hours; dpl, days after lesion. In situ hybridization reveals Olig2 mRNA up-regulation in the Thy1-APPPS (F) compared to the wild-type cortex (E). (Scale bars: 100 μm.)
However, a pronounced difference was detected in the cell type composition of Olig2+ cells in the Thy1-APPPS cortex compared to the stab-wound injury. The largest fraction of Olig2+ cells, the LM- cells in intact and stab-lesioned cortex, was only a minor component (10% of all of the Olig2+ cells) in the Thy1-APPPS cortex, mostly due to an increase in the proportion of Olig2+ oligodendrocytes (40 ± 8% versus 17 ± 3% in wild-type cortices) and astrocytes (24 ± 1% versus 11 ± 1% of wild types; 20.6 ± 5.6% of Olig2+ cells had GFAP). Thus, the huge increase in Olig2+ cells after amyloid deposition was caused by oligodendrocytes and other glial cells (90%), in contrast to the acute stab-wound containing mostly LM-/Olig2+ cells (52%). Consistent with this difference, the proliferative reaction to injury was much less pronounced in the Thy1-APPPS cortex with only a 1.7-fold increase in the number of BrdUrd-labeled cells (short pulse) as opposed to the 12-fold increase seen after stab-wound injury (see above). Indeed, only a small number of Olig2+ cells in the Thy1-APPPS cortex were double-labeled with BrdUrd after both 2 h (15 ± 3% versus 45 ± 2% in intact wild types) or 2 weeks of BrdUrd (7 ± 4% versus 22 ± 6% in intact wild types), indicating that proliferation of Olig2+ cells is even reduced upon amyloid deposition. Indeed, whereas the cortex of Thy1-APPPS mice CC1+ oligodendrocytes comprised the majority of the increased Olig2+ cells and virtually all of them displayed Olig2-positivity (92 ± 8% in transgenic versus 58 ± 4% in controls), their total number was not significantly increased and they did not incorporate BrdUrd (Fig. 8, which is published as supporting information on the PNAS web site). We conclude, therefore, that amyloid deposition induces transcriptional up-regulation of Olig2 most prominently in oligodendrocytes and does not instruct Olig2+ cell proliferation.
Discussion
The present study shows that Olig2 induction in the adult neocortex is a common feature of both acute and chronic brain lesions, whereas neurogenic fate determinants are not induced upon injury. The increase in the number of Olig2+ cells occurred upon various injury paradigms and in various regions of the CNS, such as the striatum, the spinal cord, and brain stem (40–42). Thus, the increase in the number of Olig2-positive cells is a pan-lesion phenomenon that occurs throughout the CNS.
Diverse mechanisms seemingly lead to the increase in the number of Olig2+ cells after lesion, such as transcriptional up-regulation and cell proliferation, although it cannot be excluded that the recruitment of Olig2+ progenitors/cells to the injury sites also contributes to this phenomenon. However, it is clear that distinct mechanisms are involved in the increase of the number of Olig2+ cells in distinct injury paradigms. Specific cell types (oligodendrocytes) up-regulate Olig2 at the transcriptional level upon amyloid plaque deposition in the cortex of Thy1-APPPS mice, whereas Olig2+ cells expand largely by proliferation upon acute stab-wound injury. These data reveal a striking difference in the reaction of distinct sets of glial cells to acute and chronic injury and provide not only molecular insights into the brain reaction to injury, but also allowed the discovery of previously uncharacterized cell populations reacting to injury, such as the oligodendrocytes in the amyloid deposition model or the LM- cells reacting exclusively to acute injuries.
Strikingly, however, these diverse sets of cells activate the same molecular pathway regulated by the transcription factor Olig2. Olig2 is expressed in diverse cell and precursor types during development (neuroepithelial cells, motoneuron precursors, and differentiating oligodendrocytes; refs. 33, 37, 38, 43–45) and has recently been discovered to play an essential role in transit-amplifying precursors in the adult subependymal zone (22, 39). Thus, our results could be interpreted such that Olig2 may mediate some dedifferentiation aspects of glial cells upon injury, because it is typically down-regulated in most mature cell types (neurons, astrocytes, and even partially in oligodendrocytes; refs. 33, 39, and 46). This interpretation is also consistent with the decrease in LM- cells upon suppression of Olig2 function, suggesting that these cells require Olig2 for the maintenance of this un/dedifferentiated status. However, it is also possible that some precursors that either express or up-regulate Olig2 migrate to the injury site where they then multiply. Although we saw no evidence for such migration, only direct viral-tracing experiments initiated before lesion will ultimately allow determining the origin of Olig2+ cells.
