

Abstract
The basal proton conductance of mitochondria causes mild uncoupling and may be an important contributor to metabolic rate. The molecular nature of the proton-conductance pathway is unknown. We show that the proton conductance of muscle mitochondria from mice in which isoform 1 of the adenine nucleotide translocase has been ablated is half that of wild-type controls. Overexpression of the adenine nucleotide translocase encoded by the stress-sensitive B gene in Drosophila mitochondria increases proton conductance, and underexpression decreases it, even when the carrier is fully inhibited using carboxyatractylate. We conclude that half to two-thirds of the basal proton conductance of mitochondria is catalysed by the adenine nucleotide carrier, independently of its ATP/ADP exchange or fatty-acid-dependent proton-leak functions.
Keywords: adenine nucleotide translocase (ANT) knock-out mouse, carboxyatractylate, Drosophila, proton leak
Abbreviations: ANT, adenine nucleotide translocase; CAT, carboxyatractylate; FCCP, carbonyl cyanide p-trifluoromethoxyphenylhydrazone; sesB, stress-sensitive B; TPMP, triphenylmethylphosphonium; UCP, uncoupling protein
INTRODUCTION
Oxidative phosphorylation in cells is not fully efficient. Instead, some energy is lost as heat [1]. When substrates are oxidized by the mitochondrial electron transport chain, protons are pumped out across the inner membrane, generating a protonmotive force. During oxidative phosphorylation, protons are driven back into the matrix through the F1Fo-ATP synthase and the potential energy in the protonmotive force is used to synthesize ATP. However, some protons return to the matrix through alternative leak pathways, causing mild uncoupling and lowering the efficiency.
Mitochondrial proton leak may be physiologically important. It accounts for 20–30% of the oxygen consumption of isolated resting hepatocytes [2] and about 50% of the oxygen consumption of perfused, resting skeletal muscle in rat [3]. Taking the contributions of different tissues to whole-body resting energy consumption into account, proton cycling may account for 20–25% of basal metabolic rate [3,4]. Even when hepatocytes and perfused muscle are stimulated to turn over ATP, significant proton leak occurs and proton cycling may still account for 15% of metabolic rate [5]. NMR measurements suggest similar high contributions of proton cycling in yeast grown on glucose [6], but not in mouse muscle in vivo [7], and reaching definitive quantitative conclusions about the contribution of mitochondrial proton leak to metabolic rate remains technically difficult.
At least two types of mitochondrial carrier cause significant inhibitor-sensitive inducible proton conductance when they are activated by small molecules. The first type is the UCPs (uncoupling proteins), UCP1, UCP2 and UCP3, which cause nucleotidesensitive proton leak when they are activated by fatty acids or alkenals derived from the peroxidation of membrane phospholipids [8,9]. The second type is the ANT (adenine nucleotide translocase), which exchanges ADP for ATP across the mitochondrial inner membrane and may also play an important role in the mitochondrial permeability transition pore and the mitochondrial apoptosis pathway. The ANT causes a CAT (carboxyatractylate)-sensitive proton leak when activated by fatty acids [10–12], AMP [13] or alkenals [14].
In contrast, we still do not understand the mechanism of the basal proton conductance of mitochondria (the leak pathway that is present in all mitochondria that have been studied and may make a major contribution to metabolic rate, but is insensitive to known activators or inhibitors). The activity of the basal-proton-leak pathway is different in mitochondria from different tissues [15], from mammals [16,17] and birds [18] with different body mass, in some ectotherms compared with endotherms [19–21], and when thyroid hormone levels are high [22,23]. Some of the variation can be explained by differences in mitochondrial inner-membrane surface area, but much cannot.
In all of these systems there is a striking correlation between mitochondrial proton conductance and the fatty acyl composition of inner-membrane phospholipids [17,18,20–22]. The content of n−3 polyunsaturates, particularly docosahexaenoate (C22:6,n−3), correlates with high proton conductance, and the content of monounsaturates, particularly oleate (C18:1,n−9), correlates with low proton conductance. However, the proton conductance of phospholipid vesicles prepared from mitochondrial lipids is only 2–25% of the conductance of the mitochondria that they are derived from, and does not change when the composition changes [24,25]. Therefore some factor other than membrane surface area and phospholipid composition is an important determinant of the basal proton conductance of mitochondria. Perhaps this factor is a mitochondrial membrane protein.
