

Abstract
Repeated stress can impair function in the hippocampus, a brain structure essential for learning and memory. Although behavioral evidence suggests that severe stress triggers cognitive impairment, as seen in major depression or posttraumatic stress disorder, little is known about the molecular mediators of these functional deficits in the hippocampus. We report here both pre- and postsynaptic effects of chronic stress, manifested as a reduction in the number of NMDA receptors, dendritic spines, and expression of growth-associated protein-43 in the cornu ammonis 1 region. Strikingly, the stress-induced decrease in NMDA receptors coincides spatially with sites of plasminogen activation, thereby predicting a role for tissue plasminogen activator (tPA) in this form of stress-induced plasticity. Consistent with this possibility, tPA-/- and plasminogen-/- mice are protected from stress-induced decrease in NMDA receptors and reduction in dendritic spines. At the behavioral level, these synaptic and molecular signatures of stress-induced plasticity are accompanied by impaired acquisition, but not retrieval, of hippocampal-dependent spatial learning, a deficit that is not exhibited by the tPA-/- and plasminogen-/- mice. These findings establish the tPA/plasmin system as an important mediator of the debilitating effects of prolonged stress on hippocampal function at multiple levels of neural organization.
Keywords: dendritic spines, learning, NMDA receptor
Psychological stress induces neuronal responses that can be either adaptive and directed toward maintaining homeostasis or maladaptive, leading to severe behavioral abnormalities (1). Posttraumatic stress disorder (PTSD) is a devastating disease triggered by a severe traumatic event(s) and characterized by cognitive impairment, depression, fear, and anxiety (2). Although little is known about the cellular mechanisms of PTSD, its different aspects are mediated by different brain structures (3). Animal and human studies suggest that stress-induced fear and anxiety are mediated by the amygdala (4, 5), and cognitive decline is a result of hippocampal dysfunction (6, 7). It has been hypothesized that the decrease in complexity of the hippocampal dendritic tree contributes to learning deficits (8), but molecular mechanisms underlying this dendritic plasticity are poorly understood.
One molecule strategically positioned to control neuronal activity, dendritic remodeling, and learning is the NMDA receptor. It is located on dendritic spines and is critically involved in spine motility (9) and experience-induced neuronal plasticity (10). Although the decrease in the number of NMDA receptors leads to memory deficits (10), overexpression of some of its subunits results in more efficient learning (11).
There is evidence that NMDA receptor function is linked to stress-induced neuronal and cognitive changes, because stress-induced remodeling is blocked by NMDA-receptor antagonists (12). The NMDA receptor has numerous ligands and modulators, and it is likely that the above processes may involve a number of them. Such a modulatory role has been attributed to the tissue plasminogen activator (tPA)/plasmin system (13, 14). tPA facilitates NMDA receptor signaling (13, 14) and is involved in numerous aspects of brain function, including stress-induced anxiety (15), dendritic remodeling (16, 17), hippocampus-dependent learning, and long-term potentiation (18, 19). Here we show that chronic stress causes a decrease in the number of NMDA receptors and dendritic spines in the hippocampus. These changes colocalize with sites of accelerated plasmin generation, result in impairment of spatial learning, and are attenuated in mice in which either the tPA or plasminogen gene has been disrupted.
Materials and Methods
Restraint Stress. Eight- to 12-week-old wild-type C57/BL6 (tPA+/+, plasminogen+/+) and homozygous tissue-plasminogen activator or plasminogen knockout mice (tPA-/- and plasminogen-/-) back-crossed to C57/BL6 for at least nine generations were used for the experiments. Experiments were performed during the light period of the circadian cycle. Control animals were left undisturbed, and stressed animals were subjected to daily 6-h restraint stress for 3 weeks in a separate room. The sessions consisted of 6-h per day (10 a.m.-4 p.m.) restraint of the mice in wire mesh restrainers secured at the head and tail ends with clips. During restraint sessions, the mice were placed in their home cages.
In Situ Zymography. In situ zymography was performed according to Sappino et al. (20). Mice were anesthetized and transcardially perfused with ice-cold saline, and their brains were removed, frozen, and cut at 15-μm-thick sections. The overlay mixture (10 mM Tris/10 mg/ml agarose/2% skim milk/4 μg/ml human Glu-plasminogen) was prepared at 42°C, and 300 μl was applied onto prewarmed brain sections mounted on glass slides and spread evenly under glass coverslips. The slides were incubated at 37°C in humid chambers, and thereafter the developed zymograms were examined under dark-field illumination.
To identify sites of accelerated plasmin generation, both human Glu-plasminogen (4 μg/ml) and recombinant tPA (240 ng/ml) were added to the overlay mixture, incubated for 20-30 min, and photographed.
