

Abstract
The MAPK (mitogen-activated protein kinase) pathway is one of the most important and intensively studied signalling pathways. It is at the heart of a molecular-signalling network that governs the growth, proliferation, differentiation and survival of many, if not all, cell types. It is de-regulated in various diseases, ranging from cancer to immunological, inflammatory and degenerative syndromes, and thus represents an important drug target. Over recent years, the computational or mathematical modelling of biological systems has become increasingly valuable, and there is now a wide variety of mathematical models of the MAPK pathway which have led to some novel insights and predictions as to how this system functions. In the present review we give an overview of the processes involved in modelling a biological system using the popular approach of ordinary differential equations. Focusing on the MAPK pathway, we introduce the features and functions of the pathway itself before comparing the available models and describing what new biological insights they have led to.
Keywords: computational modelling, extracellular-signal-regulated kinase (ERK), mitogen-activated protein kinase (MAPK), receptor tyrosine kinase, signalling network, systems biology
Abbreviations: CD domain, common docking domain; EGF(R), epidermal growth factor (receptor); ERK, extracellular-signal-regulated kinase; FBA, Flux Balance Analysis; FGF, fibroblast growth factor; GAP, GTPase-activating protein; Grb2, growth-factor-receptor-bound protein 2; JIP, JNK (c-Jun N-terminal kinase)-interacting protein; KSR, kinase suppressor of Ras; MAPK, mitogen-activated protein kinase; MAPKK/MKK, MAPK kinase; MAPKKK/MKKK, MAPKK kinase; MCA, Metabolic Control Analysis; MEK, MAPK/ERK kinase; MKP, MAPK phosphatase; MP1, MEK partner 1; NGF, nerve growth factor; ODE, ordinary differential equation; PDE4, phosphodiesterase 4; PDGF, platelet-derived growth factor; PKA, cAMP-activated protein kinase; PLCγ, phospholipase Cγ; RSK, ribosomal S6 kinase; RTK, receptor tyrosine kinase; SBML, Systems Biology Markup Language; Shc, Src homology and collagen homology; SOS, son of sevenless; STAT5, signal transduction and activator of transcription 5
THE GENERAL STRUCTURE OF MAPK PATHWAYS
In molecular biology the MAPK (mitogen-activated protein kinase) pathway is considered to be a paradigm for signal transduction, as it occupies a central role in key cellular processes and is evolutionarily conserved. Various manifestations of the MAPK pathway are found in all eukaryotic cells so far examined and have been studied extensively in a multitude of organisms, ranging from yeast to humans. On the basis of the substantial body of data available in the literature, this pathway has frequently been the system of choice for computational modelling of biological signal transduction over the last decade.
The term ‘MAPK pathway’ refers to a module of three kinases which are activated by sequentially phosphorylating each other in response to a diverse range of stimuli, such as cytokines, growth factors, neurotransmitters, cellular stress and cell adherence. Accordingly, the pathway plays a pivotal role in many key cellular processes, ranging from growth control in all its variations, cell differentiation and survival to cellular adaptation to chemical and physical stress (for reviews, see [1–3]).
The MAPK pathway employs one of the most generic signalling designs found in biological signal transduction, namely that of a cycle formed by a kinase phosphorylating a target protein and an opposing phosphatase that is in charge of dephosphorylating the target (Figure 1). This type of protein phosphorylation presents a fundamental mechanism by which the activities of numerous enzymes, receptors, transporters, docking and scaffolding proteins are regulated.
Figure 1. Kinase/phosphatase signalling reaction.

A phospho group is transferred from a nucleotide to a serine, threonine or tyrosine residue of a target protein (T). The reaction is catalysed by a kinase. The phosphorylated target protein (T–P) is in turn dephosphorylated by a phosphatase to complete the cycle. Phosphorylation of a protein often entails conformational changes that modulate the function of the protein. Phosphorylation of MEK, for example, activates the kinase domain of the enzyme. MEK then catalyses the phosphorylation and, as a consequence thereof, activation of MAPK.
The general layout of the MAPK pathway consists of three kinase/phosphatase cycles built into a three-tiered cascade, consisting of a MAPK, which is activated via phosphorylation by a MAPKK/MKK (MAPK kinase), which in turn is phosphorylated by a MAPKKK/MKKK (MAPKK kinase) (Figure 2). MAPKs are deactivated by a family of phosphatases termed MKPs (MAPK phosphatases) [4].
Figure 2. General structure of the three-tiered cascade of the MAPK pathway.

A signal is propagated along this cascade by sequential phosphorylation as described in the text.
Most activators of the MAPK pathway initiate signalling by activating receptors in the cell membrane, which assemble into receptor signalling complexes and activate a MAPKKK typically through a small GTPase. Signal transduction within the MAPK-pathway module appears to be fairly specific and is often perceived as a pathway with a linear structure. The number of known MAPK effectors, however, is very large and diverse, including mainly transcription factors, protein kinases and cytoskeletal proteins. Upon activation, MAPK can translocate from the cytoplasm to the nucleus, where it regulates gene transcription through affecting chromatin structure and modifying the activity of transcription factors [5]. Eukaryotic cells contain at least 12 different MAPKKKs, seven MAPKKs and eight MAPKs, which can be attributed to at least four functionally distinct MAPK modules. MAPK modules have evolved by gene duplication, and several closely related modules, delivering specific biological responses, are co-expressed in a particular cell [2]. Currently it is not entirely clear exactly to what extent kinases of different MAPK modules are specific within their module or whether they are able to cross-talk within other MAPK modules or with other targets [3,6]. However, the fact that only relatively few MAPKs receive and integrate a plethora of extracellular stimuli to control a widely diverse range of cellular processes implies that considerable versatility and specificity is built into MAPK signalling. Some of the mechanisms which explain how specificity is achieved in MAPK signalling have recently come to light:
The duration and amplitude of MAPK phosphorylation is critical for the control of distinctive processes, such as proliferation or differentiation, in PC12 cells [7] or the activation of c-Fos [8].
Specific binding interactions, mediated by the CD (common docking) domain on MAPKs and scaffolds such as JIP [JNK (c-Jun N-terminal kinase)-interacting protein], KSR (kinase suppressor of Ras) or MP1 {MEK [MAPK/ERK (extracellular-signal-regulated protein kinase) kinase] partner 1}, tether several components of a MAPK module into a protein complex [9,10].
Cross-talk with other signalling pathways could modulate MAPK signalling; examples including regulation of the serine/threonine-protein kinase Raf by PKA (cAMP-activ- ated protein kinase) and PDE4 (phosphodiesterase 4) by ERK [11].
THE ERK/MAPK PATHWAY
In the following we will focus on the Raf/MEK/ERK pathway, because the majority of computational MAPK models address this particular MAPK module. Signal transduction along the Raf/MEK/ERK pathway (Figure 3) begins with the activation of the small GTPases Ras (and possibly Rap) by receptor tyrosine kinases, G-protein-coupled receptors and/or integrins [12]. These membrane proteins assemble large signalling complexes upon activation, which recruit and activate Ras proteins by inducing the exchange of Ras-bound GDP with GTP, converting Ras into its activated conformation. This process is mediated by the interaction of Ras with GDP/GTP-exchange factors, such as SOS (son of sevenless). De-activation of Ras, on the other hand, is controlled by GAPs (GTPase-activating proteins), which significantly enhance the otherwise very low GTPase activity of Ras and thus effectively enhance the hydrolysis of GTP to GDP [13]. Upon activation, the small G-proteins recruit the MKKKs c-Raf (and, if present, A- and B-Raf) to the plasma membrane, where Raf is activated in a complicated, only partially understood, process that involves binding to Ras, phosphorylation and changes in conformation and protein interactions [12]. Raf then activates MEK-1/2 by phosphorylation of two serine residues. MEKs generally recognize only specific MAPKs as substrates. MEK-1/2 phosphorylates ERK-1/2 at threonine and tyrosine residues in a ‘TEY’ motif within its activation loop. This dual phosphorylation is correlated with ERK-1/2 activity and can be used as a measurement for the indirect quantification of ERK activation [14]. ERK is a serine/threonine kinase. Activated ERK can phosphorylate over 80 substrates in the cytoplasm and the nucleus. It can regulate gene expression by directly phosphorylating transcription factors such as Ets, Elk, and Myc, or indirectly by targeting substrates such as p90-RSK (ribosomal S6 kinase) family kinases, which can modify transcription factors and histones [15]. The term MAPK was originally synonymous with ERK, but is increasingly used to indicate the superfamily of different MAPKs. The latter usage is adopted in the text below.
Figure 3. Structure of the ERK pathway.

Upon ligand binding, RTK autophosphorylates (phosphates are shown as red circles) on tyrosine residues, which serve as docking sites for adaptor and signalling molecules. Ras and Rap1 are activated by the recruitment of guanosine-nucleotide exchange factors (SOS, C3G) via adaptor proteins (Shc and Grb2; Crk). Ras can activate Raf-1 and B-Raf; Rap1 presumably can activate B-Raf. Raf proteins phosphorylate and activate MEK-1/2, which in turn activate ERK-1/2 (indicated by black arrows). Negative-feedback loops (indicated by red lines) include the induction of MKPs by ERK as well as the inhibitory phosphorylation of Raf-1 and SOS.