If a protein is involved, then given the known inducible proton conductances of UCPs and ANT, some member of the mitochondrial anion transporter family is a plausible candidate. UCPs themselves do not contribute to the basal proton conductance of mitochondria [26,27]. Similarly, yeast mitochondria from cells overexpressing ANT have unaltered resting respiration rates [6], suggesting that the ANT does not contribute to basal proton conductance. To test whether any other mitochondrial anion transporter might be responsible, mitochondria from Saccharomyces cerevisiae strains containing independent disruptions of 21 of the 35 mitochondrial carrier family genes in yeast were examined [28]. None of the 21 carriers contributed measurably to the basal proton conductance of yeast mitochondria.
Following an unexpected initial observation that the basal proton conductance of muscle mitochondria from mice lacking the ANT1 isoform was lower than that of wild-type controls, we initiated the detailed study reported in the present paper. We re-investigate the role of ANT in basal proton conductance using mitochondria from transgenic mice and Drosophila melanogaster. We conclude that the amount of ANT present in the mitochondrial inner membrane strongly affects the basal proton conductance of the membrane and suggest that ANT is a major catalyst of the basal fatty-acid-independent proton leak in mitochondria.
EXPERIMENTAL
Mammals
Experiments with ANT1 knock-out mice used male Ant1−/− and sibling wild-type B6 mice (4–5 months old) [29]. Other mouse experiments used 10–12-week-old male CD1 mice. Female Wistar rats were 6–8 weeks old. Rabbits were female and less than 1 year old. Mice, rats and rabbits were housed under standard laboratory conditions, fed a standard chow diet ad libitum, and killed by stunning and cervical dislocation. Fresh tissues from sows and bullocks (less than 2 years old) were from a local abattoir, and fresh horse tissues were from a single 11-year-old gelding horse.
Drosophila
Six strains of D. melanogaster were used (Table 1): two wild-type strains (Dahomey and Oregon R), two ANT-overexpressing strains (ANT102E and ANT42A) and two ANT-underexpressing strains (DC701 and ras59). Oregon R and the ANT mutants were from Dr M. Ashburner, University of Cambridge, U.K., and Dahomey was from Professor L. Partridge, University College London, U.K. As determined by sequence alignment analysis, Drosophila has two adjacent ANT genes: sesB (stress-sensitive B) and ANT2. These genes are transcribed from a single promoter and the two products are generated by differential splicing of a dicistronic primary RNA transcript, with sesB transcripts much more abundant than ANT2 transcripts [30].
Table 1. Genotypes of Drosophila strains.
| Name | Genotype | Flies used | |
|---|---|---|---|
| ANT-overexpressers | ANT102E | y w; P{ANT2+, sesB+10.3}102E/(eyD) | Heterozygous female |
| ANT42A | y w; P{ANT2+, sesB+10.3}42A(CyO) | Homozygous female | |
| Wild-type | Dahomey | Wild-type | Female |
| Oregon R | Wild-type | Female | |
| ANT-underexpressers | DC701 | y cv v f; 1(1)9Ed-6[DC701]/In(1)FM7 | Heterozygous female |
| Ras59 | y cv v f; df(1)ras59/In(1)FM6 | Heterozygous female |
Mutant fly strains with ANT ablated by deletion or point mutation are homozygous and hemizygous lethal but can be studied as female heterozygotes. We used two mutants: DC701, in which Trp-112 in sesB is changed to a stop codon but ANT2 is unchanged; and ras59, in which sesB, ANT2 and several flanking genes on chromosome 1 are deleted [30]. The results from the two strains were usually indistinguishable, so results were pooled as ANT-underexpressers except where noted.
We also studied mutants in which a 10.3 kb DNA fragment, containing both sesB and ANT2, was inserted into chromosome 2 (ANT42A) or chromosome 4 (ANT102E) [30]. The results from these ANT-overexpressing strains were usually indistinguishable, so results were pooled as ANT-overexpressers. Drosophila strains were maintained as described previously [31].
Mitochondria
Mitochondria were prepared at 4 °C by differential centrifugation using whole tissues from small animals, tissue samples from large animals, or whole Drosophila. For skeletal-muscle mitochondria, hind-leg skeletal muscle was finely diced in CP-1 medium [100 mM KCl, 50 mM Tris/HCl (pH 7.4) and 2 mM EGTA], digested on ice for 4 min (mice) or 5 min (other species) in CP-1 medium [to which was added 0.5% (w/v) BSA, 5 mM MgCl2, 1 mM ATP and 2.5 units/ml protease (type VIII from Bacillus licheniformis)] and homogenized three times in a Polytron homogenizer for 3 s (mouse, rat, rabbit) or 7 s (other species). Mitochondria were isolated in CP-1 medium [15,32]. Heart mitochondria were isolated similarly by protease digestion and Polytron homogenization in STE medium [250 mM sucrose, 5 mM Tris/HCl (pH 7.4) and 2 mM EGTA] [15,33]. Liver and kidney mitochondria were isolated in STE medium using a teflon/glass Dounce homogenizer [15], after extra homogenization with a loose-fitting plunger (0.54 mm clearance) for tissue from farm animals. Drosophila mitochondria were prepared in STE containing 1% (w/v) BSA [31]. The protein concentration of mitochondrial suspensions was determined using a Bio-Rad DC protein assay kit (Drosophila) or by the biuret method (mammals) [34] with BSA as a standard.