Immunohistochemistry and Western Blotting. At indicated time-points, mice were anesthetized with 2.5% avertin. The animals were transcardially perfused with PBS and their brains removed, frozen, and mounted in Tissue-Tek OCT medium (Sakura Finetek, Torrance, CA). Fifteen-μm coronal brain sections were cut on a cryostat, collected on silane-coated slides, and stored in -80°C until analyzed. The sections were then fixed with 4% paraformaldehyde in PBS for 25 min at 4°C, rinsed with PBS, blocked with 1% BSA and 1% goat serum, and incubated with anti-NR1 primary antibody (Santa Cruz Biotechnology, 1:1,000), followed by a FITC-conjugated secondary antibody (Vector Laboratories, 1:1,000). Images were obtained by using a Zeiss Axioscope equipped with appropriate fluorescent filters and connected to a digital camera.
For Western blotting, the hippocampi were dissected and homogenized in 0.1 M Tris/0.1% Triton X-100, pH 7.2 and the protein concentration adjusted to 2 mg/ml. After electrophoresis and transfer to nitrocellulose, the membranes were blocked in 5% skim milk in PBS, washed, and probed with primary antibodies: rabbit anti-growth-associated protein-43 (GAP-43) (Chemicon International, Temecula, CA, 1:2,500), anti-NR1 (Santa Cruz Biotechnology; 1:1,000), anti-NR2A (Chemicon International, 1:1,000), anti-NR2B (Santa Cruz Biotechnology, 1:1,000), anti-brain-derived neurotrophic factor (Santa Cruz Biotechnology, 1:1,000), anti-GABAB R1 (Chemicon International, 1:1,000), anti-glutamic acid decarboxylase-65 (Chemicon International, 1:1,000), and, after stripping, reblotted for actin (Sigma, mouse anti-actin; 1:2,500), followed by appropriate horseradish-peroxidase-conjugated secondary antibody (Vector Laboratories, 1:1,000). For quantification, the background was digitally eliminated by using scion image (Scion, Frederick, MD), and optical densities of the bands were normalized to actin.
Golgi Staining. Animals were anesthetized, and their brains removed and processed for the Golgi-Cox technique (21). One hundred twenty-μm-thick sections were obtained by using a rotary microtome (Jung RM 2055; Leica, Rueil-Malmaison, France). Sections were collected serially, dehydrated in absolute alcohol, cleared in xylene, and coverslipped. Slides were coded before quantitative analysis. The experimenter was blind to the code, which was broken only after the analysis was completed.
Analysis of Dendritic Spine Density. We used the NeuroLucida image analysis system (Microbrightfield, Wiliston, VT) attached to an Olympus (Melville, NY) BX61 microscope (×100, 1.3 numerical aperture, Olympus BX61), for analysis of spine density in cornu ammonis (CA)1 pyramidal neurons that were selected on the basis of morphological criteria described in earlier studies (22). All protrusions, irrespective of their morphological characteristics, were counted as spines if they were in direct continuity with the dendritic shaft. For the purpose of this study, dendrites directly originating from the main apical shaft of CA1 pyramidal neurons were classified as primary dendrites. Starting from the origin of the branch and continuing away from the cell soma, spines were counted along a 50-μm stretch of the dendrite in 10 consecutive steps of 5 μm each.
Morris Water Maze. Training of stress-naïve mice in the Morris water maze was carried out in a circular pool (140 cm in diameter) between 10 a.m. and 4 p.m. in three trials per day, with each trial lasting a maximum of 120 sec with a 1-hr intertrial interval. To eliminate mice with vision problems, on day one, the animals were trained to locate a visible platform, and the ones that were unable to do so within 120 sec in three consecutive trials were eliminated from the study. On days two and three (spatial phase), the mice were trained to locate a hidden platform in the training quadrant. Probe trials (120 sec), during which the platform was removed, were performed on the fourth day to assess retention of the previously acquired information. After the procedures were completed, the animals were subjected to 21 days of restraint stress. To investigate the effect of stress on spatial learning, the procedure was repeated, but the animals were retrained to learn a different platform location. Animal movements were recorded and analyzed by an investigator unaware of the genotype and treatment.
Statistical Analysis. Data are presented as mean ± SEM. Between-group comparisons were performed with factorial ANOVA followed by Tukey posthoc comparison. P values <0.05 were considered significant. Numbers of animals in each experiment and the level of statistical significance are presented in figure legends.