Although high expression levels of Olig2 in precursor cells may well be beneficial to expand their potential (see ref. 39), its later down-regulation is crucial to allow the progression along the neuronal lineage, as observed in the developing spinal cord and in the adult olfactory bulb (39, 40, 46, 47). Indeed, here we could show that antagonizing Olig2 function after stab lesion enabled a small, but significant, number of infected cells to generate neuroblasts. Because many antineurogenic factors are released upon acute brain injury (30, 48–50), the small number of neuroblasts is no surprise. However, these data show as a proof-of-principle that suppression of antineurogenic determinants allows the progression of at least some precursors in the adult lesioned mammalian cortex toward a neurogenic fate. In fact, high levels of Olig2 may also be responsible for the exclusive glial fate of neurosphere cells transplanted into the injured brain (22, 50–53).
Our data further unravel a key mechanism of Olig2 function upon brain lesion, namely to repress neurogenic factors such as Pax6, a transcription factor previously shown to be sufficient to convert at least some astrocytes into neurons in vitro (17). Here, we could show that up-regulation of Pax6 by either Olig2VP16 or Pax6 containing retroviral vectors is also sufficient in vivo to instruct neurogenesis from cells that normally do not generate neurons. However, although in vitro the cells responding to Pax6 were clearly astrocytes (17), the source of the newly generated neurons is not clear in vivo. Whereas we could show that neurogenesis can occur in the adult injured mammalian cortex, a major challenge is to improve survival and further differentiation of the newborn neurons because their numbers strongly decreased until 30 days after viral injection. This decrease is most likely due to cell death, but viral construct silencing during neuronal maturation cannot be excluded. Further steps, therefore, are required to translate the initial neurogenesis toward long-lasting neuronal repair. Taken together, this work identified a central step toward endogenous repair by highlighting a molecular mechanism interfering with endogenous neurogenesis from precursors reacting to brain injury.
Supplementary Material
Acknowledgments
We thank Ulrich Dirnagl for helpful suggestions on the manuscript, Charles Stiles (Harvard Medical School) for the anti-olig2 antibody, and Barbara Del Grande and Lana Polero for excellent secretarial help. The anti-nestin antibody was obtained by the Developmental Studies Hybridoma Bank. This work was supported by the German Research Foundation (to M.G.) and by fellowships of The Von Humboldt Foundation and the Deutscher Akademischer Austauschdienst (to A.B.).
Author contributions: A.B. and M.G. designed research; A.B. and D.E. performed research; M.R.V., G.F.H., M.J., and D.R. contributed new reagents/analytic tools; and A.B. and M.G. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: dcx, doublecortin; dpi, days after injection; dpl, days after lesion; GFAP, glial fibrillary acidic protein; GM, gray matter; LM-, lineage marker-negative; MCAO, middle cerebral artery occlusion; Olig2+, Olig2-immunopositive.
References
- 1.Doetsch, F., Caille, I., Lim, D. A., Garcia-Verdugo, J. M. & Alvarez-Buylla, A. (1999) Cell 97, 703-716. [DOI] [PubMed] [Google Scholar]
- 2.Garcia, A. D., Doan, N. B., Imura, T., Bush, T. G. & Sofroniew, M. V. (2004) Nat. Neurosci. 7, 1233-1241. [DOI] [PubMed] [Google Scholar]
- 3.Fawcett, J. W. & Asher, R. A. (1999) Brain Res. Bull. 49, 377-391. [DOI] [PubMed] [Google Scholar]
- 4.Ridet, J. L., Malhotra, S. K., Privat, A. & Gage, F. H. (1997) Trends Neurosci. 20, 570-577. [DOI] [PubMed] [Google Scholar]