Western blots
Samples of mitochondria were solubilized and 20 μg of protein per lane was run on SDS/PAGE (12% gels) with 10× normal SDS concentration in loading buffer. Proteins were blotted on to nitrocellulose and blocked for 1 h at 37 °C with Tris-buffered saline, 0.05% (v/v) Tween 20 (TBST) with 10% (w/v) non-fat dry milk. Primary antibody (rabbit polyclonal anti-ANT, non-isotype specific; a gift from Professor H. Schmid, Hormel Institute, University of Minnesota, MN, U.S.A.) was used at 1:1000 dilution in TBST+5% milk. Secondary antibody (Pharmacia horseradish-peroxidase-linked goat anti-rabbit) was used at 1:5000 dilution. Detection was by ECL® (Amersham Biosciences) with a 1 min exposure.
Mitochondrial respiration and measurement of ANT content by CAT titre
CAT (Calbiochem) is a tight binding, specific inhibitor of ANT, so the minimum amount of CAT required to lower state-3 respiration (ADP actively phosphorylated) to the state-4 rate (no ADP phosphorylated) equals the amount of ANT present. Example graphs are shown in Figure 4(A). Respiration was measured in a Clark oxygen electrode (Rank Brothers, Cambridge, U.K.) at 37 °C (mammals) or 25 °C (Drosophila) and calibrated with air-saturated assay medium [120 mM KCl, 5 mM KH2PO4, 3 mM Hepes, 1 mM EGTA, 2 mM MgCl2 and 0.3% (w/v) defatted BSA, pH 7.2], assumed to contain 406 nmol of O/ml at 37 °C and 479 nmol of O/ml at 25 °C [35]. Mitochondria were resuspended to 0.2–1.0 mg of protein/ml (mammals) or 0.2 mg of protein/ml (Drosophila) in assay medium supplemented with the complex 1 inhibitor rotenone (5 μM) and the substrate succinate (4 mM) (mammals) or α-glycerol phosphate (10 mM) (Drosophila). Excess ADP (1 mM) was added to establish state 3, then respiration was successively inhibited by small additions of CAT [up to 5 nmol/mg of protein (mammals) or 15 nmol/mg of protein (Drosophila)] until state 4 was well established. To check that mitochondria were in state 4, more ADP was added, followed by approx. 2 μM FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone) to confirm that the electron-transport chain was still functional. Respiration rate was plotted against CAT added and the minimum CAT titre was calculated as the intercept between the steepest slope and the state-4 rate (plus CAT). Results are presented as nmol of CAT/mg of protein; it is generally thought that one CAT binds per ANT dimer [36].
Figure 4. ANT content of mitochondria from Drosophila expressing different amounts of ANT.

(A) CAT titre of respiration. State-3 respiration was titrated by successive additions of CAT to bring the mitochondria into a CAT-inhibited state 4. ANT content was measured as the CAT titre where the steepest slope in the titration crosses the state-4 rate (broken lines). Results of a single representative determination are shown for mitochondria from an ANT-overexpressing strain (▲), a wild-type strain ( ) and an ANT-underexpressing strain (○). (B) ANT contents measured by CAT titre. Replicate titrations were performed on each preparation and averaged. Values are means±S.E.M. from 19 (mutants) or 21 (wild-type) independent preparations. *P<0.05 compared with wild-type.
) and an ANT-underexpressing strain (○). (B) ANT contents measured by CAT titre. Replicate titrations were performed on each preparation and averaged. Values are means±S.E.M. from 19 (mutants) or 21 (wild-type) independent preparations. *P<0.05 compared with wild-type.