Results
Severe stress can impair synaptic plasticity (23, 24) and learning (25), but the molecular machinery responsible for this effect is not fully understood. One possible mechanism is a modulation of NMDA receptors, which relay the majority of the excitatory signal in the central nervous system. NMDA receptors are not only indispensable for experience-induced synaptic plasticity (10) and learning (26) but are also modified by experience (27). To investigate whether long-term stress alters NMDA receptors in the hippocampus, we subjected wild-type mice to 21 days of daily restraint and determined the number and composition of NMDA receptor subunits by Western blotting. We found that chronic stress caused a decrease in NR1, NR2A, and NR2B subunits (to 27 ± 3%, 29 ± 6%, and 43 ± 5% of nonstressed controls, respectively; P < 0.001; Fig. 1A and Fig. 2 A-C). We did not observe a similar decrease after a single 6-hr session of restraint stress (data not shown). On the other hand, chronic stress did not affect the expression of brain-derived neurotrophic factor, glutamic acid decarboxylase-65, and GABAB receptors in the same animals (Fig. 6, which is published as supporting information on the PNAS web site), indicating that protein transcription/translation in the hippocampus was not generally suppressed.
Fig. 1.
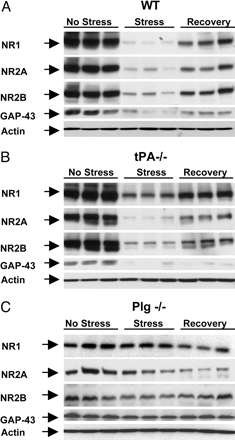
Stress-induced decrease in NMDA receptor subunits and GAP-43 is plasminogen-dependent. Wild-type, tPA-/-, and plasminogen-/- (n = 4, n = 4, and n = 3 per time point for each genotype, respectively) mice were subjected to chronic restraint stress, their hippocampi dissected, and the levels of individual NMDA receptor subunits as well as GAP-43 were determined by Western blotting. Stress caused a decrease in the levels of most NMDA receptor subunits (A), which were partially normalized after 10 days of recovery. Stress-induced changes were attenuated by deletion of the tPA gene (B) and almost completely prevented in plasminogen-/- mice (C). Differences in optical densities among the genotypes in control conditions are due to longer exposure times needed to visualize bands in wild-type and tPA-/- mice after stress.
Fig. 2.

Quantification of stress-induced changes in NMDA receptor subunits and GAP-43. Animals (wild type, n = 4; tPA-/-, n = 4; plasminogen-/-, n = 3 for each time point) were subjected to chronic restraint stress, their hippocampi dissected, and the levels of individual NMDA receptor subunits and GAP-43 were determined by Western blotting. The membranes were stripped and reblotted for actin for loading control. Changes in the expression of NR1 (A), NR2A (B), NR2B (C), and GAP-43 (D) after chronic stress or stress followed by 10 days of recovery are shown and compared with the levels observed in stress-naïve control mice. Stress-induced changes were attenuated in tPA-/- mice and almost completely prevented in plasminogen-/- animals. For quantification, the background was digitally eliminated by using scion image, and optical densities of the bands were normalized to actin. *, P < 0.05; #, P < 0.001; * in D, P < 0.05 vs. plasminogen-/-.
To investigate whether the decrease in NMDA receptor expression and the changes in its subunit composition were reversible, the animals were allowed 10 days of recovery after stress. Western blotting revealed that the observed changes in the NR1 and NR2B, but not the NR2A, subunits were partially reversed after stress was terminated (to 47 ± 3% and 51 ± 6% of control values for NR1 and NR2B, respectively; Fig. 1 A and Fig. 2 A and C).
This decrease in NMDA receptor expression could, in principle, be mediated by a variety of molecular mechanisms. The tPA/plasmin system, a recently identified modulator of NMDA receptor function (13, 14), offers an attractive candidate mechanism. At physiological concentrations, tPA is known to interact with NMDA receptors through nonproteolytic mechanisms, whereas plasmin can actually degrade these receptors (14, 28). To investigate whether tPA is indeed involved in stress-induced decrease in NMDA receptors, we subjected tPA-/- mice to chronic restraint stress. Western blotting revealed that the decrease in the NR1 subunit observed in wild-type mice (27 ± 3% of control) was attenuated in tPA-/- mice (to 42 ± 3% of the control values; P < 0.001; Figs. 1B and 2 A), indicating that the decrease in NR1 was partly tPA-dependent. The decrease in NR2A or NR2B after stress was not affected by the lack of tPA (to 25 ± 2% and 38 ± 2% of nonstress values, respectively; P < 0.001; Figs. 1 and 2 B and C).
To further investigate whether plasminogen activation to plasmin was involved in NMDA receptor decrease, we subjected plasminogen-/- mice to the same treatment. Deletion of the plasminogen gene attenuated stress-induced changes in NMDA receptor subunits' expression even further than tPA deletion (93 ± 2%, 69 ± 8%, and 72 ± 3% of control values for the NR1, NR2A, and NR2B subunits, respectively; Figs. 1C and 2 A-C). These results indicate that activation of plasminogen to the broad spectrum protease plasmin is an important step in stress-mediated decrease in NMDA receptors.