COMPUTATIONAL MODELLING
Systems biology is concerned with the study of biological systems in terms of their underlying network structure rather than simply their individual molecular components. A biological system can be anything from a simple biochemical reaction cycle, to a gene regulatory network or signal transduction pathway, to a cell, tissue or an entire organism. At the heart of systems biology is mathematical modelling – the process of translating a biological system into a mathematical model for subsequent computer simulation and analysis. In order to be useful and applicable to biological questions, models have to faithfully describe the biological system and be able to make predictions about its behaviour. Thus, while the basis of a model is the topological representation of its components and their links, it is the description of the biological system's dynamic behaviour which equips the model with predictive power [16]. However, it is important to emphasize that a model is not a real or exact representation of the biological system; rather, it is a simplified description to assist in analysis and to help us to better understand the real world. A mathematical model can be used to generate new insights, make testable predictions, identify gaps in our biochemical knowledge, test conditions that may be difficult to study in the laboratory and help identify what is right and wrong with our hypotheses; for example, could the proposed mechanism give the observed results? Ultimately the purpose of a model is to increase our understanding of the real biological system, to identify the key components and processes and to predict biological behaviour, such as what effect a particular drug will have on the system, or which components need to be disturbed for the system to behave in a specific way.
ODEs (ORDINARY DIFFERENTIAL EQUATIONS)
One of the most commonly used approaches to modelling biological systems is that of ODEs. The technical definition of a differential equation is an equation involving one or more unknown functions and their derivatives. Essentially, a differential equation describes how a property of interest, such as [A] – the concentration of A, changes over time; this is usually expressed by describing how the rate of change of the concentration is related to the concentration at that moment. For example, consider the simple reaction below which depicts the decay (or conversion) of A into B:
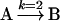 |
(1) |
This reaction is a plain uncatalysed reaction that can be modelled using Mass Action kinetics; k represents the rate constant of the reaction which, in this example, is equal to 2 mM/s. Therefore, the reaction proceeds at the following rate:
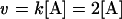 |
(2) |
As can be seen, the rate of the reaction (v) is dependent on [A] – the greater the value of [A], the higher the rate will be, and therefore the faster A will be consumed and the faster B will be produced. From the above equations it is relatively straightforward to construct differential equations representing the rate of change in [A] and [B] over time:
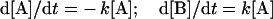 |
(3) |
In order to simulate the above reaction we also need to know the initial (time zero) values of [A] and [B], which in this example are set to be equal to 5 and 0 mM respectively. Simulation uses numerical methods to solve the differential equations and approximate the change in concentration of all the species in a system over time. One of the simplest methods of numerical integration is Euler's method, the basic idea of which is to approximate a curve with a series of straight lines tangential to the curve. The rate of change is the same as the slope of a line drawn as a tangent to the curve at that time; therefore, a series of these tangent lines can be used to follow the function over time. Euler's method is a point–slope type method, since it uses an initial concentration (the point) and a differential equation (the slope) to monitor a species concentration over time. In practice this method involves using this initial slope for a very small finite step-size (Δt) in the time direction. The method is then repeated for another step size using the result from the previous step as the new starting point. For example, the simple reaction above (eqn 1) was simulated using Euler's method over two time steps (Δt) of 0.01 s; the calculations used to compute [A] at these two time points are shown below (the extension of this example to longer periods of times is simple):
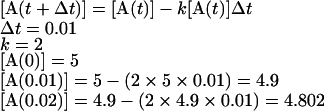 |
(4) |
Euler's method described above is a very basic method of numerical integration. However, there are many more advanced methods, such as the Runge-Kutta, Rosenbrock and Richardson extrapolation; for more information on the different methods of numerical integration, see [17]. Furthermore, although the above example reaction (eqn 1) is both small and simple, differential equations can easily be used to model large biological systems involving many more species and reactions as well as more complex reaction mechanisms such as those obeying Michaelis-Menten kinetics. It is also noteworthy that, when modelling a system comprising several biochemical species, each represented by a differential equation, ODE methods require simultaneous solving of all the differential equations that represent the system.
THE MODELLING PROCESS
One way to view the actual process of modelling the behaviour of a biological system is to consider it as five distinct steps (Figure 4).
Figure 4. The five steps of modelling.

This diagram depicts the five steps involved in modelling a biological system. The first step is identifying the biological system to model, followed by actually defining the model to represent the system, simulating the model and validating the simulation results. If the model is valid, it can be analysed further; if it is not, the definition step is revisited, where the model is checked for various types of errors. For more information on each of the steps, see the text.
Step 1: system delimitation
This step involves selecting the biological system to be modelled, e.g. the EGF (epidermal growth factor)- or NGF (nerve growth factor)-activated ERK cascade. More importantly, it also includes identifying the biological question the model aims to answer, e.g. how do EGF and NGF produce differing responses in ERK activation?
Step 2: definition
This is the key step in the modelling process and is by no means trivial. It involves defining the model to represent the biological system of interest. Essentially, this involves drawing a detailed topological chart of the system that shows all the species (e.g. proteins) involved, what reactions they can participate in and where. The kinetic types for each reaction then need to be defined and the parameter data (e.g. rate constants and initial concentrations) assigned to give a set of detailed kinetic reactions. This definition step can involve exhaustive searches of the scientific literature to see what is already known about the system and what parameter data are already available, as well as performing laboratory experiments to provide data or the use of computational techniques to estimate missing parameters (see, for example, [18]). It is important to note that many biological processes are very complex and not fully understood. Therefore defining a model often involves making simplifying assumptions that reduce complex and poorly understood processes into simpler ones that can still represent the biological processes well enough to explain the observed data.
Step 3: simulation
After the model has been defined, the next step is to translate the kinetic reactions into a set of differential equations that describe how the concentration of each species in the system changes over time; this set of differential equations is then simulated (or solved) over a desired period of time. This is a relatively straightforward step, as there are many software tools available for the generation and simulation of models of biological systems that are based on differential equations (see below for more details on simulation tools).
Step 4: validation
Simulating a model typically returns a table of data or a curve showing how each species' concentration varies over time. These data must then be validated against available experimental data. If the model behaves as the experimental data suggest, then the model can be analysed further. If it does not, then the definition step must be revisited, where the model is checked for errors, such as incorrect kinetics or parameter data, over-simplifications of processes and perhaps missing components. In essence, this is a ‘debugging’ loop involving model definition, simulation and validation, where the model is refined in order to obtain behaviour which conforms with experimental observations. Sometimes the refinement already reveals useful information about the system. For instance, refining the mathematical model of STAT5 (signal transduction and activator of transcription 5) phosphorylation in response to erythropoetin-receptor stimulation uncovered the fact that the model could capture the experimental kinetics of STAT5 phosphorylation when a time delay was introduced to account for the shuttling of STAT5 between the nucleus and cytosol. This indicated that STAT5 cycles between the nucleus and cytosol, a phenomenon which subsequently was proven experimentally [19]. The validation step is crucial if a reliable model is to be generated; if the model's results do not match known biology, we cannot trust predictions about unknown biology.
Step 5: analysis
After the model has been validated, it can be analysed and the simulation results interpreted. Analysis can come in various forms, from the simple examination of the species' concentration graphs to more complex statistical analyses. Sensitivity analysis is a commonly used approach that studies the response of system variables to changes in parameter values, and can therefore be used to identify the key reactions and species, as well as monitoring how robust a model is. Essentially, sensitivity analysis works by varying the data for a parameter by a small amount and analysing what effect this has on a specific system variable, such as the peak height or duration of the phosphorylated ERK signal; a small change to a key parameter datum is likely to have a large effect on the system variable. Different parameters can have widely different sensitivities, and the sensitivity of a specific parameter can also vary depending on which system variable is considered. On the other hand, sensitivity analysis can also be used to estimate the impact of uncertainty in parameters on system variables. Robustness is a very important factor in biological systems, as it allows a system to absorb fairly large perturbations and still function reasonably well; this is because the functionally important behaviour of a system has a certain degree of resilience to damage. If a system variable has a low sensitivity with respect to a parameter, it is robust to alterations in that parameter, however caused. The structural robustness of a model can also be analysed by monitoring how it performs when parts of it are removed; for example, how does the system behave if a specific species, reaction or entire pathway is removed. This can be useful, because there is often redundancy in biological systems where multiple pathways are available for the production or activation of a certain protein. Overall, the analysis step aims to generate new predictions and hypotheses about biological processes that were not known or unproven before, thus increasing our overall understanding of the system itself.