Proton-leak kinetics
The kinetics of the mitochondrial proton leak were measured by determining the dependence of the respiration rate required to drive proton leak (in the presence of oligomycin to inhibit ATP synthesis) on mitochondrial membrane potential, measured using an electrode sensitive to the potential-sensitive probe, TPMP (triphenylmethylphosphonium), in the presence of nigericin to dissipate the pH gradient, as described previously [37,38]. Briefly, mitochondria were resuspended at 0.5–1.0 mg of protein/ml in assay medium containing oligomycin (1 μg/ml), nigericin (100 ng/ml) and rotenone (5 μM; absent with glutamate/malate as substrate). The TPMP-sensitive electrode was calibrated with sequential additions of TPMP up to 2 μM (skeletal muscle and heart) or 5 μM (liver and kidney), and 4 mM succinate (or 3.2 mM glutamate plus 0.8 mM malate) was added to initiate respiration. Membrane potential and respiration were progressively inhibited through successive steady states with the complex II inhibitor, malonate, up to 6 mM (or with the complex IV inhibitor, cyanide, up to 400 μM when glutamate plus malate was substrate). Finally FCCP (5 μM) was added to dissipate the membrane potential and release all the TPMP+ from the mitochondria, allowing correction for any small baseline drift. Respiration at each steady state was plotted against the appropriate membrane potential to display the dependence of proton-leak rate on potential. When glutamate plus malate was used as substrate, respiration rates were multiplied by 1.66 to convert them into the equivalent respiration rates with succinate.
The proton-leak kinetics in Drosophila mitochondria were measured in the same way, using mitochondria at 0.2 mg of protein/ml, TPMP at 5 μM, and α-glycerol phosphate (10 mM) as the substrate. Respiration was inhibited by the addition of cyanide up to 110 μM.
TPMP binding
Corrections for TPMP+ binding to mitochondria were applied. The matrix volume of Drosophila mitochondria, determined using radiolabelled mannitol and water [37], was 0.67±0.04 μl/mg of protein (n=9). The TPMP-binding correction for Drosophila mitochondria, measured using radiolabelled TPMP and Rb (by method ‘A’ from [37]), was 0.24±0.01 (n=9). For mammalian mitochondria from different tissues, the TPMP-binding corrections determined (using method ‘B’ from [37]) in rat were taken from [15]. In liver mitochondria, the TPMP-binding corrections for horse and sheep do not differ from rat, whereas the mouse value may be lower and the rabbit value may be higher [16]. This possible species dependency was ignored, making direct comparisons between the panels in Figure 10 less accurate.
Figure 10. Correlation between proton conductance and ANT content in mammalian mitochondria.

Mitochondria were isolated from liver (◆), skeletal muscle (■), heart (●) and kidney (▲) of the species indicated. The dependence of proton-leak rate (measured as the respiration rate driving proton leak in the presence of oligomycin) on membrane potential was determined as in Figure 3(A) and the interpolated rates at the highest common potential (120 mV) were calculated using mean data. Error bars represent the weighted mean of the respiration rate S.E.M. (or range when n=2) values of the flanking experimental points. ANT content was determined by CAT titre as in Figure 1(B); values are means±S.E.M. (or range when n=2). The numbers of independent preparations were one (mouse and horse tissues, and rat heart), two (pig tissues, rat liver and ox heart), three (ox liver, kidney and skeletal muscle) or four (rabbit tissues and rat skeletal muscle).
Phospholipids
Mitochondrial lipids were extracted using chloroform or methanol and phospholipid content was determined by phosphorus analysis [17]. For fatty acyl group analysis, phospholipids were isolated by silicic-acid chromatography. Fatty-acid methyl esters were prepared, purified on a Florisil column, and separated by gas chromatography using a flame ionization detector. Mol% of each fatty acid (fa) was calculated using the equation below [18,24]:
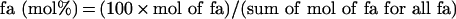 |
RESULTS
ANT content of skeletal-muscle mitochondria from ANT1 knock-out mice
Skeletal-muscle mitochondria were isolated from wild-type and ANT1 knock-out mice and the ANT content was determined by two different assays. Using a non-isotype-specific polyclonal antibody to ANT, Western blots showed that ANT was present in mitochondria from wild-type mice, but was below the limit of detection in mitochondria from the knock-out mice (Figure 1A).
Figure 1. ANT content of skeletal-muscle mitochondria from wild-type and ANT1 knock-out mice.

(A) Western blot of different preparations of mitochondria using polyclonal isotype-unspecific anti-ANT antibody. WT, wild-type; KO, ANT1 knock-out. (B) CAT titre of state-3 respiration rate to determine ANT content in mitochondria from wild-type and ANT1 knock-out mice (ANT KO). Replicate titrations were performed on each preparation and averaged. Values are mean and range or S.E.M. from two (control) or three (ANT KO) independent preparations. *P<0.05 compared with wild-type.