NMDA receptors play a pivotal role in neuronal plasticity. To investigate how the observed changes in NMDA receptors affected neurons, we examined the expression of GAP-43, a marker for synaptic plasticity (29). Stress dramatically decreased GAP-43 expression in the hippocampus of wild-type mice (to 36 ± 9% of control; P < 0.001; Figs. 1 A and 2D), and it was partially normalized after 10 days of recovery (to 47 ± 5% of control values). To investigate whether the changes were tPA- or plasmin-dependent, we determined GAP-43 levels in tPA-/- and plasminogen -/- mice. Although tPA-/- animals showed an even greater reduction in GAP-43 than wild-type animals (20 ± 6% of control; P < 0.001; Figs. 1B and 2D), the stress-related changes were mostly prevented in plasminogen -/- mice (87 ± 1% of control; Figs. 1C and 2D). These results point to plasmin as an important modulator of stress-induced plasticity and indicate that changes in GAP-43 expression closely followed those of NMDA receptors.
The hippocampus can be divided into several regions, which have different, albeit overlapping, functions. Because NMDA receptors are widely expressed within the hippocampus, we investigated whether the observed decrease in NMDA receptors was confined to any particular hippocampal region. To examine the spatial distribution of NMDA receptors after stress, we performed immunohistochemistry for NR1, which is an indispensable subunit enabling the formation of a functional channel. Although the decrease in the expression of NR1 subunit after stress was evident across all three hippocampal subregions (CA1, CA3, and dentate gyrus), it was most pronounced in the CA1 region (Fig. 3 A and B).
Fig. 3.

Stress-induced changes in NMDA receptors are most pronounced in CA1 and spatially coincide with plasminogen activation. The expression of the NR1 subunit of NMDA receptor in wild-type mice (representative picture in A) is reduced by chronic stress (B), and these changes are most pronounced in the CA1 region (arrows). Although tPA activity on conventional in situ zymography is predominant in the mossy fiber pathway (black zones in D), the rate of activation of plasminogen to plasmin by tPA is the highest in CA1 (arrows in C), as unmasked by addition of tPA to overlay gel (see Materials and Methods). Because plasmin has the ability to degrade NMDA receptors, this pattern of plasminogen activation is consistent with stress-induced decrease in their NR1 subunits.
To investigate whether this pattern of spatial expression of NMDA receptors after stress could be attributed to plasmin generation within CA1, we performed in situ zymography. This assay was originally designed to measure the endogenous tPA activity present in a brain section through histologically visualized activation of exogenous plasminogen (20) (Fig. 3D). We did not observe any consistent changes in tPA levels after chronic stress in the hippocampus using conventional in situ zymography (data not shown). Although tPA levels were the highest in the mossy fiber pathway, plasminogen activation was also observed in other hippocampal and cortical regions after longer incubation times. Moreover, tPA can be induced in CA1 region after various forms of stimulation (30, 31). These small amounts of tPA could be sufficient to produce physiologically relevant responses, because it has been shown in other tissues that plasmin generation by tPA can be dramatically facilitated by a presence of specific cell surface plasminogen-activating system/receptor (32, 33). To identify the specific areas that undergo facilitated plasmin activation by tPA, we added exogenous tPA and plasminogen into the overlay gel. When the mixture was applied onto brain sections, plasmin was preferentially generated within neuronal layers and was prominent in the CA1 region and the dentate gyrus (Fig. 3C). This spatial pattern of plasminogen activation is consistent with data showing a prominent role for plasmin in mediating the decrease in NMDA receptor expression after stress.
The majority of synaptic NMDA receptors are located on dendritic spines, which constitute the postsynaptic terminal. Dendritic spines are highly motile protrusions that display a considerable level of experience-dependent structural plasticity (9). Long-term stress causes a reduction in dendritic arborization (34) and alters synaptic terminal structure in the hippocampus (7). To investigate whether the changes in dendritic spines paralleled those observed in NMDA receptor expression and plasmin generation, we performed Golgi staining and determined the number of dendritic spines in CA1 hippocampal neurons after stress. Stress caused a decrease in dendritic spines (reduction by 9 ± 2%; P < 0.05; Fig. 4), consistent with the changes in synaptic structure reported in other hippocampal regions (7, 34). To investigate whether the observed changes were tPA-dependent, we subjected tPA-/- mice to the same procedure. We found that tPA-/- mice had fewer spines than their wild-type counterparts (by 20 ± 1%; P < 0.001), but stress did not alter the number of spines in these animals (102 ± 1% of prestress values; P > 0.05; Fig. 4 A and B). These results provide evidence that stress-induced changes in the hippocampus have molecular (NMDA receptors and GAP-43), as well as morphological (dendritic plasticity) components, which are mediated by the tPA/plasmin system.
Fig. 4.