COMPUTATIONAL TOOLS
Currently, there a number of software tools available for the simulation and analysis of differential-equation-based models of biological systems, such as Gepasi [20], E-CELL [21], Virtual Cell [22], GENESIS [23] combined with Kinetikit [24], Jarnac combined with JDesigner [25], Mathematica (Wolfram Research; http://www.wolfram.com/) and Matlab (Mathworks; www.mathworks.com) (for a recent review of simulation tools, see [26]). The majority of these tools have a graphical interface that permits the user to enter the biochemical reactions and kinetic constants, which the tool then uses to automatically generate the corresponding ODEs and simulate the model. Some have a number of advanced features to visualize and analyse models, display simulation results and also estimate missing parameter data. Furthermore, the majority of tools now support SBML (Systems Biology Markup Language; www.sbml.org) [27], which is concerned with introducing a standard representation of models of biological systems. This enables models to be shared, evaluated and developed co-operatively, as well as enabling the use of multiple tools without having to rewrite models for each tool; a full list of SBML-compatible modelling tools is available from the SBML website (www.sbml.org). A recommended website for the novice is the pathway model repository of the Silicon Cell project (http://www.jjj.bio.vu.nl/index.html; [28]). Via an interactive webpage, pathway models can be viewed and the effects of changes in kinetic parameters can be simulated.
ERK MODELS
The ERK cascade is one of the most important cell-signalling pathways and has been the subject of intensive study in the laboratory and, more recently, through mathematical-modelling techniques. Early mathematical models of the ERK cascade focused on investigating the properties and behaviour of the core cascade itself. The first model was published in 1996 by Huang and Ferrell [29] and showed that the ERK cascade exhibited ultrasensitivity, i.e. a non-linear sigmoid activation curve, with the degree of ultrasensitivity increasing as one moves down the cascade. This was quickly followed by two models in 1997 [30,31] that showed that the activating dual phosphorylation of ERK itself was accomplished via a two-collision, distributive mechanism whereby MEK phosphorylates one site, dissociates, and then has to rebind to phosphorylate the second site. This generates a pool of largely singly phosphorylated, i.e. inactive, ERK molecules, which appears as a gentle response curve. When enough singly phosphorylated ERK molecules have accumulated, most further phosphorylation events produce doubly phosphorylated, i.e. active, ERK, causing the slope of the activation curve to increase sharply and steeply. This provided a mechanistic basis for the ultrasensitivity of ERK activation and explained how ERK can convert graded inputs into switch-like outputs [31]. Then in 1998, Ferrell and Machleder [32], using Xenopus oocytes, showed that, because of ultrasensitivity, ERK is activated essentially in an all-or-none fashion in individual cells when they are treated with increasing concentrations of progesterone. Thus the apparently graded concentration-dependent response curve observed when a whole cell population was analysed was actually composed of increasing numbers of responders compared with non-responders on the level of the individual cells. Over the past decade, an ever-increasing number of models of the ERK cascade have been developed, growing in both size and complexity through the years. Models now routinely incorporate growth-factor receptors and the plethora of adaptor proteins that can bind to them and subsequently activate the ERK cascade. Currently, there are over 30 mathematical models that in some way incorporate the ERK cascade (Figure 5). These models have been used to investigate various aspects of the biological behaviour of this system, such as bistable feedback loops [24,33], oscillations [34], feedback inhibition [35], autocrine loops [36,37], scaffold proteins [38,39], feedback effects [40], temperature-dependence [41], receptor internalization [42], signal specificity [43], receptor expression [44], robustness [45], cross-talk [46], receptor trafficking [47,48], memory [49], bistability and hysteresis [50], Ras activation [51], receptor regeneration [52], receptor comparison [53] and temporal dynamics [54] (for a recent review of the relationships between some of these ERK models, see [55], or, for more general reviews of models of cell-signalling pathways, see [56–58]).
Figure 5. Timeline of ERK models.

This diagram is a timeline of mathematical models that, in some way, incorporate the ERK cascade. Models are represented as ovals labelled with the name of the first author and located above the year in which they were published. White ovals represent models of the core ERK cascade, whereas grey ovals represent larger models generally, including growth-factor receptors, adaptor proteins as well as the ERK cascade itself. Models highlighted in black are the models we have selected for discussion in detail below (for brevity, only the first author is named). 1996: Huang [29]; 1997: Burack [30], Ferrell [31]; 1998: Ferrell [32]; 1999: Bhalla [24], Kholodenko [60]; 2000: Brightman [35], Kholodenko [34], Levchenko: [38]; 2001: Asthagiri [40], González [88]; 2002: Bhalla [33], Moehren [41], Schoeberl [42], Shvartsman [36], Somsen [39], Swain [43]; 2003: Aksan [44], Hatakeyama [46], Hendriks [47], Resat [48], Bluthgen [45], Cho [89], Xiong [49]; 2004: Maly [37], Markevich [51], Oliveira [78], Qiu [52], Yamada [53], Chapman [90], Markevich [50]; 2005: Aksan [91], Perez-Jimenez [92], Oney [93], Sasagawa [54].
The most common growth factor receptor that is currently incorporated into models of the ERK cascade is the EGFR (EGF receptor) (for a recent review of models of the EGFR system itself, see [59]). This is because the EGFR system has been well-studied, is present at substantial levels in various cell types, and good antibodies and molecular reagents are widely available, enabling a range of quantitative studies to be performed. We have selected three popular models of the ERK cascade encompassing the EGFR system for discussion in detail below; we review what each model considers and, more importantly, what biological insights and predictions they have led to. Our selection of models is a good representation of the existing models; they are spread over the timeline, are ODE-based and represent the same biological system and are therefore directly comparable (additional information on the models, including links to simulation files, is available at http://www.brc.dcs.gla.ac.uk/~rorton/mapk/).
Model 1: Kholodenko et al. [60]
In 1999, Kholodenko et al. [60] developed an ODE-based mathematical model of the EGFR signalling network to investigate the short-term pattern of cellular responses to EGF in isolated rat hepatocytes. The model consists of 25 reactions involving 23 different species (Figure 6) and includes three adaptor proteins that can directly interact with phosphotyrosine residues on EGFR [namely Shc (Src homology and collagen homology), Grb2 (growth-factor-receptor-bound protein 2) and PLCγ (phospholipase Cγ)]. The kinetic parameters in the model were based on the scientific literature and/or derived from basic physical-chemical quantities. In order to effectively validate the model before analysis, a number of ‘wet’ laboratory experiments were performed such as time courses of EGFR phosphorylation and EGF-induced tyrosine phosphorylation of adaptor proteins. The simulation was then compared with these data to show that the model gives a good fit to the experimentally observed time courses.
Figure 6. Kholodenko et al. [60] model diagram.

This schematic representation of a model of EGFR signalling mediated by adaptor and target proteins is taken from Figure 1 of [60] and is reproduced with the permission of the American Society for Biochemistry and Molecular Biology. © 1999.
Analysis of the model showed that the rapid, and short-lived, pattern of EGFR phosphorylation can be explained by the fact that bound adaptor proteins protect receptor phosphotyrosine residues against dephosphorylation by tonically active phosphatases, rather than having to invoke the activation of tyrosine phosphatases by the receptor [61]. The protection of phosphotyrosine residues from dephosphorylation is transient, as bound adaptors turn over and allow dephosphorylation of receptors. This kinetic model also explains why the levels of phosphorylated adaptors such as Shc, which becomes phosphorylated when recruited to the receptor, stay high even when receptor phosphorylation already has declined. Sensitivity analysis of the model showed that the dynamics of the EGFR signalling pathway appeared to be robust to significant changes in many of the rate constants of the protein interactions involved. Interestingly, however, the time course of phosphorylation/activation responses to EGF appeared to be more sensitive to variations in the relative concentrations of adaptor proteins than to most variations of the kinetic constants. In particular, the Shc/Grb2 ratio was suggested to be an important controlling factor of the kinetics of the EGFR signalling response. This finding can be explained by the competition between adaptor molecules during the formation of multiprotein signalling complexes. It also highlights the fact that changes in the expression levels of adaptor proteins are potent modulators of EGFR signalling, providing a rational basis for the observation that many adaptor proteins, when overexpressed, or stabilized through mutations, can usurp the EGFR mitogenic signalling pathways efficiently and act as oncogenes. Obviously, this finding also has implications for normal physiology, because various cell types and also cells in different functional states can show considerable variation in the abundance of signalling proteins.
Technically this model does not include the core ERK cascade of Raf, MEK and ERK in its description, as it only goes down as far as the Ras guanine-nucleotide exchange factor SOS. However, this model was a pacemaker for the field in several ways. It included the feedback between theoretical prediction and experimental validation, which now is deemed essential for modern systems biology. It also was one of the first models to incorporate the EGFR with its associated adaptor proteins, and it does predict a transient recruitment of SOS to EGFR at the plasma membrane where Ras is located. This transient recruitment of SOS is therefore predicted to give rise to a transient activation of Ras and the ERK cascade, as expected for an EGF response. No wonder, then, that this model has been used as a basis for many other models of the EGFR system which do include the core ERK cascade (see, for example [42,46]).
Model 2: Brightman and Fell [35]
In 2000, Brightman and Fell [35] developed an ODE-based mathematical model of the EGF signal-transduction pathway in PC12 cells to investigate the factors influencing the kinetics of ERK cascade activation. Their model consisted of 30 reactions involving 29 species (Figure 7) and includes a self-contained module of the activation and internalization of EGFRs induced by EGF. Through the phosphorylation of Shc, activated receptors can then initiate an intracellular signal-transduction pathway that results in the activation of Ras and, ultimately, a cytosolic ERK cascade comprising Raf, MEK and ERK. Feedback regulation of the pathway is mediated by the inhibitory phosphorylation of SOS, which causes the dissociation of the Shc–Grb2–SOS complex. The kinetic constants of reactions and initial concentrations of species were largely based on a range of measured or estimated values published in the existing scientific literature.