The amount of functional ANT present in mouse mitochondria was assessed more quantitatively by titration with the specific high-affinity inhibitor, CAT. CAT titre showed that the ANT content of skeletal-muscle mitochondria isolated from ANT1 knock-out mice was measurable; 0.31±0.23 nmol/mg of protein compared with 1.99±0.02 nmol/mg of protein (P<0.05) in skeletal-muscle mitochondria from wild-type animals (Figure 1B). We calculate that ANT makes up 6% of total mitochondrial protein in wild-type mouse skeletal-muscle mitochondria and approx. 1% in ANT1 knock-out mice.
Respiration rates of skeletal-muscle mitochondria from ANT1 knock-out mice
Mitochondria from wild-type mice were coupled, with respiratory control ratios (the state-3 rate with ADP present divided by the state-4 rate after added ADP is consumed) of 4.0 with succinate as substrate (Figure 2A). Unexpectedly, mitochondria from ANT1 knock-out mice were also able to increase their respiration rate when ADP was added, and gave a respiratory control ratio of 1.7 (Figure 2A). This observation suggests that the small residual ANT content in the ANT1 knock-outs provides ADP to the mitochondrial matrix at a rate sufficient to stimulate oxidative phosphorylation marginally.
Figure 2. Respiration rates of skeletal-muscle mitochondria from wild-type and ANT1 knock-out (ANT KO) mice.

(A) Respiratory control ratios with 4 mM succinate as substrate (state-3 rate with 700 μM ADP added/state-4 rate). (B) Respiration rates in state 3 (solid bars) and state 4 (part-shaded bars). Replicate measurements were performed on each preparation and averaged. Values are means±S.E.M. from three independent preparations. *P<0.05 compared with wild-type in the same condition.
As expected, when the respiration rates of mitochondria synthesizing ATP at maximum rate (state 3) were measured, they were severely compromised in the mitochondria from the knock-out mice (Figure 2B). More surprisingly, the respiration rates when ATP was not being synthesized and the ANT should not have been turning over (state 4) were also significantly depressed in the knock-outs (Figure 2B). This effect might have been caused by a depression in the ability of the knock-out mitochondria to oxidize substrate, or by a decrease in the proton conductance of the inner membrane (or both), since both reactions exert significant control over the state-4 rate [39].
Proton conductance of skeletal-muscle mitochondria from ANT1 knock-out mice
The kinetics of the proton leak in isolated mitochondria can be assayed by measuring the proton-leak rate (measured as the respiration rate required to drive proton leak, in the presence of oligomycin to prevent ATP synthesis) as a function of its driving force, mitochondrial membrane potential (measured with an electrode sensitive to the potential-sensitive probe, TPMP+). Figure 3(A) shows the normal non-linear response of proton-leak rate to membrane potential in skeletal-muscle mitochondria from wild-type mice. Experiments were carried out in the presence of BSA and 2 mM Mg to minimize known fatty-acid-dependent [12] and Mg-inhibited [38] proton-leak pathways to try to ensure that only basal proton leak was measured.
Figure 3. Proton leak of skeletal-muscle mitochondria from wild-type and ANT1 knock-out mice.

(A) Dependence of proton-leak rate (measured as the respiration rate driving proton leak in the presence of oligomycin) on membrane potential in mitochondria from wild-type () and ANT1 knockout (○) mice. The broken line indicates the highest common potential (151.4 mV). (B) Relationship between proton-leak rate at the highest common potential [measured as the respiration rate driving proton leak at 151.4 mV in (A)] and ANT content (from Figure 1B). Replicate measurements were performed on each preparation and averaged. Values are means±S.E.M. from three independent preparations. For the interpolated value of respiration (in B), the error shown is the weighted mean of the respiration-rate errors of the flanking experimental points in (A).
The proton-leak kinetics were altered in mitochondria from ANT1 knock-out mice: at any membrane potential between 75 mV and 150 mV, the proton-leak rate was about half that of wild-type controls, showing that the proton conductance of the inner membrane was halved in mitochondria from ANT1 knock-out mice. There was no sign of any impairment in substrate oxidation activity in the ANT1 knock-outs, which would have been apparent as an inability to maintain a higher membrane potential in state 4, despite the lower proton conductance. On the contrary, measurements of mRNA levels suggest that respiratory capacity may be up-regulated in these knock-outs [40].
The contribution of ANT to the proton conductance of mouse skeletal-muscle mitochondria is shown more explicitly in Figure 3(B), where the proton-leak rate at the highest potential common to both types of mitochondria in Figure 3(A) (151.4 mV) is plotted against the ANT content determined by CAT titre in Figure 1(B). The state-4 proton leak in wild-type mitochondria is driven by a respiration rate of 77 nmol of O/min per mg of protein. Figure 3(B) suggests that approx. half of this proton leak is dependent on the presence of ANT, whereas the remaining leak, driven by a respiration rate of about 35 nmol of O/min per mg of protein, is caused by other unidentified proton-leak pathways.