- 5.Hartfuss, E., Galli, R., Heins, N. & Götz, M. (2001) Dev. Biol. 229, 15-30. [DOI] [PubMed] [Google Scholar]
- 6.Malatesta, P., Hack, M. A., Hartfuss, E., Kettenmann, H., Klinkert, W., Kirchhoff, F. & Götz, M. (2003) Neuron 37, 751-764. [DOI] [PubMed] [Google Scholar]
- 7.Mori, T., Buffo, A. & Götz, M. (2005) in Current Topics in Developmental Biology, ed. Schatted, G. (Elsevier Life Sciences, San Diego) 69, 67-99. [DOI] [PubMed] [Google Scholar]
- 8.Doetsch, F., Garcia-Verdugo, J. M. & Alvarez-Buylla, A. (1999) Proc. Natl. Acad. Sci. USA 96, 11619-11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gates, M. A., Thomas, L. B., Howard, E. M., Laywell, E. D., Sajin, B., Faissner, A., Götz, B., Silver, J. & Steindler, D. A. (1995) J. Comp. Neurol. 361, 249-266. [DOI] [PubMed] [Google Scholar]
- 10.Temple, S. & Alvarez-Buylla, A. (1999) Curr. Opin. Neurobiol. 9, 135-141. [DOI] [PubMed] [Google Scholar]
- 11.Seri, B., Garcia-Verdugo, J. M., McEwen, B. S. & Alvarez-Buylla, A. (2001) J. Neurosci. 21, 7153-7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seri, B., Garcia-Verdugo, J. M., Collado-Morente, L., McEwen, B. S. & Alvarez-Buylla, A. (2004) J. Comp. Neurol. 478, 359-378. [DOI] [PubMed] [Google Scholar]
- 13.Doetsch, F., Petreanu, L., Caille, I., Garcia-Verdugo, J. M. & Alvarez-Buylla, A. (2002) Neuron 36, 1021-1034. [DOI] [PubMed] [Google Scholar]
- 14.Malatesta, P., Hartfuss, E. & Götz, M. (2000) Development (Cambridge, U.K.) 127, 5253-5263. [DOI] [PubMed] [Google Scholar]
- 15.Noctor, S. C., Flint, A. C., Weissman, T. A., Dammerman, R. S. & Kriegstein, A. R. (2001) Nature 409, 714-720. [DOI] [PubMed] [Google Scholar]
- 16.Miyata, T., Kawaguchi, A., Okano, H. & Ogawa, M. (2001) Neuron 31, 727-741. [DOI] [PubMed] [Google Scholar]
- 17.Heins, N., Malatesta, P., Cecconi, F., Nakafuku, M., Tucker, K. L., Hack, M. A., Chapouton, P., Barde, Y. A. & Götz, M. (2002) Nat. Neurosci. 5, 308-315. [DOI] [PubMed] [Google Scholar]
- 18.Laywell, E. D., Rakic, P., Kukekov, V. G., Holland, E. C. & Steindler, D. A. (2000) Proc. Natl. Acad. Sci. USA 97, 13883-13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belachew, S., Chittajallu, R., Aguirre, A. A., Yuan, X., Kirby, M., Anderson, S. & Gallo, V. (2003) J. Cell Biol. 161, 169-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aguirre, A. A., Chittajallu, R., Belachew, S. & Gallo, V. (2004) J. Cell Biol. 165, 575-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alonso, G. (2005) Glia 49, 318-338. [DOI] [PubMed] [Google Scholar]
- 22.Hack, M. A., Sugimori, M., Lundberg, C., Nakafuku, M. & Götz, M. (2004) Mol. Cell Neurosci. 25, 664-678. [DOI] [PubMed] [Google Scholar]
- 23.Parras, C. M., Galli, R., Britz, O., Soares, S., Galichet, C., Battiste, J., Johnson, J. E., Nakafuku, M., Vescovi, A. & Guillemot, F. (2004) EMBO J. 23, 4495-4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Toresson, H., Potter, S. S. & Campbell, K. (2000) Development (Cambridge, U.K.) 127, 4361-4371. [DOI] [PubMed] [Google Scholar]
- 25.Vosko, M. R., Burggraff, D., Liebetrau, M., Wunderlich, N., Jager, G., Groger, M., Plesnila, N. & Hamann, G. F. Neurol. Res. 27, in press. [DOI] [PubMed]
- 26.Hata, R., Mies, G., Wiessner, C., Fritze, K., Hesselbarth, D., Brinker, G. & Hossmann, K. A. (1998) J. Cereb. Blood Flow Metab. 18, 367-375. [DOI] [PubMed] [Google Scholar]
