

Abstract
During apoptosis, engagement of the mitochondrial pathway involves a decisive event characterized by the release of mitochondrial intermembrane space proteins, such as cytochrome c. This permeabilization of the mitochondrial outer membrane depends on activation and oligomerization of multidomain Bcl-2-family proteins Bax or Bak. Although specific members of the Bcl-2 family can activate these proapoptotic proteins, we found that heat directly activated Bax or Bak to induce cytochrome c release. A preparation of mitochondria heated at 43°C released cytochrome c in association with Bak oligomerization, and Bcl-xL prevented these events. Similarly, heat induced the oligomerization of recombinant Bax, conferring an ability to permeabilize mitochondria. Compared with wild-type cells, bax–/–bak–/– mouse embryonic fibroblasts and mitochondria isolated from these cells were resistant to heat-induced cytochrome c release. Cytosol from untreated cells inhibited heat-activated Bax or Bak; however, depletion of cytosolic Bcl-xL ablated this protection. Although mitochondria heated in the presence of cytosol did not release cytochrome c, they displayed a dramatic increase in sensitivity to permeabilization by the BH3-only protein Bid. Additionally, a peptide corresponding to the BH3 domain of Puma counteracted the inhibitory effect of cytosol and permitted heat-activated Bak to permeabilize the mitochondria. Therefore, heat represents a condition under which multidomain proapoptotic proteins are activated, and this activation is regulated by both antiapoptotic and BH3-only members of the Bcl-2 family. Our results support an emerging paradigm, wherein the activation of Bax or Bak and the blockade of antiapoptotic Bcl-2 proteins are pivotal steps in the mitochondrial pathway of apoptosis.
Keywords: BH3-only, oligomerization, mitochondria, Bcl-xL, cytochrome c
The Bcl-2 family of proteins plays a key role in regulating apoptosis at the level of mitochondrial cytochrome c release (1). Once released from mitochondria, cytochrome c interacts with Apaf-1, leading to caspase 9 activation and subsequent cleavage and activation of caspase 3, spurring the demise of the cell (2). Antiapoptotic Bcl-2 proteins, such as Bcl-2 and Bcl-xL, function to prevent cytochrome c release by counteracting the proapoptotic family members, which are divided into two subgroups based on the presence of Bcl-2 homology (BH) domains: the BH3-only family and the BH123 multidomain proteins. The BH3-only family activates the multidomain proteins, namely Bax and Bak, either directly or indirectly by engaging the antiapoptotic proteins (3–6). The exact mechanism of direct activation remains unclear, but it appears that Bax and Bak interact with certain BH3-only molecules, such as Bid, inducing them to undergo conformational changes, oligomerize, and permeabilize membranes (3, 5, 7, 8). Alternatively, the indirect mechanism involves BH3-only-family members binding to and occupying the antiapoptotic proteins, thereby derepressing Bax and Bak (5, 6). Neutralization or removal of antiapoptotic proteins may be necessary to initiate proapoptotic signals and, ultimately, Bax or Bak activation (9). However, release from antiapoptotic molecules may not be sufficient to activate Bax or Bak without an additional activation step. These findings lead to an emerging model, where not only do apoptotic signals often converge on the multidomain proteins, but the activation of these proteins is regulated on multiple levels to determine precisely when to engage an apoptotic program and commit a cell to die.
Once activated, the multidomain proapoptotic molecules Bax and Bak permeabilize the mitochondrial outer membrane to allow release of cytochrome c (10). Bax and Bak are required for apoptosis through the mitochondrial pathway as bax–/–bak–/– double-knockout (DKO) cells are resistant to several apoptotic stimuli (10). Although Bax and Bak are generally activated by BH3-only proteins, Bax activation has been artificially achieved in vitro by detergent-induced conformational changes and oligomerization (11). Thus, direct activation of the multidomain proteins may be possible with a conformation-altering stimulus. Cells subjected to hyperthermic conditions face a barrage of protein damage. In addition to denaturing proteins, heat may induce conformational changes that expose previously concealed regions (12). This scenario may mirror the ability of the BH3-only protein Bid to alter Bax conformation by exposing a normally buried hydrophobic domain, presumably resulting in oligomerization and subsequent membrane insertion of Bax (3, 13). Therefore, if heat can directly induce a relevant conformational change, it may be possible to activate Bax and Bak independently of BH3-only proteins.
In this study, we observed that heat directly altered the multidomain proteins Bax and Bak, resulting in oligomerization and activation. Cells and mitochondria lacking Bax and Bak were resistant to heat-induced cytochrome c release and apoptosis, suggesting that these proteins were required for heat-induced apoptosis. Furthermore, whereas cytosolic antiapoptotic proteins inhibited heat-activated Bax and Bak, additional BH3-only molecules derepressed the protective effect of cytosol and allowed heat-induced cytochrome c release to proceed. Thus, although heat directly activated both Bax and Bak to permeabilize the mitochondrial outer membrane and release cytochrome c, this effect was sensitive to regulation by other cytosolic Bcl-2-family members.