These observations suggest that ANT catalyses a fatty-acid-independent proton conductance that accounts for about half of the basal proton conductance of mouse skeletal-muscle mitochondria. However, the experimental errors were quite large, so this conclusion is only provisional. To determine whether the effect of ANT on proton conductance is mouse-specific or more general, we next developed a model in which ANT was over- or under-expressed in fruit flies.
sesB encodes a functional ANT activity in Drosophila mitochondria
There are two ANT genes in Drosophila: sesB and ANT2. We first examined whether these genes code for functional ANT. Ablation of sesB in the DC701 mutant flies or deletion of both sesB and ANT2 in the ras59 mutant flies gave similar results, so the pheno-types we report here are probably caused almost entirely by the ANT encoded by sesB.
Figure 4(A) shows representative results of titrations of the amount of functional ANT using CAT in Drosophila strains with different sesB gene dosage. The titre of CAT needed to fully inhibit state-3 respiration to the state-4 rate was greatest in ANT-overexpressing flies, intermediate in wild-type flies and lowest in ANT-underexpressing flies, strongly indicating that sesB codes for the Drosophila ANT, as predicted from sequence alignments [30].
Figure 4(B) shows the average results from many preparations of Drosophila mitochondria. ANT content in flies was high (presumably because many of the mitochondria are derived from metabolically very active flight muscle); ANT made up 10% of total mitochondrial protein in wild-type flies, 17% in ANT-overexpressers and 7% in ANT-underexpressers. The amount of ANT was roughly proportional to gene dosage, indicating that ANT expression relative to other mitochondrial proteins in Drosophila is not subject to strong feedback control. These results show that the three groups of flies have different amounts of functional ANT (see also [14]) and that Drosophila mitochondria provide an appropriate model in which to study the effects of ANT content on proton conductance.
Respiration rates of Drosophila mitochondria expressing different amounts of ANT
Mitochondria from all groups of Drosophila strains were reasonably well coupled, with respiratory control ratios between about 3 and 5 (Figure 5A). Unlike mitochondria from ANT1 knockout mice, mitochondria from ANT-underexpressing flies were not less coupled than wild-type mitochondria, suggesting that mitochondria from ANT-underexpressing flies were less compromised by the more modest relative drop in ANT content than those from ANT1 knock-out mice.
Figure 5. Respiration rates of mitochondria from Drosophila expressing different amounts of ANT.

(A) Respiratory control ratios with 10 mM α-glycerol phosphate as substrate (state-3 rate with 1 mM ADP added/state-4 rate). (B) Respiration rates in state 3 (solid bars) and state 4 (part-shaded bars). Replicate measurements were performed on each preparation and averaged. Values are means±S.E.M. from 8 (ANT-overexpressers), 9 (wild-type) or 11 (ANT-underexpressers) independent preparations. *P<0.05 compared with wild-type in the same condition.
Surprisingly, when the respiration rates of Drosophila mitochondria in state 3 were measured, they were lower in mitochondria from ANT-overexpressers (not higher, as might have been expected) (Figure 5B). This observation suggests that ANT overexpression leads to a compensatory down-regulation of the capacity for substrate oxidation (possibly because of competition for import machinery or space in the mitochondrial inner membrane). Further evidence for down-regulation of substrate oxidation appears below. The respiration rates in state 4 were not different (Figure 5B). Since both proton conductance and substrate oxidation exert significant control over the state-4 rate [39], the lack of effect of ANT-expression level on state-4 respiration rate may mean that neither proton conductance nor substrate oxidation capacity changed as ANT content varied, or that changes in proton conductance were masked by compensatory changes in substrate oxidation capacity. The experiments below support the second alternative.
Proton conductance of Drosophila mitochondria expressing different amounts of ANT
The dependence of proton-leak rate on membrane potential was measured in mitochondria isolated from each group of flies. As before, oligomycin, BSA and 2 mM Mg were present to minimize proton conductance by other known pathways. Figure 6(A) shows that the proton-leak kinetics were different in the three types of mitochondria: at any membrane potential between 120 mV and 160 mV, the proton-leak rate was about 30% higher in the ANT-overexpressers than in the wild-type controls, and 30% lower in the ANT-underexpressers than in the wild-type.
Figure 6. Proton leak of mitochondria from Drosophila expressing different amounts of ANT.