Figure 7. Brightman and Fell [35] model diagram.

This schematic representation of the Brightman and Fell model of EGF signal transduction is taken from Figure 1 of [35]. Reprinted by permission of the Federation of European Biochemical Societies. © 2000. In this schema, GS represents the Grb2–SOS complex.
The analysis of this model indicated that negative feedback inhibition of the ERK cascade was the most important factor in determining whether the cascade activation was transient or sustained, and that differences in feedback regulation were likely to underlie the characteristic patterns of EGF- and NGF-induced ERK activation in PC12 cells. In the model, EGF initially activates Ras and the ERK cascade via formation of the Shc–Grb2–SOS signalling complex. This signal is rapidly terminated through the negative feedback phosphorylation of SOS, resulting in the dissociation of the Shc–Grb2–SOS complex. However, the analysis showed that the dephosphorylation of SOS (reaction number 28 in Figure 5) was one of the most important steps in determining the duration of the signal, and that only a 40-fold increase in the rate of this reaction generated a time course of ERK cascade activation similar to that observed when PC12 cells are stimulated with NGF rather than EGF. This suggests that NGF, but not EGF, could enhance phosphatase activity towards phosphorylated SOS, resulting in sustained signalling through the Shc–Grb2–SOS complex and therefore a sustained activation of MEK and ERK. This model drew attention to the importance of the regulation of the Ras family proteins as critical regulators of the kinetics of ERK signalling. An elegant and elaborate analysis confirming and significantly extending these results was recently presented [54], showing that the activation kinetics of Ras and its relative, Rap1, can account for the differences in ERK activation in response to EGF as compared with NGF.
Model 3: Schoeberl et al. [42]
In 2002, Schoeberl et al. [42] developed an ODE-based mathematical model describing the dynamics of the EGF signal-transduction pathway to investigate the effects of receptor internalization on the ERK cascade, and also the signal–response relationship between the binding of EGF to its receptor at the cell surface and the activation of downstream proteins in the signalling cascade. Their model consists of 125 reactions involving 94 species (Figure 8), advancing model building by including two principal pathways of Ras activation, Shc-dependent and Shc-independent, as well as EGFR internalization by endocytosis. Receptor internalization is comprehensively represented in the model, with receptor species able to become internalized via two distinct routes. The majority of kinetic parameters were based on values published in the scientific literature, and initial concentrations were either compiled from the literature or based on laboratory experiments.
Figure 8. Schoeberl et al. [42] model diagram.

This schematic representation of the Schoeberl et al. model of the EGF-receptor-induced ERK kinase cascade is taken from Figure 1 of [42] and is reproduced with the permission of Nature Biotechnology (http://www.nature.com/). © 2002.
This model is one of the most comprehensive available, as it includes a large range of dynamic processes. A main conclusion of the modelling is that it is the initial rate of change of receptor activation that determines the cellular response to EGF. On varying the concentration of EGF, analysis of the model suggested that the cell maintains a high sensitivity over a relatively broad EGF concentration range and that the initial velocities of EGFR activation, rather than the peak maxima, are important for signal propagation. The model was also used to investigate the roles of internalized and cell-surface receptors in generating a cellular response, leading to the conclusion that EGFR internalization has a dual role: (1) signal attenuation by protection from prolonged external EGF stimulation at high EGF concentrations; and (2) signal amplification after internalization at low EGF concentrations. The analysis of sensitivity showed that the model was robust to variation in the parameters and initial conditions.
MODEL COMPARISON
It is noteworthy that, although the three models discussed above are describing the same biochemical system, namely the EGF-regulated ERK pathway, they are all different and yet they all seem to be able to explain the observed data and also make interesting predictions about the system's behaviour. This leads to the inevitable question of how is it possible for different models of the same system to all be both correct and reliable? This is a very important question, as the implications are enormous. Does it condemn modelling to the realm of reproducible artefacts, or does it demonstrate that modelling can give valid answers even in the light of different approaches, incompleteness and imperfect data? We do not know the definite answer yet, but all indications point to the latter possibility. Although the three models do differ, they do not do so to a great extent, and many of the differences can be accounted for by simplifications and where the model boundaries lie; a direct comparison of the size and features of the three models is presented in Table 1.
Table 1. Comparison of the size and features of the selected models.
This Table contains a direct comparison between the three selected models: Kholodenko et al. [60], Brightman and Fell [35] and Schoeberl et al. [42]. The comparison criteria are defined as follows (more details can be found in the text below): cell line, the cell line the model is based on; reactions, the number of reactions in the model; species, the number of biochemical species in the model; receptor complex strategy, whether adaptor proteins have to bind to the EGFR for the signal to propagate; receptor internalization, the process of internalization of receptors from the plasma membrane; receptor degradation, the process of degradation of internalized receptor; Shc-dependent pathway, the pathway comprising Shc, Grb2 and SOS leading to the activation of Ras; Shc-independent pathway, the pathway comprising Grb2 and SOS leading to the activation of Ras (i.e. Shc not required); PLCγ pathway, the pathway comprising the reactions of the adaptor protein PLCγ; core ERK cascade, the Raf, MEK and ERK cascade; negative-feedback loop, the negative feedback phosphorylation of SOS by ERKPP.
| Size or feature | Kholodenko et al. [60] | Brightman and Fell [35] | Schoeberl et al. [42] |
|---|---|---|---|
| Cell line | Rat hepatocytes | PC12 | HeLa |
| Reactions | 25 | 30 | 125 |
| Species | 23 | 29 | 94 |
| Receptor complex strategy | Yes | No | Yes |
| Receptor internalization | No | Yes | Yes |
| Receptor degradation | No | No | Yes |
| Shc-dependent pathway | Yes | Yes | Yes |
| Shc-independent pathway | Yes | No | Yes |
| PLCγ pathway | Yes | No | No |
| Core ERK cascade | No | Yes | Yes |
| Negative-feedback loop | No | Yes | No |
One interesting aspect is that the Brightman and Fell [35] model utilizes a different strategy of dealing with activated receptors when compared with the Kholodenko et al. [60] and Schoeberl et al. [42] models. In the Brightman and Fell [35] model, activated receptors simply catalyse the phosphorylation of Shc. However, the other two models utilize a more realistic receptor complex strategy where adaptor proteins such as Shc have to bind to activated receptors and stay bound in order for the signal to propagate. These two different strategies have led to different predictions as to the basis of the transient signal response to EGF. In the Brightman and Fell [35] model, Shc is phosphorylated by activated receptors, enabling it to bind Grb2 and SOS before activating Ras and the core ERK cascade; the Shc–Grb2–SOS complex is a functional complex and does not need to bind to the receptor. The transient nature of the EGF response is in part caused by the negative-feedback phosphorylation of SOS by ERKPP (activated ERK), resulting in the dissociation of the Shc–Grb2–SOS complex and therefore stopping the activation of Ras and the core ERK cascade. In contrast, in the Kholodenko [60] model, Shc followed by Grb2 and SOS bind rapidly to activated receptors, which would enable the activation of the membrane-bound Ras and the core ERK cascade. However, the receptor-bound Shc–Grb2–SOS complex then dissociates and there is a build up of this unbound complex. As unbound receptors are exposed to phosphatases and are rapidly deactivated, a transient response can result without the need for a negative feedback loop.
The boundary of a model is essentially the point or points at which the model stops; the model does not consider and therefore define the biological processes beyond these points. The decision of where the boundary lies is typically determined by what biological processes and questions the model is intended to investigate and answer. A good illustration of model boundaries is given by the comparison of the Kholodenko et al. [60] model with the Schoeberl et al. [42] one. Both models have a similar general structure, as they essentially have the same Shc-dependent and Shc-independent pathways and utilize a receptor-complex strategy. However, the Kholodenko et al. [60] model includes the PLCγ pathway, whereas the Schoeberl et al. [42] model includes the ERK cascade and receptor internalization. This is because the Kholodenko et al. [60] model was designed to investigate the short-term pattern of cellular responses to EGF through EGFR and its adaptor proteins; therefore only the receptor proximal events were included. In contrast, the Schoeberl et al. [42] model was designed to investigate the downstream effects of EGFR activation as well as the effects of receptor internalization on the ERK cascade This illustrates an experimental-design strategy for reductionism that is familiar to the biologist: depending on the question that is the focus of the investigation, the experimental system is simplified in the way that is most appropriate to address the question at hand.