Methods
Cell Culture, Immunoblotting, Mitochondrial Swelling, Annexin V Staining, Preparation of Cytosolic Extracts (CEs), and Immunodepletion. Details are provided in Supporting Methods, which is published as supporting information on the PNAS web site.
Isolation of Mitochondria. As described in ref. 14, mouse liver was homogenized in mitochondria isolation buffer (MIB: 200 mM manitol/68 mM sucrose/10 mM Hepes-KOH, pH 7.4/10 mM KCl/1 mM EDTA/1 mM EGTA/0.1% BSA/complete proteinase inhibitors (Roche Applied Science, Indianapolis)] by using a Teflon Dounce homogenizer and recovered by centrifugation. To further purify mitochondria from light membranes, the heavy-membrane preparation was layered on Percoll (Sigma) (30% in 225 mM manitol, 25 mM Hepes, and 1 mM EGTA) and centrifuged at 25,000 × g for 30 min to pellet the pure mitochondria. Mitochondria from cultured cells were isolated by Dounce homogenization using a mitochondria isolation kit (Pierce) or MIB buffer and recovered by centrifugation.
Heat Treatment. Cells were heat treated by replacing media with 43°C preheated media and incubating the cells for 1 h in a 43°C water bath. Alternatively, isolated mitochondria (100 μgin50 μl of mitochondrial assay buffer: MIB supplemented with 5 mM succinate, 100 mM KCl, 2 mM ATP, 10 μM phosphocreatine, and 10 μg/ml creatine kinase) were placed in a thin-walled tube and heated at 43°C in a thermocycler (Applied Biosystems). After heating, the contents were transferred to an Eppendorf tube and incubated in a 37°C water bath for 1 h.
Cytochrome c Release Assays. Intact cells. Cells were harvested, resuspended in two pellet volumes of lysis buffer [80 mM KCl, 250 mM sucrose, and 50 μg/ml digitonin (Sigma), 1 mM DTT, and complete proteinase inhibitors (Roche Applied Science)], and placed on ice for 10 min until >95% Trypan-blue positive. Samples were centrifuged at 10,000 × g for 5 min, and cytosols were transferred to new tubes. The pellet was resuspended in wash buffer (20 mM Hepes-KOH and 250 mM KCl), centrifuged again, and the wash added to the respective cytosols. To extract mitochondrial proteins, the pellet was resuspended in lysis buffer, lysed by freeze/thaw, and centrifuged at 20,000 × g for 10 min. The supernatant, containing mitochondrial proteins, and the cytosols were analyzed by Western blot. Isolated mitochondria. After appropriate incubations, the mitochondria were centrifuged at 5,500 × g for 10 min. The supernatant was transferred, and the mitochondria pellet was resuspended in 1× SDS/PAGE sample buffer and analyzed by Western blot.
Production of Recombinant Proteins. Full-length (FL) Bax, Bax ΔC, active N/C Bid, Bcl-xL, and Bcl-xLΔC were prepared as described in ref. 8 and refs. 14–16. Additional details are provided in Supporting Methods.
Protein Crosslinking. Bismaleimidohexane (BMH, Pierce) was dissolved in DMSO (Sigma) and added to mitochondria at a final concentration of 0.1–1 mM for 30 min at room temperature. The mitochondria were pelleted, resuspended in SDS/PAGE sample buffer containing β-mercaptoethanol to quench the crosslinking reaction, and analyzed by Western blot.
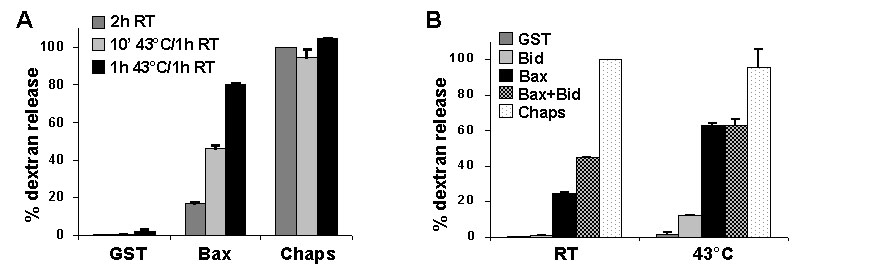
Liposome-Permeabilization Assay. Synthetic liposomes loaded with fluorescein-dextran (8) were incubated with FL Bax at either 43°C or room temperature for 1 h, followed by incubation for 1 h at room temperature. Where indicated, FL Bcl-xL was also included. Dextran release was quantitated as described in ref. 8, where GST control protein represents baseline, and 1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (Chaps) gives maximum release.