- 27.Bondolfi, L., Calhoun, M., Ermini, F., Kuhn, H. G., Wiederhold, K. H., Walker, L., Staufenbiel, M. & Jucker, M. (2002) J. Neurosci. 22, 515-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moehlmann, T., Winkler, E., Xia, X., Edbauer, D., Murrell, J., Capell, A., Kaether, C., Zheng, H., Ghetti, B., Haass, C. & Steiner, H. (2002) Proc. Natl. Acad. Sci. USA 99, 8025-8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nimmerjahn, A., Kirchhoff, F. & Helmchen, F. (2005) Science 308, 1314-1318. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto, S., Nagao, M., Sugimori, M., Kosako, H., Nakatomi, H., Yamamoto, N., Takebayashi, H., Nabeshima, Y., Kitamura, T., Weinmaster, G., et al. (2001) J. Neurosci. 21, 9814-9823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu, Q. R., Sun, T., Zhu, Z., Ma, N., Garcia, M., Stiles, C. D. & Rowitch, D. H. (2002) Cell 109, 75-86. [DOI] [PubMed] [Google Scholar]
- 32.Arnett, H. A., Fancy, S. P., Alberta, J. A., Zhao, C., Plant, S. R., Kaing, S., Raine, C. S., Rowitch, D. H., Franklin, R. J. & Stiles, C. D. (2004) Science 306, 2111-2115. [DOI] [PubMed] [Google Scholar]
- 33.Liu, Y. & Rao, M. S. (2004) Glia 45, 67-74. [DOI] [PubMed] [Google Scholar]
- 34.Stolt, C. C., Lommes, P., Friedrich, R. P. & Wegner, M. (2004) Development (Cambridge, U.K.) 131, 2349-2358. [DOI] [PubMed] [Google Scholar]
- 35.Price, J. (1987) Development (Cambridge, U.K.) 101, 409-419. [DOI] [PubMed] [Google Scholar]
- 36.Hajihosseini, M., Iavachev, L. & Price, J. (1993) EMBO J. 12, 4969-4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mizuguchi, R., Sugimori, M., Takebayashi, H., Kosako, H., Nagao, M., Yoshida, S., Nabeshima, Y., Shimamura, K. & Nakafuku, M. (2001) Neuron 31, 757-771. [DOI] [PubMed] [Google Scholar]
- 38.Novitch, B. G., Chen, A. I. & Jessell, T. M. (2001) Neuron 31, 773-789. [DOI] [PubMed] [Google Scholar]
- 39.Hack, M. A., Saghatelyan, A., de Chevigny, A., Pfeifer, A., Ashery-Padan, R., Lledo, P. M. & Götz, M. (2005) Nat. Neurosci. 8, 865-872. [DOI] [PubMed] [Google Scholar]
- 40.Fancy, S. P., Zhao, C. & Franklin, R. J. (2004) Mol. Cell Neurosci. 27, 247-254. [DOI] [PubMed] [Google Scholar]
- 41.Han, S. S., Liu, Y., Tyler-Polsz, C., Rao, M. S. & Fischer, I. (2004) Glia 45, 1-16. [DOI] [PubMed] [Google Scholar]
- 42.Talbott, J. F., Loy, D. N., Liu, Y., Qiu, M. S., Bunge, M. B., Rao, M. S. & Whittemore, S. R. (2005) Exp. Neurol. 192, 11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takebayashi, H., Yoshida, S., Sugimori, M., Kosako, H., Kominami, R., Nakafuku, M. & Nabeshima, Y. (2000) Mech. Dev. 99, 143-148. [DOI] [PubMed] [Google Scholar]
- 44.Takebayashi, H., Nabeshima, Y., Yoshida, S., Chisaka, O., Ikenaka, K. & Nabeshima, Y. (2002) Curr. Biol. 12, 1157-1163. [DOI] [PubMed] [Google Scholar]
- 45.Zhou, Q. & Anderson, D. J. (2002) Cell 109, 61-73. [DOI] [PubMed] [Google Scholar]
- 46.Lee, S. K., Lee, B., Ruiz, E. C. & Pfaff, S. L. (2005) Genes Dev. 19, 282-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu, Q. R., Cai, L., Rowitch, D., Cepko, C. L. & Stiles, C. D. (2001) Nat. Neurosci. 4, 973-974. [DOI] [PubMed] [Google Scholar]
- 48.Monje, M. L., Toda, H. & Palmer, T. D. (2003) Science 302, 1760-1765. [DOI] [PubMed] [Google Scholar]
- 49.Ekdahl, C. T., Claasen, J.-H., Bonde, S., Kokaia, Z. & Lindvall, O. (2003) Proc. Natl. Acad. Sci. USA 100, 13632-13637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lim, D. A., Tramontin, A. D., Trevejo, J. M., Herrera, D. G., Garcia-Verdugo, J. M. & Alvarez-Buylla, A. (2000) Neuron 28, 713-726. [DOI] [PubMed] [Google Scholar]
- 51.Cao, Q., Benton, R. L. & Whittemore, S. R. (2002) J. Neurosci. Res. 68, 501-510. [DOI] [PubMed] [Google Scholar]
- 52.Eriksson, C., Bjorklund, A. & Wictorin, K. (2003) Exp. Neurol. 184, 615-635. [DOI] [PubMed] [Google Scholar]
- 53.Hofstetter, C. P., Holmstrom, N. A., Lilja, J. A., Schweinhardt, P., Hao, J., Spenger, C., Wiesenfeld-Hallin, Z., Kurpad, S. N., Frisen, J. & Olson, L. (2005) Nat. Neurosci. 8, 346-353. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}