Simplifications in a model can come in many forms and typically involve the simplification of biological processes or events to reduce the number of reactions needed in the model while still being able to represent the biological processes well enough to explain the observed data. The classic example of a simplification is the activation of Raf by Ras-GTP. Ras-GTP recruits Raf from the cytosol to the plasma membrane, where it is activated through a still not completely known process that involves interaction with adaptor proteins, lipids and changes in phosphorylation. However, this complex process has been effectively represented in many models of the ERK pathway as a simple two-step process:
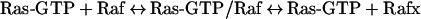 |
Rafx represents the active form of Raf. Other simplifications can be found when examining, for example, the Schoeberl et al. [42] model:
Only one member of the EGFR family is considered
EGFR dimers are considered as single molecules
The binding of the adaptor proteins Shc and Grb2 to EGFR is assumed to be competitive
GAP must be bound to the EGFR before any other adaptor proteins can bind
There may also be some biological inaccuracies in the models discussed. In the Brightman and Fell [35] model, singly phosphorylated MEK is able to phosphorylate ERK; this is probably irrelevant, as singly phosphorylated MEK species have not been observed. Moreover, mutating one of the two phosphorylation sites in MEK abrogates activation, suggesting that only doubly phosphorylated MEK is active [62]. In the Schoeberl et al. [42] model there is an apparent inactive active form of Ras called Ras-GTP* (species 43/71 in Figure 8). The role of this Ras-GTP* is to limit the number of Raf molecules that Ras-GTP can activate to one. However, this is probably an artificial and incorrect assumption, as Ras is either active in its GTP-bound state or inactive in its GDP-bound state. There is no ‘in-between’ situation, and there is no evidence that the number of Raf molecules that Ras-GTP can activate is limited (for a review of Raf activation, see [63]). Although these are not major errors, it could be viewed as a concern that models with apparent errors can still explain the observed data and be used to make valid predictions about the biological system. However, as robustness seems to be an inherent and ubiquitous property of biological systems [64], the robustness of the models in tolerating small errors may simply reflect the biological property. From a pragmatic point of view, it could be viewed as an advantage, as models, despite not being perfect, can be used to suggest interesting new hypotheses and explanations for the observed data that challenge our current understanding, which is by no means complete. Importantly, these predictions need to be verified experimentally in the laboratory.
After a model has been completed, it can easily be modified or expanded upon and used in the development of new models of the same or related biological systems. For example, the Kholodenko et al. [60] model has been used as a basis for many other models of the EGFR system [41,46,48,51]. The Brightman and Fell [35] model has since been analysed further by Babu et al. [65], who investigated the effects of a number of MEK inhibitors in this system, and it was also used in the development of other models of the ERK cascade [44,52]. The Schoeberl et al. [42] model has since been analysed further in a number of studies [66–69]; for example, Gong and Zhao [66] investigated the relevance of the Shc-dependent and Shc-independent pathways; it has also been combined with data on metalloprotease activation to build a model of autocrine signal transduction by cancer cells exposed to ionizing radiation [70].
ALTERNATIVE RECEPTOR SYSTEMS
Although the EGFR is the most common growth-factor-receptor system modelled together with the ERK cascade, other growth-factor receptors have also been successfully modelled. Bhalla et al. [33] in 2002 developed an ODE-based model of the PDGF (platelet-derived growth factor)-activated ERK cascade, revealing novel and interesting design principles of this network, in particular that ERK activation can operate in different states depending on the history of the cell. The first application of PDGF to a naïve cell induces a bistable, switch-like ERK activation response, where even a brief stimulus results in sustained ERK activity. As part of this response, the expression of MKPs is induced. Thus, when the cells are restimulated with PDGF after the initial ERK activation has returned to baseline, there is a higher level of MKPs in the cell, causing ERK activation to proceed in a monostable fashion with the ERK activity increasing proportionally with the dose of PDGF. In 2004, Qiu et al. [52] developed an ODE-based model of the NGF-activated ERK cascade and suggested that the sustained behaviour of the response was mainly due to a continual regeneration of NGF receptors. Also in 2004, Yamada et al. [53] developed ODE-based models of the EGF and FGF (fibroblast growth factor)-activated ERK cascade and proposed that the protein FRS2 (FGF-receptor substrate 2) plays a key role in generating the sustained behaviour of the FGF response by recruiting more SOS to the plasma membrane; furthermore, they found that the negative-feedback system in the model did not profoundly affect the time course of ERK activation. In one of the most recent (2005) models, Sasagawa et al. [54] developed an extensive ODE-based model of the EGF- and NGF-activated ERK cascade in PC12 cells. This model is among the most comprehensive to date, as it includes both the Ras and Rap1 pathways to ERK activation and combines theoretical predictions with experimental validation. The model was used to investigate how EGF and NGF encode transient and sustained dynamics of ERK activation respectively. A salient finding was that the transient activation of ERK depended on the rate of receptor activation rather than the concentration of the growth factors. In contrast, sustained ERK activation by NGF depended on the final concentration of NGF, but not on the temporal rate of increase in NGF concentration. These diverse response modes are due to differences in Ras and Rap1 inactivation. Ras, which mediates transient ERK activation, is inactivated by the receptor-induced recruitment of Ras-GAP to the membrane, resulting in a stringent temporal regulation of Ras activity. In contrast, Rap1, which in this model is responsible for sustained ERK activation via stimulation of B-Raf, is inactivated by Rap-GAP, which is not regulated by growth factors, but functions constitutively. Thus the activation of Rap1 becomes a function of the NGF concentration. In this model the Ras and Rap1 pathways capture the temporal rate of increase and concentration of growth factors and encode these distinct properties into transient and sustained ERK activation respectively.
ALTERNATIVE MODELLING METHODS
Although ODEs are commonly used to model biological systems such as the ERK cascade, they have one major drawback, and that is they are reliant on high-frequency sampling and absolute parameter data being available, such as detailed kinetic rates and absolute initial concentrations. However, a lot of the data generated by biologists, including data generated from high-throughput techniques, are not directly amenable to modelling, as they often contains sparse time series, are qualitative rather than quantitative, and show relative changes rather than changes in absolute concentrations. Furthermore, as there is only very little standardization of measurements, data from different laboratories usually can only be compared in a semiquantitative or qualitative fashion [16]. However, although high frequency and absolute data is required if one wants to produce a near-exact replica of the experimental system, it is often not necessary to know every single parameter with high accuracy, due to issues such as variable sensitivity. Therefore ODE-based models can readily be used to assess whether the system is capable of showing specific qualitative features such as oscillations. In addition, there are also a number of techniques to estimate missing parameter data in a model (see, for example, [18]), which typically work by varying the missing parameters values until the expected behaviour is obtained. An alternative method, developed by Brown et al. [71] in 2004, used an ensemble approach to model complex signalling networks, namely the NGF- and EGF-activated ERK cascade; these models also included both the Ras and Rap1 pathways. Instead of using kinetic parameters, which are commonly not available, the ensemble method was used to match the model to experimental time courses of the activities of signalling molecules, which generates an ensemble of weighted parameters which can then be used to analyse the model. This model was used to evaluate the importance of different regulatory loops in generating a sustained activation of ERK. Furthermore, this approach suggested that only a small fraction of parameter combinations are likely to be well constrained and that the few well-constrained parameters reveal critical focal points in the signalling network. Two additional approaches are FBA (Flux Balance Analysis; [72]) and MCA (Metabolic Control analysis; [73]). FBA is an approach to constrain a metabolic network based on the stoichiometry of the metabolic reactions and does not require kinetic information. MCA is a quantitative sensitivity analysis of fluxes and concentrations; the relative control exerted by each step on a system variable is measured by applying a perturbation to the step and measuring the effect on the variable of interest after the system has settled to a new steady state. It is important to note that FBA and MCA are essentially analyses of the steady state and are therefore less suited to the dynamic aspects of signal transduction; however, MCA has recently been extended to the dynamics of signal transduction [74].
There are also a number of alternative approaches to ODEs that can be used to model and analyse biological systems. In 2003, Resat et al. [48] developed one of the largest models of the EGFR system to date using a probability weighted–dynamic Monte Carlo stochastic simulation. They developed an integrated model of both the trafficking and signalling components of the EGFR system that consisted of hundreds of distinct endocytic compartments and about 13000 reactions that occurred over a broad spatio-temporal range. The signalling component of this model consisted of the ODE model developed by Kholodenko et al. [60], highlighting that ODE-based models can be utilized by other modelling techniques. Pi-calculus [75] has successfully been used to model biological systems, with molecules and their domains represented by computational processes, and reactions by communication and rearranging the communication channels. Further developments in this area include stochastic Pi-calculus [76] and the hybrid system BioSPi (http://www.wisdom.weizmann.ac.il/~biospi/).
Petri nets are another class of innovative modelling notations used to analyse biological systems [77]. In this approach, biological networks are represented by intuitively readable, but strictly formalized, graphical diagrams (of molecules and connecting reactions) that are directly subjected to algebraic analysis. For example, in 2004, Oliveira et al. [78] constructed an algebraic–combinatorial model of the SOS compartment of the EGFR system by using a Petri-net approach. Extended Petri-net approaches that incorporate both discrete and continuous state transitions (hybrid function Petri nets) combine the powerful simulation capabilities of differential equations with the formal logical analysis available in the Petri-net computations [79–83]. The Biochemical Abstract Machine BIOCHAM [84,85] is a programming environment for modelling biochemical systems, making simulations and querying the model in temporal logic, which allows the behaviour of a model to be checked against biological predictions expressed in logical statements. The interface is based on a simple language for representing biochemical networks. BIOCHAM provides mechanisms to reason about the ‘reachability’ of certain states, the existence of stable states and some types of temporal behaviour (e.g. oscillations). An alternative algebraic modelling and analysis approach was proposed by Calder et al. [86], where the stochastic process algebra PEPA (Performance Evaluation Process Algebra) was used to model the ERK signalling pathway. The main advantage of algebraic modelling techniques such as these lies in their ability to systematically reason over structural properties such as the interaction of sub-networks (for example cross-talk) and the behavioural equivalence of different networks.