Results
Heat Induces Cytochrome c Release from Isolated Mitochondria. As a direct physiological stress, heat has the capability to alter protein conformation and function (12). To test the ability of heat to directly alter organelle function, mouse liver mitochondria were enriched, heated at 43°C, and assayed for cytochrome c release. Mitochondria heated for 15 or 30 min at 43°C released cytochrome c into the supernatant after an additional incubation at 37°C for 1 h (Fig. 1A). As a positive control for cytochrome c release, recombinant active N/C Bid, a BH3-only protein that directly activates the multidomain protein Bak on mitochondria (7), was added to the mitochondria preparation for 1 h at 37°C. Further enrichment of mitochondria by removing light membranes (i.e., endoplasmic reticulum) had no effect on heat-induced cytochrome c release, and heating light membranes alone did not induce cytochrome c release from mitochondria (see Fig. 7, which is published as supporting information on the PNAS web site). Mitochondria from Jurkat cells heated at 43°C also released almost all detectable cytochrome c (see Fig. 8A, which is published as supporting information on the PNAS web site). To ascertain whether heat-induced cytochrome c release was simply due to damage of the mitochondrial membranes, the presence of Hsp60, a chaperone protein that localizes to the mitochondrial matrix, was assessed. Mitochondria were heated at 43°C for 15, 30, or 60 min and incubated for an additional hour at 37°C. Even with heat treatment that caused complete release of cytochrome c (i.e., 30 and 60 min), Hsp60 remained in the mitochondria (Fig. 1B), indicating that, although the outer membrane was permeabilized, the inner mitochondrial membrane remained intact. After digitonin treatment to disrupt the inner membrane, Hsp60 was detected in the supernatant, confirming that the protein remains soluble after heat treatment (Fig. 8B). Moreover, in contrast to calcium-treated mitochondria, heated mitochondria did not swell as a means to rupture the outer membrane to release cytochrome c (Fig. 1C).
Fig. 1.

Heating enriched mitochondria induces cytochrome c release without swelling or release of matrix proteins. (A) Mitochondria were enriched from mouse liver and heated at 43°C for the specified times. Where indicated, heating was followed by incubation at 37°C for 1 h. Active N/C Bid (45 nM) was incubated with mitochondria for 1 h at 37°C. The mitochondrial pellet and supernatant were probed for the presence of cytochrome c by Western blot. (B) Enriched liver mitochondria were heated at 43°C for the indicated amounts of time, followed by incubation for 1 h at 37°C. Cytochrome c and Hsp60 were detected by Western blot. (C) After heating liver mitochondria at 43°C for the indicated amounts of time, mitochondrial swelling was assessed for 30 min at 37°C by absorbance at OD 520. Untreated mitochondria (untreated mito) and a sample without mitochondria (no mito) were also included. As a positive control, 100 mM Ca2+ was added to a sample of enriched mitochondria to induce swelling.
Bcl-xL Blocks Heat-Induced Cytochrome c Release and Bak Oligomerization. Because members of the Bcl-2 family regulate the release of cytochrome c from mitochondria, we investigated whether antiapoptotic Bcl-xL could prevent heat-induced cytochrome c release. Recombinant Bcl-xL or an inactive mutant Bcl-xL(G138A) (17) was heated with enriched mouse liver mitochondria for 60 min at 43°C, followed by a 37°C incubation for an additional hour. The presence of Bcl-xL prevented heat-induced cytochrome c release, whereas mutant Bcl-xL(G138A) had no effect (Fig. 2A). Of note, heating Bcl-xL at 43°C did not alter its function, suggesting that heat-induced effects may target specific proteins. Bcl-xL is known to inhibit Bax- and Bak-mediated cytochrome c release, but, of the multidomain proapoptotic Bcl-2 members, only Bak is constitutively localized to the mitochondrial membrane, whereas Bax resides in the cytosol and translocates to the mitochondria upon activation (7, 18). Because heat directly induced cytochrome c release from enriched liver mitochondria, which lack Bax (7), we tested whether heat could oligomerize Bak to allow for mitochondrial membrane permeabilization. Heated mitochondria treated with crosslinker, but not its vehicle control, displayed multiple high-molecular-weight Bak bands indicative of oligomerization (Fig. 2B) (7). To determine whether Bcl-xL prevents heat-induced Bak oligomerization, recombinant WT or mutant Bcl-xL was added to mitochondria and heated at 43°C for 1 h. Active N/C Bid was added to mitochondria for 1 h at 37°C as a positive control for Bak oligomerization. High-molecular-weight oligomers of Bak appeared after heating mitochondria in the presence of mutant Bcl-xL but were absent when mitochondria were heated with WT Bcl-xL (Fig. 2C). The oligomers formed by heating mitochondria were of similar size to those produced by active N/C Bid. This method, however, does not exclude the possibility that other mitochondrial proteins are present in the complexes. Thus, heating isolated mitochondria induced Bak oligomerization and cytochrome c release over time, both of which were prevented by the presence of WT Bcl-xL but not mutant Bcl-xL(G138A).
Fig. 2.