Stress-induced decrease in the number of spines on CA1 pyramidal neurons is tPA-dependent. (A) Photomicrographs of representative segments of Golgi stain-impregnated apical dendritic branches demonstrating decrease in the number of spines in wild-type (WT, Left) but not mutant (tPA-/-, Right) mice. (Scale bar, 10 μm.) WT No Stress and WT Stress, n = 23 neurons, n = 4 animals; tPA-/- No Stress, n = 24 neurons, n = 4 animals; tPA-/- Stress, n = 29 neurons, n = 4 animals. (B) Percent change in spine density elicited by stress in CA1 neurons from wild-type (Left, normalized to unstressed wild type) and tPA-/- mice (Right, normalized to unstressed tPA-/-). *, P < 0.05.
The changes we observed were most pronounced in the CA1 region of the hippocampus, an area previously implicated in spatial learning. To investigate whether stress-induced changes in neuronal molecular machinery and in their morphology have behavioral consequences, we subjected wild-type mice to chronic stress and measured their spatial learning by using the Morris water maze test (35). Stress impaired the ability of mice to locate a hidden platform, and this effect was most pronounced during the third trial (latency to find a hidden platform increased to 385 ± 75% of prestress values; P < 0.01; Fig. 5 A and C), but once they learned the correct location, the memory trace persisted, and the retrieval was not impaired (Fig. 5 B and D).
Fig. 5.

Stress-induced impairment in spatial learning depends on both tPA and plasminogen. Wild-type (n = 9), tPA-/- (n = 9), and plasminogen-/- (n = 8) mice were trained to locate a hidden platform located in the training quadrant submerged 1 cm below water surface. (A) Six learning trials (120-sec cutoff time) were carried out in 2 days. Probe trial (120 sec; B), during which the platform was removed, was performed on the next day to assess retention of the previously acquired information. After the procedures were completed, the animals were subjected to 21 days of restraint stress and their learning abilities reinvestigated (C and D), but this time the animals were trained to learn a different platform location. Stress caused learning impairment in wild-type but not in tPA-/- or plasminogen-/- mice (C). However, memory retrieval was not affected by stress (D). Numbers inside columns in B and D represent percent time the mice spent in the platform quadrant during the probe trial. **, P < 0.01.
Next we investigated whether the learning impairment caused by stress could be prevented by deletion of either tPA or plasminogen, similar to that observed with the molecular and morphological changes. Therefore, we subjected tPA-/- and plasminogen-/- mice to stress and measured their spatial learning in the Morris water maze. First, we found that the baseline ability to learn this task was not affected by the lack of tPA or plasminogen (Fig. 5 A and B), which indicates that tPA-/- or plasminogen-/- mice do not have functional abnormalities within the hippocampus that would impair their learning ability. However, in contrast to wild-type mice, tPA-/- and plasminogen-/- animals did not show learning impairment after stress (Fig. 5 C and D).
Discussion
Our results indicate that repeated restraint stress decreases NMDA receptor and GAP43 expression and dendritic spine density in the hippocampus. The effects of repeated stress on these measures are found within the CA1 region and are associated with an impairment of the ability of an animal to learn a spatial memory task. These changes are mediated by the tPA/plasmin system, because they are attenuated by deletion of critical elements of this system. Stress did not decrease brain-derived neurotrophic factor, glutamic acid decarboxylase-65, or GABAB receptor levels.
Acute stress causes transient changes in synaptic functional plasticity (24), whereas chronic stress leads to longer-lasting morphological changes such as dendrite remodeling, suppression of neurogenesis, and spine reduction (8, 36, 37). There is also the possibility that structural and functional alterations become permanent when the duration or intensity of stress increases (38). Structural remodeling usually displays a region-specific characteristic. For example, it has been shown that the basolateral nucleus of the amygdala responds to chronic stress with dendritic growth (38), whereas a simplification of dendritic tree is observed in the hippocampus, particularly in the CA3 region (34). Morphological remodeling within the amygdala can cause pathological anxiety, whereas reshaping hippocampal circuitries may result in cognitive impairment (37). However, the molecular mechanisms behind these effects are not fully understood.
One obvious candidate mediator of stress-induced neuronal plasticity is the NMDA receptor, the blockade of which inhibits stress-induced dendritic shrinkage (12). NMDA receptors are heteromeric ion channels composed of at least one NR1 subunit and several NR2 subunits (39). Our present work demonstrates that stress down-regulates all NMDA receptor subunits in the CA1 region of the hippocampus, the area crucial in spatial learning. The changes in NMDA receptors were reversible, because they gradually normalized during recovery. Stress-induced changes in NMDA receptors were specific, because we did not observe similar alterations with the level of glutamic acid decarboxylase-65, brain-derived neurotrophic factor, or GABAB receptors. Altogether, these findings are in line with the ability of stress to inhibit neuronal plasticity (23, 24) and to cause cognitive impairment (25).