FUTURE DIRECTIONS
Signalling pathways have traditionally been drawn as separate linear entities, reflecting the history of how they were discovered rather than their functional context. However, signalling pathways are extensively interconnected and embedded in networks with common protein components and a multitude of links and crosstalk between pathways. Owing to the complexity of these networks, computational methods are required in order to explain in detail how they function and predict possible behaviours. Over recent years, the computational or mathematical modelling of biological systems has become increasingly valuable and can provide useful information to understand their behaviour. The ERK cascade is currently one of the most popular systems to be modelled, as it is one of the most intensely studied signalling pathways and has also been implicated in various diseases. Currently, there are many differential-equation-based models of the ERK cascade that have been used to investigate various aspects of its biological behaviour, and these have led to some novel insights and predictions as to how this system functions. However, no two models appear to be the same topologically or dynamically, as they consider different biological processes and components, use different representations and have different kinetic properties. Over the years, models have increased in both size and complexity. The first (1996) model of the ERK cascade [29] only considered the core cascade itself, whereas the most recent (2005) model [54] considered both the EGF and NGF receptors, numerous adaptor proteins and both the Ras and Rap1 pathways leading to the activation of the ERK cascade, as well as numerous feedback loops.
Coupled with the increase in the number, size and complexity of mathematical models is an increase in the number of techniques and software tools available to simulate and analyse them. Although differential equation methods are currently the most widely used, there are a number of good alternatives available and a number of promising alternatives in development. The adoption of some sort of unifying standard, such as SBML, for all published models of biological systems would now be a welcome development, enabling models to be shared and evaluated with ease and thus eliminating the need to painstakingly recreate models based on static tables of supplementary data and compared with simulation graphs in papers. To this end, the BioModels database (www.ebi.ac.uk/biomodels; [87]) was recently launched and aims to be a curated database for the deposition of models of biological systems in an SBML format. Similarly, the recently founded Receptor Tyrosine Kinase (RTK) Consortium (http://www.rtkconsort.org) is an international research effort to advance the understanding of RTK signalling systems, including the MAPK pathways, through a combination of mathematical modelling and quantitative biological experimentation. Thus, with the first steps accomplished, we can now expect quantitative biology to rapidly gain momentum.
Acknowledgments
We acknowledge support through the Department of Trade and Industry Beacons Project ‘BPS: Biochemical Pathway Simulator’.
References
- 1.Cobb M. H. MAP kinase pathways. Prog. Biophys. Mol. Biol. 1999;71:479–500. doi: 10.1016/s0079-6107(98)00056-x. [DOI] [PubMed] [Google Scholar]
- 2.Widmann C., Gibson S., Jarpe M. B., Johnson G. L. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol. Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- 3.Chang L., Karin M. Mammalian MAP kinase signalling cascades. Nature (London) 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 4.Farooq A., Zhou M. M. Structure and regulation of MAPK phosphatases. Cell. Signalling. 2004;16:769–779. doi: 10.1016/j.cellsig.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 5.Murphy L. O., MacKeigan J. P., Blenis J. A network of immediate early gene products propagates subtle differences in mitogen-activated protein kinase signal amplitude and duration. Mol. Cell. Biol. 2004;24:144–153. doi: 10.1128/MCB.24.1.144-153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hindley A., Kolch W. Extracellular signal regulated kinase (ERK)/mitogen activated protein kinase (MAPK)-independent functions of Raf kinases. J. Cell Sci. 2002;115:1575–1581. doi: 10.1242/jcs.115.8.1575. [DOI] [PubMed] [Google Scholar]
- 7.Stork P. J. ERK signaling: duration, duration, duration. Cell Cycle. 2002;1:315–317. doi: 10.4161/cc.1.5.145. [DOI] [PubMed] [Google Scholar]
- 8.Murphy L. O., Smith S., Chen R. H., Fingar D. C., Blenis J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat. Cell Biol. 2002;4:556–564. doi: 10.1038/ncb822. [DOI] [PubMed] [Google Scholar]
- 9.Tanoue T., Nishida E. Docking interactions in the mitogen-activated protein kinase cascades. Pharmacol. Ther. 2002;93:193–202. doi: 10.1016/s0163-7258(02)00188-2. [DOI] [PubMed] [Google Scholar]
- 10.Pouyssegur J., Lenormand P. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Eur. J. Biochem. 2003;270:3291–3299. doi: 10.1046/j.1432-1033.2003.03707.x. [DOI] [PubMed] [Google Scholar]
- 11.Houslay M. D., Kolch W. Cell-type specific integration of cross-talk between extracellular signal-regulated kinase and cAMP signaling. Mol. Pharmacol. 2000;58:659–668. [PubMed] [Google Scholar]
- 12.Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem. J. 2000;351:289–305. [PMC free article] [PubMed] [Google Scholar]
- 13.Bernards A. GAPs galore! A survey of putative Ras superfamily GTPase activating proteins in man and Drosophila. Biochim. Biophys. Acta. 2003;1603:47–82. doi: 10.1016/s0304-419x(02)00082-3. [DOI] [PubMed] [Google Scholar]
- 14.Yung Y., Dolginov Y., Yao Z., Rubinfeld H., Michael D., Hanoch T., Roubini E., Lando Z., Zharhary D., Seger R. Detection of ERK activation by a novel monoclonal antibody. FEBS Lett. 1997;408:292–296. doi: 10.1016/s0014-5793(97)00442-0. [DOI] [PubMed] [Google Scholar]
- 15.Davie J. R., Spencer V. A. Signal transduction pathways and the modification of chromatin structure. Prog. Nucleic Acid Res. Mol. Biol. 2001;65:299–340. doi: 10.1016/s0079-6603(00)65008-0. [DOI] [PubMed] [Google Scholar]
- 16.Kolch W., Calder M., Gilbert D. When kinases meet mathematics: the systems biology of MAPK signalling. FEBS Lett. 2005;579:1891–1895. doi: 10.1016/j.febslet.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 17.Press W. H., Teukolsky S. A., Vetterling W. T., Flannery B. P. Cambridge, U.K.: Cambridge University Press; 2002. Numerical Recipes in C: The Art of Scientific Computing. [Google Scholar]
- 18.Mendes P., Kell D. Non-linear optimization of biochemical pathways: applications to metabolic engineering and parameter estimation. Bioinformatics. 1998;14:869–883. doi: 10.1093/bioinformatics/14.10.869. [DOI] [PubMed] [Google Scholar]
- 19.Swameye I., Muller T. G., Timmer J., Sandra O., Klingmuller U. Identification of nucleocytoplasmic cycling as a remote sensor in cellular signaling by databased modeling. Proc. Natl. Acad. Sci. U.S.A. 2003;100:1028–1033. doi: 10.1073/pnas.0237333100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mendes P. GEPASI: a software package for modelling the dynamics, steady states and control of biochemical and other systems. Comput. Appl. Biosci. 1993;9:563–571. doi: 10.1093/bioinformatics/9.5.563. [DOI] [PubMed] [Google Scholar]
- 21.Tomita M., Hashimoto K., Takahashi K., Shimizu T. S., Matsuzaki Y., Miyoshi F., Saito K., Tanida S., Yugi K., Venter J. C., Hutchison C. A., 3rd E-CELL: software environment for whole-cell simulation. Bioinformatics. 1999;15:72–84. doi: 10.1093/bioinformatics/15.1.72. [DOI] [PubMed] [Google Scholar]
- 22.Schaff J., Fink C. C., Slepchenko B., Carson J. H., Loew L. M. A general computational framework for modeling cellular structure and function. Biophys. J. 1997;73:1135–1146. doi: 10.1016/S0006-3495(97)78146-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bower J. M., Beeman D., editors. 2nd edn. New York: Springer-Verlag; 1998. The Book of GENESIS: Exploring Realistic Neural Models with the GEneral NEural SImulation System. [Google Scholar]