Heat-induced cytochrome c release is regulated by Bcl-2 proteins. (A) Mouse-liver mitochondria were enriched and heated with 200 μg/ml recombinant WT Bcl-xLΔC or mutant Bcl-xL(G138A)ΔC (mut) at 43°C for 1 h. After an additional incubation at 37°C for 1 h, cytochrome c release from the mitochondria (pellet) to the supernatant was assayed by Western blot. (B) Enriched mitochondria, heated at 43°C as indicated, were treated with 1 mM bismaleimidohexane (BMH) crosslinker or DMSO (vehicle control) and analyzed by Western blot using an anti-Bak antibody. (C) Mitochondria were either heated with 200 μg/ml WT or mutant (mut) Bcl-xLΔC at 43°C for 1 h or incubated with 45 nM active N/C Bid at 37°C for 1 h. Samples were treated and assessed for Bak oligomerization as in B. Arrows indicate crosslinked oligomers.
Heat Induces Oligomerization of Recombinant Bax. To further explore the idea that heat may generate a physical change in multidomain proapoptotic proteins resulting in activation, purified recombinant Bax (see Fig. 9, which is published as supporting information on the PNAS web site) was heated at 43°C and added to freshly isolated mouse-liver mitochondria for 1 h at 37°C. In contrast to untreated BaxΔC, which did not induce cytochrome c release at the concentrations used, heated BaxΔC acquired cytochrome-c-releasing activity after heating at 43°C for 60 min (Fig. 3 A and B). BaxΔC heated at 90°C, however, was not capable of inducing cytochrome c release (Fig. 3B), and BaxΔC heated at 40°C for 60 min did not induce cytochrome c release (data not shown). Similar to BaxΔC, recombinant FL Bax heated at 43°C for 60 min released cytochrome c from enriched liver mitochondria (Fig. 3B), indicating that Bax may be directly activated by heat. To determine whether this effect of heat on Bax is independent of other proteins, liposomes composed of mitochondrial lipids (5, 8) were incubated with or without Bax and subjected to heat. Heating FL Bax with dextran-filled liposomes induced dextran release that was inhibited by the presence of Bcl-xL (Fig. 3C). The dextran release increased over time at 43°C and was comparable with the release seen with activating Bax by N/C Bid (see Fig. 10, which is published as supporting information on the PNAS web site). It was recently reported that heat treatment of cells for 1 h at 43°C resulted in Bax activation, demonstrated by an antibody that specifically recognizes an active conformation of Bax (19). These data suggest that heating Bax at 43°C induces a conformational change that activates it and bestows an ability to release cytochrome c from mitochondria. Accordingly, heating recombinant BaxΔC at 43°C for 1 h caused its oligomerization (Fig. 3D), even in the absence of lipid vesicles, which are required for Bid-induced Bax oligomerization in the absence of mitochondria (8). Moreover, when BaxΔC was heated at 43°C for 1 h in the presence of Bcl-xL, WT Bcl-xL inhibited the heat-induced Bax oligomerization but mutant Bcl-xL(G138A) did not (Fig. 3E). This result corresponded to the ability of WT Bcl-xL (but not the mutant protein) to prevent cytochrome c release induced by heated Bax (Fig. 5C). Therefore, heating Bax at 43°C directly induced its oligomerization and activation to release cytochrome c from mitochondria or dextrans from liposomes, and these events were suppressed by Bcl-xL.
Fig. 3.

Heating recombinant Bax induces its oligomerization and ability to release cytochrome c. (A) BaxΔC (15 μM) was heated at 43°C for increasing amounts of time and added to enriched mouse-liver mitochondria for 1 h at 37°C. The mitochondria (pellet) and the supernatant were assessed for the presence of cytochrome c by Western blot. (B) BaxΔC was heated for 1 h at 43°C or, where indicated, at 90°C for 5 min. FL Bax (120 nM) was heated at 43°C for 1 h. After heating, Bax was added to liver mitochondria for 1 h at 37°C, and the samples were assayed as in A. (C) Liposomes loaded with fluorescent dextrans were incubated with 120 nM FL Bax for 1 h at 43°C, followed by incubation for 1 h at room temperature. Where indicated, 40 μg/ml Bcl-xL was included in the incubation. Dextran release was calculated relative to liposomes incubated for 2 h at room temperature (RT). (D) BaxΔC was heated at 43°C for 1 h, with or without lipid vesicles, and treated with 0.1 mM bismaleimidohexane crosslinking agent or DMSO (vehicle control). Western blot analysis demonstrated the presence of Bax oligomers, indicated by arrows. (E) BaxΔC was heated with WT or mutant (mut) Bcl-xLΔC at 43°C for 1 h. Samples were treated and assessed for Bax oligomerization as in C.
Fig. 5.