(A) Dependence of proton-leak rate (measured as the respiration rate driving proton leak in the presence of oligomycin) on membrane potential in mitochondria from ANT-overexpressers (▲), wild-type () and ANT-underexpressers (○). The broken line indicates the highest common potential (145.7 mV). (B) Relationship between proton-leak rate at the highest common potential [measured as the respiration rate driving proton leak at 145.7 mV in (A)] and ANT content (from Figure 4B). Replicate measurements were performed on each preparation and averaged. Values are means±S.E.M. from 26 (ANT-overexpressers and wild-type) or 23 (ANT-underexpressers) independent preparations. For the interpolated values of respiration (in B), the error shown is the weighted mean of the respiration-rate errors of the flanking experimental points in (A).
The effects of ANT-expression level on substrate oxidation capacity are also clear in Figure 6(A). These secondary changes in substrate oxidation capacity masked the effect of the changed ANT content on respiratory control ratios in Figure 5(A) and on state-3 rates in Figure 5(B), and masked the effects of the proton-conductance change on state-4 rates in Figure 5(B). Figure 6(A) shows that despite having an increased proton conductance and lower membrane potential, mitochondria from ANT-overexpressers were unable to increase their state-4 respiration rate compared with controls as would have been expected if their substrate oxidation capacity was not diminished by ANT overexpression. Conversely, despite having a lower proton conductance and a higher membrane potential, mitochondria from ANT-underexpressers did not decrease their state-4 respiration rate as would have been expected if their substrate oxidation capacity had not been up-regulated by ANT underexpression. These considerations highlight the importance of measuring proton conductance, rather than relying on inferences from state-4 rates as is often the case.
The contribution of ANT to the proton conductance of Drosophila mitochondria is shown explicitly in Figure 6(B), where respiration rates driving proton leak at the highest potential common to all three types of mitochondria in Figure 6(A) (145.7 mV) are plotted against ANT content determined by CAT titre in Figure 4(B). As with mouse-muscle mitochondria (Figure 3B), proton conductance varies as ANT changes, but the dependence in flies (Figure 6B) has less associated experimental error and is much clearer. The state-4 proton leak in wild-type mitochondria is driven by a respiration rate of 152 nmol of O/min per mg of protein. Extrapolation to zero ANT suggests that 53 nmol of O/min per mg of protein is independent of the presence of ANT (Figure 6B), so approximately two-thirds of the proton conductance of wild-type Drosophila mitochondria is dependent on the presence of ANT, while the remaining third is caused by other unidentified proton-leak pathways. The ANT-dependent proton conductance in Drosophila mitochondria was higher than that in mouse skeletal-muscle mitochondria, perhaps reflecting the higher ANT content in fly (10% of total protein) compared with mouse-muscle (6%) mitochondria.
These observations using Drosophila mitochondria strongly support the conclusion from the mouse ANT1 knock-outs that ANT catalyses a fatty-acid-independent proton conductance that accounts for a large proportion of the proton conductance of mitochondria.
Effect of inhibition of ANT function on the proton conductance of Drosophila mitochondria expressing different amounts of ANT
The experiments described above were carried out in the presence of 0.3% (w/v) defatted BSA, to bind endogenous fatty acids and minimize their activation of the known fatty-acid-dependent proton leak catalysed by ANT [10,11]. To check that the effect of ANT on proton conductance was not related to ANT activity, either through the normal ADP/ATP exchange or by residual contaminating fatty acids even in the presence of albumin, we studied the effect of inhibition of the ANT using CAT. Surprisingly, CAT lowered the proton conductance of Drosophila mitochondria (Figure 7).
Figure 7. Effect of CAT on the proton leak of mitochondria from wild-type Drosophila.

Dependence of proton-leak rate (measured as the respiration rate driving proton leak in the presence of oligomycin) on membrane potential in the absence () or presence ( ) of 50 nmol of CAT/mg of mitochondrial protein. Replicate measurements were performed on each preparation and averaged. Values are means±S.E.M. from four independent preparations.
) of 50 nmol of CAT/mg of mitochondrial protein. Replicate measurements were performed on each preparation and averaged. Values are means±S.E.M. from four independent preparations.
To investigate whether the effect of ANT content on basal proton conductance was dependent on the activity of ANT, the experiment in Figure 6 was repeated in the presence of saturating CAT. Figures 8(A) and 8(B) show a subset of four of the 23–26 experiments in Figures 6(A) and 6(B). The results in the subset have greater error bars than the full set, but show the same effects. Figure 8(C) shows the proton-leak kinetics of the same subset, but measured in the presence of saturating CAT to prevent fatty-acid- or turnover-dependent proton leak through ANT. The results were the same as in the absence of CAT: mitochondria from ANT-overexpressers had higher proton conductance and mitochondria from ANT-underexpressers had lower proton conductance than wild-type controls (Figure 8C). The basal proton conductance was still a linear function of ANT content, with ANT-dependent proton leak accounting for 60–80% of the total conductance (Figure 8D). Thus there is a component of the proton conductance in Drosophila mitochondria that requires ANT to be uninhibited (Figure 7), but when the ANT is fully inhibited by CAT, the remaining basal proton conductance is predominantly caused by the presence of inactive ANT in the membrane.