- 24.Bhalla U. S., Iyengar R. Emergent properties of networks of biological signaling pathways. Science. 1999;283:381–387. doi: 10.1126/science.283.5400.381. [DOI] [PubMed] [Google Scholar]
- 25.Sauro H. M., Hucka M., Finney A., Wellock C., Bolouri H., Doyle J., Kitano H. Next generation simulation tools: the Systems Biology Workbench and BioSPICE integration. Omics. 2003;7:355–372. doi: 10.1089/153623103322637670. [DOI] [PubMed] [Google Scholar]
- 26.Pettinen A., Aho T., Smolander O. P., Manninen T., Saarinen A., Taattola K. L., Yli-Harja O., Linne M. L. Simulation tools for biochemical networks: evaluation of performance and usability. Bioinformatics. 2005;21:357–363. doi: 10.1093/bioinformatics/bti018. [DOI] [PubMed] [Google Scholar]
- 27.Hucka M., Finney A., Sauro H. M., Bolouri H., Doyle J. C., Kitano H., Arkin A. P., Bornstein B. J., Bray D., Cornish-Bowden A., et al. The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics. 2003;19:524–531. doi: 10.1093/bioinformatics/btg015. [DOI] [PubMed] [Google Scholar]
- 28.Olivier B. G., Snoep J. L. Web-based kinetic modelling using JWS Online. Bioinformatics. 2004;20:2143–2144. doi: 10.1093/bioinformatics/bth200. [DOI] [PubMed] [Google Scholar]
- 29.Huang C. Y., Ferrell J. E., Jr Ultrasensitivity in the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. U.S.A. 1996;93:10078–10083. doi: 10.1073/pnas.93.19.10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burack W. R., Sturgill T. W. The activating dual phosphorylation of MAPK by MEK is nonprocessive. Biochemistry. 1997;36:5929–5933. doi: 10.1021/bi970535d. [DOI] [PubMed] [Google Scholar]
- 31.Ferrell J. E., Jr, Bhatt R. R. Mechanistic studies of the dual phosphorylation of mitogen-activated protein kinase. J. Biol. Chem. 1997;272:19008–19016. doi: 10.1074/jbc.272.30.19008. [DOI] [PubMed] [Google Scholar]
- 32.Ferrell J. E., Jr, Machleder E. M. The biochemical basis of an all-or-none cell fate switch in Xenopus oocytes. Science. 1998;280:895–898. doi: 10.1126/science.280.5365.895. [DOI] [PubMed] [Google Scholar]
- 33.Bhalla U. S., Ram P. T., Iyengar R. MAP kinase phosphatase as a locus of flexibility in a mitogen-activated protein kinase signaling network. Science. 2002;297:1018–1023. doi: 10.1126/science.1068873. [DOI] [PubMed] [Google Scholar]
- 34.Kholodenko B. N. Negative feedback and ultrasensitivity can bring about oscillations in the mitogen-activated protein kinase cascades. Eur. J. Biochem. 2000;267:1583–1588. doi: 10.1046/j.1432-1327.2000.01197.x. [DOI] [PubMed] [Google Scholar]
- 35.Brightman F. A., Fell D. A. Differential feedback regulation of the MAPK cascade underlies the quantitative differences in EGF and NGF signalling in PC12 cells. FEBS Lett. 2000;482:169–174. doi: 10.1016/s0014-5793(00)02037-8. [DOI] [PubMed] [Google Scholar]
- 36.Shvartsman S. Y., Hagan M. P., Yacoub A., Dent P., Wiley H. S., Lauffenburger D. A. Autocrine loops with positive feedback enable context-dependent cell signaling. Am. J. Physiol. Cell Physiol. 2002;282:C545–C559. doi: 10.1152/ajpcell.00260.2001. [DOI] [PubMed] [Google Scholar]
- 37.Maly I. V., Wiley H. S., Lauffenburger D. A. Self-organization of polarized cell signaling via autocrine circuits: computational model analysis. Biophys. J. 2004;86:10–22. doi: 10.1016/s0006-3495(04)74079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levchenko A., Bruck J., Sternberg P. W. Scaffold proteins may biphasically affect the levels of mitogen-activated protein kinase signaling and reduce its threshold properties. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5818–5823. doi: 10.1073/pnas.97.11.5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Somsen O. J., Siderius M., Bauer F. F., Snoep J. L., Westerhoff H. V. Selectivity in overlapping MAP kinase cascades. J. Theor. Biol. 2002;218:343–354. doi: 10.1006/jtbi.2002.3082. [DOI] [PubMed] [Google Scholar]
- 40.Asthagiri A. R., Lauffenburger D. A. A computational study of feedback effects on signal dynamics in a mitogen-activated protein kinase (MAPK) pathway model. Biotechnol. Prog. 2001;17:227–239. doi: 10.1021/bp010009k. [DOI] [PubMed] [Google Scholar]
- 41.Moehren G., Markevich N., Demin O., Kiyatkin A., Goryanin I., Hoek J. B., Kholodenko B. N. Temperature dependence of the epidermal growth factor receptor signaling network can be accounted for by a kinetic model. Biochemistry. 2002;41:306–320. doi: 10.1021/bi011506c. [DOI] [PubMed] [Google Scholar]
- 42.Schoeberl B., Eichler-Jonsson C., Gilles E. D., Muller G. Computational modeling of the dynamics of the MAP kinase cascade activated by surface and internalized EGF receptors. Nat. Biotechnol. 2002;20:370–375. doi: 10.1038/nbt0402-370. [DOI] [PubMed] [Google Scholar]
- 43.Swain P. S., Siggia E. D. The role of proofreading in signal transduction specificity. Biophys. J. 2002;82:2928–2933. doi: 10.1016/S0006-3495(02)75633-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aksan I., Kurnaz M. L. A computer-based model for the regulation of mitogen activated protein kinase (MAPK) activation. J. Recept. Signal Transduct. Res. 2003;23:197–209. doi: 10.1081/rrs-120025203. [DOI] [PubMed] [Google Scholar]
- 45.Bluthgen N., Herzel H. How robust are switches in intracellular signaling cascades? J. Theor. Biol. 2003;225:293–300. doi: 10.1016/s0022-5193(03)00247-9. [DOI] [PubMed] [Google Scholar]
- 46.Hatakeyama M., Kimura S., Naka T., Kawasaki T., Yumoto N., Ichikawa M., Kim J. H., Saito K., Saeki M., Shirouzu M., et al. A computational model on the modulation of mitogen-activated protein kinase (MAPK) and Akt pathways in heregulin-induced ErbB signalling. Biochem. J. 2003;373:451–463. doi: 10.1042/BJ20021824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hendriks B. S., Opresko L. K., Wiley H. S., Lauffenburger D. Quantitative analysis of HER2-mediated effects on HER2 and epidermal growth factor receptor endocytosis: distribution of homo- and heterodimers depends on relative HER2 levels. J. Biol. Chem. 2003;278:23343–23351. doi: 10.1074/jbc.M300477200. [DOI] [PubMed] [Google Scholar]
- 48.Resat H., Ewald J. A., Dixon D. A., Wiley H. S. An integrated model of epidermal growth factor receptor trafficking and signal transduction. Biophys. J. 2003;85:730–743. doi: 10.1016/s0006-3495(03)74516-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiong W., Ferrell J. E., Jr A positive-feedback-based bistable ‘memory module’ that governs a cell fate decision. Nature (London) 2003;426:460–465. doi: 10.1038/nature02089. [DOI] [PubMed] [Google Scholar]
- 50.Markevich N. I., Hoek J. B., Kholodenko B. N. Signaling switches and bistability arising from multisite phosphorylation in protein kinase cascades. J. Cell Biol. 2004;164:353–359. doi: 10.1083/jcb.200308060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Markevich N. I., Moehren G., Demin O. V., Kiyatkin A., Hoek J. B., Kholodenko B. N. Signal processing at the Ras circuit: what shapes Ras activation patterns? Syst. Biol. 2004;1:104–113. doi: 10.1049/sb:20045003. [DOI] [PubMed] [Google Scholar]
- 52.Qiu D., Mao L., Kikuchi S., Tomita M. Sustained MAPK activation is dependent on continual NGF receptor regeneration. Dev. Growth Differ. 2004;46:393–403. doi: 10.1111/j.1440-169x.2004.00756.x. [DOI] [PubMed] [Google Scholar]
- 53.Yamada S., Taketomi T., Yoshimura A. Model analysis of difference between EGF pathway and FGF pathway. Biochem. Biophys. Res. Commun. 2004;314:1113–1120. doi: 10.1016/j.bbrc.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 54.Sasagawa S., Ozaki Y., Fujita K., Kuroda S. Prediction and validation of the distinct dynamics of transient and sustained ERK activation. Nat. Cell Biol. 2005;7:365–373. doi: 10.1038/ncb1233. [DOI] [PubMed] [Google Scholar]
- 55.Vayttaden S. J., Ajay S. M., Bhalla U. S. A spectrum of models of signaling pathways. Chembiochem. 2004;5:1365–1374. doi: 10.1002/cbic.200400127. [DOI] [PubMed] [Google Scholar]