Cytosol from untreated cells buffers the effect of heat on enriched mitochondria and Bax. (A) CE (4 mg/ml) from untreated Jurkat cells was heated with mouse-liver mitochondria at 43°C for 30 or 60 min, followed by incubation for 1 h at 37°C. The mitochondria (pellet) and supernatant were probed for cytochrome c by Western blot. (B) Jurkat CE was immunodepleted with anti-Bcl-x antibody (↓xL) or control antibody (↓Ig), and the presence of Bcl-xL was detected by Western blotting. Each extract (4 mg/ml) was heated with mitochondria at 43°C for 60 min, and cytochrome c release was determined by Western blot. (C) Recombinant BaxΔC (15 μM) was heated at 43°C for 1 h, either alone or with 200 μg/ml WT, mutant (mut) Bcl-xLΔC, or 4 mg/ml Jurkat CE and added to mitochondria. After an additional incubation at 37°C for 1 h, mitochondria (pellet) and supernatant were probed for cytochrome c by Western blot.
Bax and Bak Are Required for Heat-Induced Cytochrome c Release. It is well documented in cells that, whereas mild heat stress induces thermotolerance, heat at higher temperatures (i.e., 43°C) induces apoptosis (20). To test the involvement of Bax and Bak in apoptosis induced by lethal heat shock, DKO mouse embryonic fibroblasts (MEFs) (10) were used. WT or DKO MEFs were exposed to sublethal (40°C) or lethal (43°C) heat for 1 h then incubated at 37°C for 5 h and evaluated for apoptosis. Compared with WT MEFs that were lethally heat stressed, apoptosis of heated DKO MEFs was dramatically reduced (Fig. 4A), indicating that Bax and/or Bak was required for heat-induced apoptosis. Unlike WT MEFs that release cytochrome c into the cytosol after lethal (but not sublethal) heat shock, DKO MEFs maintained cytochrome c in the mitochondrial pellet (Fig. 4B). The presence of a mitochondrial outer-membrane protein VDAC only in the pellet fraction confirmed that the cytosolic fraction was not contaminated with mitochondria. Similarly, enriched mitochondria from untreated WT or DKO MEFs were heated at 43°C for increasing amounts of time, followed by a 1-h incubation at 37°C. Whereas WT mitochondria fully released cytochrome c in response to 30 min of heat treatment, DKO mitochondria did not release cytochrome c, even after 60 min at 43°C (Fig. 4C). Hsp60 remained at the pellet of all samples (see Fig. 11, which is published as supporting information on the PNAS web site). Enriched mitochondria from DKO MEFs were heated for 1 h at 43°C in the presence or absence of FL Bax, followed by incubation at 37°C for 1 h. Only DKO mitochondria heated in the presence of Bax released cytochrome c (Fig. 4D). These data indicate that Bak and Bax have a requisite role in heat-induced cytochrome c release and apoptosis.
Fig. 4.

Cells and mitochondria lacking multidomain Bax and Bak are resistant to heat. (A) WT or DKO MEFs were incubated for 1 h at the indicated temperatures, followed by incubation at 37°C for an additional 5 h. Samples were incubated with annexinV-FITC and analyzed for apoptosis by flow cytometry. (B) Cells were heated as in A, harvested, and treated with digitonin. The cytosol and mitochondria (pellet) were analyzed by Western blot for the presence of cytochrome c and VDAC. A nonspecific (ns) band is displayed as a loading control. (C) Mitochondria enriched from WT (WT mito) or DKO (DKO mito) MEFs were heated at 43°C for the indicated times, followed by incubation for 1 h at 37°C. Cytochrome c release from the mitochondria (pellet) to supernatant was assayed by Western blot. (D) DKO mitochondria were enriched and incubated with 50 nM FL Bax on ice or at 43°C for 1 h, followed by incubation for 1 h at 37°C. Samples were assayed as in C.
Cytosol Inhibits Heat-Induced Cytochrome c Release. Intriguingly, we observed that, whereas heat-induced cytochrome c release from isolated mitochondria occurred within 1 h, it took several hours to detect this event in cells, suggesting that the presence of cytosol may delay heat-induced apoptotic events. To explore this idea, untreated cytosol was heated with enriched mitochondria at 43°C for 30 or 60 min, followed by incubation for 1 h at 37°C. Mitochondria heated at 43°C in the presence of CE were protected from heat-induced cytochrome c release compared with mitochondria heated without cytosol (Fig. 5A). Because recombinant Bcl-xL also counteracted heat-induced cytochrome c release (Fig. 2 A), untreated CE was immunodepleted of Bcl-xL and assessed for a loss of heat-buffering capacity. The absence of Bcl-xL after depletion with a Bcl-x antibody, but not a control Ig antibody, was confirmed by Western blot (Fig. 5B). Mitochondria were heated at 43°C for 15, 30, or 60 min without CE or for 60 min with nondepleted extract, Bcl-xL-immunodepleted extract, or control Ig-immunodepleted extract and evaluated for cytochrome c release. Depleting Bcl-xL from the CE removed the buffering factor and allowed cytochrome c to be completely released from mitochondria heated for 60 min (Fig. 5B). Because Bcl-xL appears to be a component of cytosol responsible for inhibiting the effects of heat, we evaluated the ability of cytosol, as well as recombinant Bcl-xL, to block the cytochrome-c-releasing activity of heated Bax. Recombinant BaxΔC was heated at 43°C for 60 min, either alone or in the presence of WT Bcl-xL, mutant Bcl-xL(G138A), or CE, then added to freshly isolated mitochondria for 1 h at 37°C and assayed for cytochrome c release. BaxΔC heated in the presence of WT Bcl-xL or cytosol did not induce cytochrome c release from mitochondria compared with BaxΔC heated alone or with mutant Bcl-xL (Fig. 5C). Therefore, cytosolic Bcl-xL can buffer the direct effect of heat on Bax and Bak.