There are several mechanisms that could be responsible for the decrease in NMDA receptors in our study. For example, it might be caused by either a down-regulation of individual NMDA receptor subunits in response to excessive stimulation with glutamate or by their proteolytic degradation. To examine the second possibility, we investigated the changes in NMDA receptors in mice that lacked individual elements of the tPA/plasmin system, a proteolytic cascade critically involved in NMDA receptor modulation/degradation (13, 14, 28). However, in our present study, a deletion of the tPA gene had only a minor effect in preventing these changes, which suggests that tPA is not the predominant protease cleaving NMDA receptors after stress. Alternatively, other plasminogen activators (e.g., urokinase PA) could be involved, and/or plasmin could be an ultimate protease causing the degradation of NMDA receptors. That was indeed the case, because the down-regulation of NMDA receptors caused by stress was prevented by a deletion of the plasminogen gene. These studies provide further evidence that, whereas the effect of tPA on NMDA receptor cleavage is controversial (28, 40), plasmin is capable of their proteolytic degradation (14, 28).
We have previously found that stress modulated the expression and phosphorylation of molecules centrally involved in neuronal plasticity in the amygdala (15). Acute (15), but not chronic (unpublished observation), stress increased the expression of GAP-43 in the amygdala, whereas in the present study, only chronic (but not acute) stress reduced GAP-43 expression in the hippocampus. These findings are consistent with different roles of the hippocampus and amygdala in fear and anxiety and provide further evidence that these regions are differentially regulated by stress (38).
Although the down-regulation of GAP-43 after chronic stress is likely to be a consequence of the decrease in NMDA receptors, the role of the tPA/plasmin system in this process is more difficult to reconcile. To our surprise, the deletion of the tPA gene not only failed to prevent stress-induced decrease in GAP-43 but also reduced it even further. One possibility is that tPA could stimulate the expression of GAP-43 by acting on the NR2B subunit of NMDA receptors (14). On the other hand, plasminogen deletion almost eliminated the decrease in GAP-43 mediated by stress. This could be a consequence of protection against down-regulation of NMDA receptors rendered by plasminogen deletion. Therefore, it is conceivable that both proteolytic and nonproteolytic actions of tPA and plasmin could mediate the effects of stress on neuronal plasticity.
In addition to changes in protein expression, neuronal plasticity can be manifested at the structural level as a change in shape or number of dendritic spines (9). The tPA/plasmin system has been recently identified as an important player in spine remodeling related to experience-dependent visual cortex plasticity (16, 17). To investigate whether tPA mediates spine plasticity in the hippocampus, we subjected wild-type and tPA-/- mice to chronic stress. We have found that the number of spines in CA1 neurons decreased after stress, and this was prevented by the deletion of the tPA gene. Is this effect plasminogen-dependent? Further experiments are needed to clarify this important point. A recent study suggested that plasmin is involved in spine plasticity in the visual cortex through extracellular matrix degradation, making the environment more permissive for structural rearrangements (16, 17). However, it is possible that both tPA and plasmin could be involved, acting in either a nonproteolytic and/or proteolytic manner.
We have found that stress induced decrease in NMDA receptors and GAP-43, and the number of dendritic spines is associated with less efficient learning. Learning impairment was most evident during the early stages of learning and gradually disappeared with more training. Additionally, once the memory trace was formed, it persisted, and the retrieval was not affected. It is especially important to note that the detrimental effect of stress on learning was relatively minor compared with the effect of stress on markers of plasticity. What could be the reason for this dissociation? Further experiments are needed to determine whether stress preferentially depleted the membrane (active) or intracellular (inactive) pool of NMDA receptors.
However, the impairment of acquisition rather than consolidation phase by stress is consistent with earlier reports that long-term consolidation does not depend on the hippocampus. It has been suggested that it takes place in the cortex (22, 41), where the tPA/plasmin system plays a relatively minor role in adulthood. Thus, the effect of repeated restraint stress on learning in our study is consistent with the observed small decrease in number of spines observed in the CA1 region of the hippocampus, as well as with studies that have shown that repeated stress impairs hippocampal-dependent memory (8).
Moreover, both tPA and plasminogen deletion resulted in the abolition of the stress reduction in learning efficiency, indicating that both gene products play a role in stress effects on hippocampal function. This is consistent with the fact that both the decrease in NMDA receptors and dendritic spines and the learning deficits were either attenuated or prevented by deletion of either tPA or plasminogen. Indeed, we have shown by modified zymography that there is an increase in the potential for a high rate of plasmin generation within the CA1 neurons. Further experiments will clarify whether the rate of plasmin generation is regulated by stress in the CA1 region, and how it contributes to the decrease in NMDA receptors.