- 56.Bhalla U. S. Models of cell signaling pathways. Curr. Opin. Genet. Dev. 2004;14:375–381. doi: 10.1016/j.gde.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 57.Sauro H. M., Kholodenko B. N. Quantitative analysis of signaling networks. Prog. Biophys. Mol. Biol. 2004;86:5–43. doi: 10.1016/j.pbiomolbio.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 58.Eungdamrong N. J., Iyengar R. Modeling cell signaling networks. Biol. Cell. 2004;96:355–362. doi: 10.1016/j.biolcel.2004.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wiley H. S., Shvartsman S. Y., Lauffenburger D. A. Computational modeling of the EGF-receptor system: a paradigm for systems biology. Trends Cell Biol. 2003;13:43–50. doi: 10.1016/s0962-8924(02)00009-0. [DOI] [PubMed] [Google Scholar]
- 60.Kholodenko B. N., Demin O. V., Moehren G., Hoek J. B. Quantification of short term signaling by the epidermal growth factor receptor. J. Biol. Chem. 1999;274:30169–30181. doi: 10.1074/jbc.274.42.30169. [DOI] [PubMed] [Google Scholar]
- 61.Saso K., Moehren G., Higashi K., Hoek J. B. Differential inhibition of epidermal growth factor signaling pathways in rat hepatocytes by long-term ethanol treatment. Gastroenterology. 1997;112:2073–2088. doi: 10.1053/gast.1997.v112.pm9178701. [DOI] [PubMed] [Google Scholar]
- 62.Alessi D. R., Saito Y., Campbell D. G., Cohen P., Sithanandam G., Rapp U., Ashworth A., Marshall C. J., Cowley S. Identification of the sites in MAP kinase kinase-1 phosphorylated by p74raf-1. EMBO J. 1994;13:1610–1619. doi: 10.1002/j.1460-2075.1994.tb06424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dhillon A. S., Kolch W. Untying the regulation of the Raf-1 kinase. Arch. Biochem. Biophys. 2002;404:3–9. doi: 10.1016/s0003-9861(02)00244-8. [DOI] [PubMed] [Google Scholar]
- 64.Kitano H. Biological robustness. Nat. Rev. Genet. 2004;5:826–837. doi: 10.1038/nrg1471. [DOI] [PubMed] [Google Scholar]
- 65.Babu C. V., Yoon S., Nam H. S., Yoo Y. S. Simulation and sensitivity analysis of phosphorylation of EGFR signal transduction pathway in PC12 cell model. Syst. Biol. 2004;2:213–221. doi: 10.1049/sb:20045023. [DOI] [PubMed] [Google Scholar]
- 66.Gong Y., Zhao X. Shc-dependent pathway is redundant but dominant in MAPK cascade activation by EGF receptors: a modeling inference. FEBS Lett. 2003;554:467–472. doi: 10.1016/s0014-5793(03)01174-8. [DOI] [PubMed] [Google Scholar]
- 67.Conzelmann H., Saez-Rodriguez J., Sauter T., Bullinger E., Allgower F., Gilles E. D. Reduction of mathematical models of signal transduction networks: simulation-based approach applied to EGF receptor signalling. Syst. Biol. 2004;1:159–169. doi: 10.1049/sb:20045011. [DOI] [PubMed] [Google Scholar]
- 68.Liu G., Swihart M. T., Neelamegham S. Sensitivity, principal component and flux analysis applied to signal transduction: the case of epidermal growth factor mediated signaling. Bioinformatics. 2004 doi: 10.1093/bioinformatics/bti118. [DOI] [PubMed] [Google Scholar]
- 69.Mayawala K., Gelmi C. A., Edwards J. S. MAPK cascade possesses decoupled controllability of signal amplification and duration. Biophys. J. 2004;87:L01–L02. doi: 10.1529/biophysj.104.051888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miller J. H., Zheng F. Large-scale simulations of cellular signaling processes. Parallel Comput. 2004;30:1137–1149. [Google Scholar]
- 71.Brown K. S., Hill C. C., Calero G. A., Myers C. R., Lee K. H., Sethna J. P., Cerione R. A. The statistical mechanics of complex signaling networks: nerve growth factor signaling. Phys. Biol. 2004;1:184–195. doi: 10.1088/1478-3967/1/3/006. [DOI] [PubMed] [Google Scholar]
- 72.Bonarius H. P. J., Schmid G., Tramper J. Flux analysis of underdetermined metabolic networks: The quest for the missing constraints. Trends Biotechnol. 1997;15:308–314. [Google Scholar]
- 73.Kacser H., Burns J. A. The control of flux. Symp. Soc. Exp. Biol. 1973;27:65–104. [PubMed] [Google Scholar]
- 74.Hornberg J. J., Bruggeman F. J., Binder B., Geest C. R., de Vaate A. J., Lankelma J., Heinrich R., Westerhoff H. V. Principles behind the multifarious control of signal transduction. ERK phosphorylation and kinase/phosphatase control. FEBS J. 2005;272:244–258. doi: 10.1111/j.1432-1033.2004.04404.x. [DOI] [PubMed] [Google Scholar]
- 75.Regev A., Silverman W., Shapiro E. Representation and simulation of biochemical processes using Pi-calculus process algebra. Pac. Symp. Biocomput. '01. 2001:459–470. doi: 10.1142/9789814447362_0045. [DOI] [PubMed] [Google Scholar]
- 76.Priami C., Regev A., Silverman W., Shapiro E. Application of a stochastic name passing calculus to representation and simulation of molecular processes. Inform. Process. Lett. 2001;80:25–31. [Google Scholar]
- 77.Pinney J., Westhead D., McConkey G. Petri Net representations in systems biology. Biochem. Soc. Trans. 2003;31:1513–1515. doi: 10.1042/bst0311513. [DOI] [PubMed] [Google Scholar]
- 78.Oliveira J. S., Jones-Oliveira J. B., Dixon D. A., Bailey C. G., Gull D. W. Hyperdigraph–theoretic analysis of the EGFR signaling network: initial steps leading to GTP:Ras complex formation. J. Comput. Biol. 2004;11:812–842. doi: 10.1089/cmb.2004.11.812. [DOI] [PubMed] [Google Scholar]
- 79.Koch I., Heiner M. Qualitative Modelling and Analysis of Biochemical Pathways with Petri Nets: Tutorial Notes; 5th International Conference on Systems Biology, Heidelberg, 9–13 October 2004; 2004. [Google Scholar]
- 80.Heiner M., Koch I., Will J. Model validation of biological pathways using Petri nets, demonstrated for apoptosis. Biosystems. 2004;75:15–28. doi: 10.1016/j.biosystems.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 81.Matsuno H., Doi A., Nagasaki M., Miyano S. Hybrid petri net representation of gene regulatory network. Pac. Symp. Biocomput. '00. 2000:341–352. doi: 10.1142/9789814447331_0032. [DOI] [PubMed] [Google Scholar]
- 82.Popova-Zeugmann L., Heiner M., Koch I. Berlin: Humboldt-Universität; 2004. Modelling and analysis of biochemical networks with time petri nets, Informatik-Berichte der Humboldt-Universität zu Berlin, no. 170; pp. 136–143. [Google Scholar]
- 83.Goss P. J., Peccoud J. Quantitative modeling of stochastic systems in molecular biology by using stochastic Petri nets. Proc. Natl. Acad. Sci. U.S.A. 1998;95:6750–6755. doi: 10.1073/pnas.95.12.6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chabrier N., Fages F. Symbolic model checking of biochemical networks. Lecture Notes Comput. Sci. 2003;2602:149–162. [Google Scholar]
- 85.Chabrier-Rivier N., Chiaverini M., Danos V., Fages F., Schachter V. Modeling and querying biomolecular interaction networks. Theor. Comput. Sci. 2004;325:25–44. [Google Scholar]
- 86.Calder M., Gilmore S., Hillston J. Modelling the influence of RKIP on the ERK signalling pathway using the stochastic process algebra PEPA; Proc. Bio-Concur, 2nd Workshop on Concurrent Models in Molecular Biology, London, 30 August 2004; 2004. pp. 36–49. [Google Scholar]
- 87.Editorial. In pursuit of systems. Nature (London) 2005;435:1. doi: 10.1038/435001a. [DOI] [PubMed] [Google Scholar]
- 88.González P. P., Cárdenas M., Gershenson C., Lagunez J. Advances in Systems Science: Measurement. Athens, Greece: Circuits and Control, WSES Press; 2001. Integration of computational techniques for the modelling of signal transduction; pp. 400–411. [Google Scholar]
- 89.Cho K. H., Shin S. Y., Kim H. W., Wolkenhauer O., McFerran B., Kolch W. Mathematical modeling of the influence of RKIP on the ERK signaling pathway; Proceedings of the First International Workshop on Computational Systems Biology, Roverto, Italy, 24–26 February 2003; 2003. pp. 127–144. [Google Scholar]
- 90.Chapman S., Asthagiri A. R. Resistance to signal activation governs design features of the MAP kinase signaling module. Biotechnol. Bioeng. 2004;85:311–322. doi: 10.1002/bit.10836. [DOI] [PubMed] [Google Scholar]
- 91.Aksan I. Kinetic analysis of RSK2 and Elk-1 interaction on the serum response element and implications for cellular engineering. Biotechnol. Bioeng. 2005;88:890–900. doi: 10.1002/bit.20322. [DOI] [PubMed] [Google Scholar]
- 92.Perez-Jimenez M. J., Romero-Campero F. J. Modelling EGFR signalling cascade using continuous membrane systems; Proceedings of the Third International Workshop on Computational Systems Biology, Edinburgh, 3–5 April 2005; 2005. pp. 118–129. [Google Scholar]
- 93.Oney I., Kurnaz I. A., Kurnaz M. L. Cytoplasmic-to-nuclear volume ratio affects AP-1 complex formation as an indicator of cell cycle responsiveness. FEBS Lett. 2005;579:433–440. doi: 10.1016/j.febslet.2004.11.104. [DOI] [PubMed] [Google Scholar]