The Protective Effect of Cytosol Is Reversed by Addition of BH3-Only Molecules. Because BH3-only proteins interact with and impede antiapoptotic Bcl-2-family members (4, 5), we examined whether the inhibitory effect of cytosol on heated mitochondria could be neutralized by the addition of a BH3-only protein. Enriched mouse-liver mitochondria were heated with CE at 43°C for 60 min, and increasing amounts of recombinant active N/C Bid were added for 1 h at 37°C. Whereas mitochondria heated in the presence of cytosol alone did not release cytochrome c, the addition of active N/C Bid (1–500 pM) allowed heat-induced cytochrome c release (Fig. 6A). These amounts of active N/C Bid did not release cytochrome c from mitochondria that had not been heated. Therefore, whereas heat-induced alterations were buffered by cytosol, heated mitochondria were much more receptive to interactions with additional proapoptotic proteins. To delineate whether Bid derepressed the cytosolic inhibition of heated Bak or interacted directly with heated Bak, a peptide spanning the BH3 domain of Puma, which does not directly activate the multidomain proteins (5, 21), was used. Enriched mitochondria were heated at 43°C for 60 min in the presence of cytosol and treated with 10 μM Puma peptide for an additional hour at 37°C. The addition of Puma peptide neutralized the protective effect of cytosol and allowed complete heat-induced cytochrome c release (Fig. 6B). Thus, heat-activated multidomain proteins are inhibited by cytosolic antiapoptotic proteins, such as Bcl-xL, until derepressed by a BH3-only molecule.
Fig. 6.

The cytosolic inhibition of heat-induced cytochrome c release is reversed by BH3-only protein or peptide. (A) Mouse-liver mitochondria were enriched and combined with 5 mg/ml Jurkat CE and, where indicated, heated at 43°C for 60 min before the addition of increasing amounts of active N/C Bid. (B) Enriched mitochondria were heated as in A and incubated with 10 μM Puma peptide after heat treatment. All samples were incubated at 37°C for 1 h, and mitochondria (pellet) and supernatant were assessed for cytochrome c release by Western blot. The pellet fraction was also probed for Hsp60 to confirm equal loading of the samples.
Discussion
A key step in the mitochondrial pathway of apoptosis is the activation of the multidomain BH123 proteins Bak and Bax to permeabilize the mitochondrial outer membrane. Studies using recombinant proteins have shown that the activation of Bax by active Bid (22, 23) or BH3 peptides from Bid or Bim (5) is essential and sufficient to permeabilize vesicles composed of mitochondrial lipids in the absence of other proteins (5, 8). In the process, Bax oligomerizes, and such oligomerization of Bax and Bak coincides with membrane permeabilization and cytochrome c release (7, 24). Recent studies have similarly shown that purified or recombinant p53 also has the ability to activate Bax to oligomerize in lipid membranes and cause permeabilization (25). These studies support a model in which the activation of Bax or Bak by BH3-only “activator” proteins and, perhaps, other proteins (such as p53) with this activator function, is necessary and sufficient for mitochondrial outer-membrane permeabilization and the release of proapoptotic factors from the mitochondrial intermembrane space. This effect is regulated by antiapoptotic members of the Bcl-2 family that can sequester the activator protein and also bind to activated Bax and Bak to inhibit their ability to oligomerize and permeabilize membranes.
Earlier studies on Bax and Bak, however, suggested that some detergents (e.g., nonionic detergents) can directly induce oligomerization of Bax and Bak (11), and detergent-treated Bax is capable of permeabilizing membranes in the absence of any additional activation requirements (8), raising the possibility that Bax and Bak may be activated by direct physical and/or chemical agents. We observed that heat treatment of mitochondria caused mitochondrial outer-membrane permeabilization, and this effect depended on Bak on the mitochondria, which oligomerized after heat treatment. Similarly, heating of Bax induced its oligomerization and permeabilizing activity. Antiapoptotic Bcl-xL inhibited the permeabilization of mitochondria that were either heated directly or exposed to heated Bax. Furthermore, vesicles composed of mitochondrial lipids were permeabilized when heated in the presence of recombinant Bax. These observations support the idea that heat treatment, like detergent, directly activates Bax and Bak to permeabilize mitochondrial membranes.