The findings in the present study pertain to the CA1 region of the hippocampus, whereas previous studies of repeated stress effects have focused upon the CA3 region, where dendritic shrinkage is observed (8, 38, 42). Although dendritic shrinkage has also been reported after repeated stress in both dentate gyrus and CA1, the effects in CA3 are larger (42). Future studies will need to address the extent of stress-induced changes in CA1. NMDA receptors and GAP-43 observed in the present study take place in CA3 and dentate gyrus.
Conclusion
We have added an aspect to the growing body of evidence pointing to the role of the tPA/plasmin system as a mediator of various forms of synaptic plasticity in various brain regions. It is now becoming clear that tPA exerts both proteolytic and nonproteolytic effects that contribute to various aspects of brain functioning at morphological, biochemical, and functional levels. It is hoped that these findings will be useful in designing better therapies against stress-related disorders, such as posttraumatic stress disorder.
Supplementary Material
Acknowledgments
We thank Yuliya Keptsi for technical assistance and Sharath Bennur for help with the preparation of spine images. This study was supported by grants from the Medical Research Council (United Kingdom) and the Alcoholic Beverage Medical Research Foundation (to R.P.), by an International Senior Research Fellowship from the Wellcome Trust (to S.C.), and by National Institutes of Health Grants NS35704 and NS38472 (to S.S.) and MH41256 (to B.M.).
Author contributions: R.P., S.C., B.M., and S.S. designed research; R.P., B.S.S.R., and J.P.M. performed research; R.P., B.S.S.R., J.P.M., S.C., B.M., and S.S. analyzed data; and R.P., J.P.M., S.C., B.M., and S.S. wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: tPA, tissue plasminogen activator; GAP-43, growth-associated protein-43; CA, cornu ammonis.
References
- 1.Selye, H. (1936) Nature 138, 32. [Google Scholar]
- 2.Newport, D. J. & Nemeroff, C. B. (2000) Curr. Opin. Neurobiol. 10, 211-218. [DOI] [PubMed] [Google Scholar]
- 3.McEwen, B. S. (2003) Biol. Psychiatry 54, 200-207. [DOI] [PubMed] [Google Scholar]
- 4.Conrad, C. D., LeDoux, J. E., Magarinos, A. M. & McEwen, B. S. (1999) Behav. Neurosci. 113, 902-913. [DOI] [PubMed] [Google Scholar]
- 5.Nutt, D. J. & Malizia, A. L. (2004) J. Clin. Psychiatry 65, Suppl. 1, 11-17. [PubMed] [Google Scholar]
- 6.Shin, L. M., Shin, P. S., Heckers, S., Krangel, T. S., Macklin, M. L., Orr, S. P., Lasko, N., Segal, E., Makris, N., Richert, K., et al. (2004) Hippocampus 14, 292-300. [DOI] [PubMed] [Google Scholar]
- 7.Magarinos, A. M., Verdugo, J. M. & McEwen, B. S. (1997) Proc. Natl. Acad. Sci. USA 94, 14002-14008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McEwen, B. S. (1999) Annu. Rev. Neurosci. 22, 105-122. [DOI] [PubMed] [Google Scholar]
- 9.Fischer, M., Kaech, S., Wagner, U., Brinkhaus, H. & Matus, A. (2000) Nat. Neurosci. 3, 887-894. [DOI] [PubMed] [Google Scholar]
- 10.Tsien, J. Z., Huerta, P. T. & Tonegawa, S. (1996) Cell 87, 1327-1338. [DOI] [PubMed] [Google Scholar]
- 11.Tang, Y. P., Shimizu, E., Dube, G. R., Rampon, C., Kerchner, G. A., Zhuo, M., Liu, G. & Tsien, J. Z. (1999) Nature 401, 63-69. [DOI] [PubMed] [Google Scholar]
- 12.Magarinos, A. M. & McEwen, B. S. (1995) Neuroscience 69, 89-98. [DOI] [PubMed] [Google Scholar]
- 13.Nicole, O., Docagne, F., Ali, C., Margaill, I., Carmeliet, P., MacKenzie, E. T., Vivien, D. & Buisson, A. (2001) Nat. Med. 7, 59-64. [DOI] [PubMed] [Google Scholar]
- 14.Pawlak, R., Melchor, J. P., Matys, T., Skrzypiec, A. E. & Strickland, S. (2005) Proc. Natl. Acad. Sci. USA 102, 443-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pawlak, R., Magarinos, A. M., Melchor, J., McEwen, B. & Strickland, S. (2003) Nat. Neurosci. 6, 168-174. [DOI] [PubMed] [Google Scholar]
- 16.Mataga, N., Mizuguchi, Y. & Hensch, T. K. (2004) Neuron 44, 1031-1041. [DOI] [PubMed] [Google Scholar]
- 17.Oray, S., Majewska, A. & Sur, M. (2004) Neuron 44, 1021-1030. [DOI] [PubMed] [Google Scholar]
- 18.Pang, P. T., Teng, H. K., Zaitsev, E., Woo, N. T., Sakata, K., Zhen, S., Teng, K. K., Yung, W. H., Hempstead, B. L. & Lu, B. (2004) Science 306, 487-491. [DOI] [PubMed] [Google Scholar]
- 19.Pawlak, R., Nagai, N., Urano, T., Napiorkowska-Pawlak, D., Ihara, H., Takada, Y., Collen, D. & Takada, A. (2002) Neuroscience 113, 995-1001. [DOI] [PubMed] [Google Scholar]
- 20.Sappino, A. P., Madani, R., Huarte, J., Belin, D., Kiss, J. Z., Wohlwend, A. & Vassalli, J. D. (1993) J. Clin. Invest. 92, 679-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramon-Moliner, E. (1970) in Contemporary Research Methods in Neuroanatomy, eds. Nauta, W. J. H. & Ebbesson, S. O. E. (Springer, New York), pp. 32-47.