In this scenario, apoptosis induced by heat stress may simply proceed through direct activation of Bax and Bak to permeabilize the outer mitochondrial membrane. Indeed, we observed that heat stress-induced apoptosis depends on Bax or Bak, because double-knockout cells were resistant to heat. However, this conjecture is inconsistent with the kinetics of heat stress-induced apoptosis, which takes several hours in cells (26) compared with 1 h for the direct events we observed in a cell-free system. Significantly, we found that the presence of cytosol profoundly suppressed the permeabilization of mitochondria by heat or heat-treated Bax. This effect was due, at least in part, to Bcl-xL in the cytosol, but other antiapoptotic factors may also be involved. Furthermore, we established that mitochondria heated in the presence of cytosol were significantly sensitized to permeabilization upon addition of small amounts of active Bid or Puma BH3 peptide. Thus, even in the presence of cytosol, a direct effect of heat treatment was evident.
Analogous to the effect of heat on Bax and Bak activation, the transcription factor heat-shock factor 1 (HSF-1) has also been shown to be directly activated by heat through a heat-responsive domain in the protein (27). Moreover, the direct activation of Bax and Bak by heat may extend to other stimuli that denature or otherwise expose relevant interaction domains in these proteins. For instance, heavy metals, reactive oxygen, and alkalinization may be able to directly activate these proteins. Interestingly, a rise in pH has been suggested to directly activate Bax (28, 29). Our observations suggest that, if activation of Bax and Bak is a prerequisite for engaging the mitochondrial pathway of apoptosis, then this activation may not require the function of activator BH3-only and other proteins in all cases. Stressors that act directly on the proapoptotic multidomain proteins may, therefore, bypass such a step.
Our observation that cytosol inhibited the effects of heat on Bax and Bak to permeabilize mitochondria, at least partly due to Bcl-xL, is reminiscent of another study of apoptosis induced by UV light (9). In that study, a rapid activation of proapoptotic Bcl-2-family members was inhibited by the actions of Bcl-xL and Mcl-1 in the cytosol, and apoptosis proceeded only when levels of Mcl-1 decreased through proteosomal degradation. Our results suggest that activation of BH3-only proteins can similarly allow apoptosis to proceed after heat stress, perhaps by derepressing the antiapoptotic effects of cytosol. This interplay between activation of Bax and Bak and the blockade of antiapoptotic Bcl-2-family members to regulate mitochondrial outer-membrane permeabilization and engage the mitochondrial pathway represents a refinement in our understanding of apoptosis. Rather than “death by default,” the emerging view is that apoptotic death requires the activation of Bax and Bak, achieved either by other proteins (e.g., Bid, Bim, p53) or through the direct effects of heat, pH, or, perhaps, other conditions. Without the functions of these multidomain proteins, cells persist, undergoing autophagy to ensure survival (30). Heat stress, which may represent a primitive signal for apoptosis, appears to affect such activation directly.
Supplementary Material
Acknowledgments
We thank Melissa O'Leary for technical assistance, Drs. Lisa Bouchier-Hayes and Jerry Chipuk (La Jolla Institute for Allergy and Immunology) for reagents and helpful discussion, and the laboratory of Dr. S. Korsmeyer (Howard Hughes Medical Insitute, Dana–Farber Cancer Institute, Harvard Medical School, Boston) for the DKO MEFs. This work was supported by National Institutes of Health Grants AI40696, AI47891, and CA69381.
Author contributions: L.J.P., T.K., D.D.N., H.M.B., and D.R.G. designed research; L.J.P. and T.K. performed research; C.B. contributed new reagents/analytic tools; L.J.P., T.K., D.D.N., S.T., H.M.B., and D.R.G. analyzed data; and L.J.P., H.M.B., and D.R.G. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: BH, Bcl-2 homology; CE, cytosolic extract; DKO, bax–/–bak–/– double-knockout; FL, full-length; MEF, mouse embryonic fibroblast.