- 22.Hayashi, M. L., Choi, S. Y., Rao, B. S., Jung, H. Y., Lee, H. K., Zhang, D., Chattarji, S., Kirkwood, A. & Tonegawa, S. (2004) Neuron 42, 773-787. [DOI] [PubMed] [Google Scholar]
- 23.Pavlides, C., Nivon, L. G. & McEwen, B. S. (2002) Hippocampus 12, 245-257. [DOI] [PubMed] [Google Scholar]
- 24.Kim, J. J., Foy, M. R. & Thompson, R. F. (1996) Proc. Natl. Acad. Sci. USA 93, 4750-4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luine, V., Villegas, M., Martinez, C. & McEwen, B. S. (1994) Brain Res. 639, 167-170. [DOI] [PubMed] [Google Scholar]
- 26.Sakimura, K., Kutsuwada, T., Ito, I., Manabe, T., Takayama, C., Kushiya, E., Yagi, T., Aizawa, S., Inoue, Y., Sugiyama, H., et al. (1995) Nature 373, 151-155. [DOI] [PubMed] [Google Scholar]
- 27.Heynen, A. J., Quinlan, E. M., Bae, D. C. & Bear, M. F. (2000) Neuron 28, 527-536. [DOI] [PubMed] [Google Scholar]
- 28.Matys, T. & Strickland, S. (2003) Nat. Med. 9, 371-372; author reply 372-373. [DOI] [PubMed] [Google Scholar]
- 29.Benowitz, L. I. & Routtenberg, A. (1997) Trends Neurosci. 20, 84-91. [DOI] [PubMed] [Google Scholar]
- 30.Salles, F. J. & Strickland, S. (2002) J. Neurosci. 22, 2125-2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qian, Z., Gilbert, M. E., Colicos, M. A., Kandel, E. R. & Kuhl, D. (1993) Nature 361, 453-457. [DOI] [PubMed] [Google Scholar]
- 32.Ellis, V. & Whawell, S. A. (1997) Blood 90, 2312-2322. [PubMed] [Google Scholar]
- 33.Cesarman, G. M., Guevara, C. A. & Hajjar, K. A. (1994) J. Biol. Chem. 269, 21198-21203. [PubMed] [Google Scholar]
- 34.Magarinos, A. M., McEwen, B. S., Flugge, G. & Fuchs, E. (1996) J. Neurosci. 16, 3534-3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morris, R. (1984) J. Neurosci. Methods 11, 47-60. [DOI] [PubMed] [Google Scholar]
- 36.Sandi, C., Davies, H. A., Cordero, M. I., Rodriguez, J. J., Popov, V. I. & Stewart, M. G. (2003) Eur. J. Neurosci. 17, 2447-2456. [DOI] [PubMed] [Google Scholar]
- 37.McEwen, B. S. & Chattarji, S. (2004) Eur. Neuropsychopharmacol. 14, Suppl. 5, S497-S502. [DOI] [PubMed] [Google Scholar]
- 38.Vyas, A., Mitra, R., Shankaranarayana Rao, B. S. & Chattarji, S. (2002) J. Neurosci. 22, 6810-6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hardingham, G. E. & Bading, H. (2003) Trends Neurosci. 26, 81-89. [DOI] [PubMed] [Google Scholar]
- 40.Liu, D., Cheng, T., Guo, H., Fernandez, J. A., Griffin, J. H., Song, X. & Zlokovic, B. V. (2004) Nat. Med. 10, 1379-1383. [DOI] [PubMed] [Google Scholar]
- 41.Wiltgen, B. J., Brown, R. A., Talton, L. E. & Silva, A. J. (2004) Neuron 44, 101-108. [DOI] [PubMed] [Google Scholar]
- 42.Sousa, N., Lukoyanov, N. V., Madeira, M. D., Almeida, O. F. & Paula-Barbosa, M. M. (2000) Neuroscience 97, 253-266. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.