References
- 1.Danial, N. N. & Korsmeyer, S. J. (2004) Cell 116, 205–219. [DOI] [PubMed] [Google Scholar]
- 2.Li, P., Nijhawan, D., Budihardjo, I., Srinivasula, S. M., Ahmad, M., Alnemri, E. S. & Wang, X. (1997) Cell 91, 479–489. [DOI] [PubMed] [Google Scholar]
- 3.Eskes, R., Desagher, S., Antonsson, B. & Martinou, J. C. (2000) Mol. Cell. Biol. 20, 929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Letai, A., Bassik, M. C., Walensky, L. D., Sorcinelli, M. D., Weiler, S. & Korsmeyer, S. J. (2002) Cancer Cell 2, 183–192. [DOI] [PubMed] [Google Scholar]
- 5.Kuwana, T., Bouchier-Hayes, L., Chipuk, J. E., Bonzon, C., Sullivan, B. A., Green, D. R. & Newmeyer, D. D. (2005) Mol. Cell 17, 525–535. [DOI] [PubMed] [Google Scholar]
- 6.Willis, S. N., Chen, L., Dewson, G., Wei, A., Naik, E., Fletcher, J. I., Adams, J. M. & Huang, D. C. (2005) Genes Dev. 19, 1294–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wei, M. C., Lindsten, T., Mootha, V. K., Weiler, S., Gross, A., Ashiya, M., Thompson, C. B. & Korsmeyer, S. J. (2000) Genes Dev. 14, 2060–2071. [PMC free article] [PubMed] [Google Scholar]
- 8.Kuwana, T., Mackey, M. R., Perkins, G., Ellisman, M. H., Latterich, M., Schneiter, R., Green, D. R. & Newmeyer, D. D. (2002) Cell 111, 331–342. [DOI] [PubMed] [Google Scholar]
- 9.Nijhawan, D., Fang, M., Traer, E., Zhong, Q., Gao, W., Du, F. & Wang, X. (2003) Genes Dev. 17, 1475–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei, M. C., Zong, W. X., Cheng, E. H., Lindsten, T., Panoutsakopoulou, V., Ross, A. J., Roth, K. A., MacGregor, G. R., Thompson, C. B. & Korsmeyer, S. J. (2001) Science 292, 727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsu, Y. T. & Youle, R. J. (1997) J. Biol. Chem. 272, 13829–13834. [DOI] [PubMed] [Google Scholar]
- 12.Ellis, R. J. (1999) Curr. Biol. 9, R137–R139. [DOI] [PubMed] [Google Scholar]
- 13.Desagher, S., Osen-Sand, A., Nichols, A., Eskes, R., Montessuit, S., Lauper, S., Maundrell, K., Antonsson, B. & Martinou, J. C. (1999) J. Cell Biol. 144, 891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bossy-Wetzel, E. & Green, D. R. (1999) J. Biol. Chem. 274, 17484–17490. [DOI] [PubMed] [Google Scholar]
- 15.Suzuki, M., Youle, R. J. & Tjandra, N. (2000) Cell 103, 645–654. [DOI] [PubMed] [Google Scholar]
- 16.von Ahsen, O. & Newmeyer, D. D. (2000) Methods Enzymol. 322, 183–198. [DOI] [PubMed] [Google Scholar]
- 17.Muchmore, S. W., Sattler, M., Liang, H., Meadows, R. P., Harlan, J. E., Yoon, H. S., Nettesheim, D., Chang, B. S., Thompson, C. B., Wong, S. L., et al. (1996) Nature 381, 335–341. [DOI] [PubMed] [Google Scholar]
- 18.Wolter, K. G., Hsu, Y. T., Smith, C. L., Nechushtan, A., Xi, X. G. & Youle, R. J. (1997) J. Cell Biol. 139, 1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stankiewicz, A. R., Lachapelle, G., Foo, C. P. Z., Radicioni, S. M. & Mosser, D. D. (2005) J. Biol. Chem., in press. [DOI] [PubMed]
- 20.Bettaieb, A. & Averill-Bates, D. A. (2005) J. Cell. Physiol. 205, 47–57. [DOI] [PubMed] [Google Scholar]
- 21.Chipuk, J. E., Bouchier-Hayes, L., Kuwana, T., Newmeyer, D. D. & Green, D. R. (2005) Science 309, 1732–1735. [DOI] [PubMed] [Google Scholar]
- 22.Wang, K., Yin, X. M., Chao, D. T., Milliman, C. L. & Korsmeyer, S. J. (1996) Genes Dev. 10, 2859–2869. [DOI] [PubMed] [Google Scholar]
- 23.Luo, X., Budihardjo, I., Zou, H., Slaughter, C. & Wang, X. (1998) Cell 94, 481–490. [DOI] [PubMed] [Google Scholar]
- 24.Korsmeyer, S. J., Wei, M. C., Saito, M., Weiler, S., Oh, K. J. & Schlesinger, P. H. (2000) Cell Death Differ. 7, 1166–1173. [DOI] [PubMed] [Google Scholar]
- 25.Chipuk, J. E., Kuwana, T., Bouchier-Hayes, L., Droin, N. M., Newmeyer, D. D., Schuler, M. & Green, D. R. (2004) Science 303, 1010–1014. [DOI] [PubMed] [Google Scholar]
- 26.Samali, A., Holmberg, C. I., Sistonen, L. & Orrenius, S. (1999) FEBS Lett. 461, 306–310. [DOI] [PubMed] [Google Scholar]
- 27.Newton, E. M., Knauf, U., Green, M. & Kingston, R. E. (1996) Mol. Cell. Biol. 16, 839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khaled, A. R., Kim, K., Hofmeister, R., Muegge, K. & Durum, S. K. (1999) Proc. Natl. Acad. Sci. USA 96, 14476–14481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cartron, P. F., Oliver, L., Mayat, E., Meflah, K. & Vallette, F. M. (2004) FEBS Lett. 578, 41–46. [DOI] [PubMed] [Google Scholar]
- 30.Lum, J. J., Bauer, D. E., Kong, M., Harris, M. H., Li, C., Lindsten, T. & Thompson, C. B. (2005) Cell 120, 237–248. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}