Figure 8. Proton leak of mitochondria from Drosophila expressing different amounts of ANT, in the absence and presence of excess CAT.

Experiments were carried out on a small subset of the preparations used in Figure 6, using mitochondria from ANT-overexpressers (▲, control; ■, plus CAT), wild-type (, control; , plus CAT) and ANT-underexpressers (○, control; ◇, plus CAT). (A) Dependence of proton-leak rate on membrane potential in the absence of CAT. The broken line indicates the highest common potential (146.2 mV). (B) Relationship between proton-leak rate at the highest common potential in (A) and ANT content (from Figure 4B). (C) Dependence of proton-leak rate on membrane potential in the presence of 50 nmol of CAT/mg of protein. The broken line indicates the highest common potential (151.0 mV). (D) Relationship between proton-leak rate at the highest common potential in (C) and ANT content (from Figure 4B). Replicate measurements were performed on each preparation and averaged. Values are means±S.E.M. from four independent preparations. For the interpolated values of respiration in (B) and (D), the error shown is the weighted mean of the respiration-rate errors of the flanking experimental points in (A) and (C) respectively.
Effect of ANT content on membrane surface area of Drosophila mitochondria
We normally calculate the proton conductance of the inner membrane per mg of total mitochondrial protein, so that if alterations in the amount of ANT, a major membrane protein, caused alterations in inner-membrane bilayer surface area per mg of protein, the proton conductance per mg of protein might change even if the proton conductance per unit surface area was unaltered. To test this possibility, the total lipid phosphorus content of the mitochondria was measured as an index of bilayer surface area. There was no effect of overexpression or underexpression of ANT on the amount of lipid phosphorus (7 μg/mg of protein) extracted from the mitochondria.
Effect of ANT content on membrane phospholipid fatty acyl composition of Drosophila mitochondria
Natural differences in basal proton conductance of mitochondria correlate with differences in the fatty acyl composition of membrane phospholipids [20]. Figure 9 shows that fatty acyl composition was not altered in ANT-overexpressers or in DC701 ANT-underexpressers compared with wild-type controls. However, there were some significant changes in ras59 ANT-underexpressers. Since DC701 has a premature stop codon specifically in sesB and shows no fatty acyl changes, whereas ras59 has a large deletion of sesB and several adjacent genes, the alterations in fatty acyl composition in ras59 are probably unrelated to ANT underexpression. Thus fatty acid compositional changes are not responsible for the effects of ANT content on basal proton conductance.
Figure 9. Effect of ANT content on the membrane phospholipid fatty acyl composition of Drosophila mitochondria.

For details, see the Experimental section. Fatty acids are described as the ‘number of C atoms:number of double bonds (position of first double bond from the methyl end)’, thus C18:2(n−6) is linoleic acid. Values are means±S.E.M. from 11 (ANT-overexpressers, black bars; wild-type, dark grey bars) or six independent preparations (DC701 ANT-underexpressers, white bars; ras59 ANT-underexpressers, light-grey bars). *P<0.05 compared with wild type.
Correlation between ANT content and proton conductance in mammalian mitochondria
If ANT content is an important determinant of basal proton conductance in all mitochondria, then mitochondria with naturally high ANT content should also have high proton conductance. This prediction was tested by measuring proton conductance and ANT content in mitochondria isolated from four different tissues from six different mammalian species. Figure 10 shows that in each species, mitochondria with high ANT content had high proton conductance, as predicted. Skeletal muscle and heart, tissues with high oxidative capacity, yielded mitochondria with high ANT contents and high proton conductance, whereas liver and kidney, tissues with lower oxidative capacity, yielded mitochondria with lower ANT content and lower proton conductance. Some of the differences between tissues may relate to differences in membrane surface area, but lipid phosphorus per mg of protein is only about 2.5-fold higher in muscle than in liver or kidney mitochondria from rat, and accounts for only part of the difference in proton conductance between tissues [15]; therefore, the remaining differences in proton conductance per unit surface area may be caused largely by differences in ANT content.
DISCUSSION
Our results show that genetic manipulation of the amount of ANT in mitochondria from mouse skeletal muscle or whole Drosophila causes substantial changes in the proton conductance of the mitochondria. The changes do not require turnover of the carrier, as they are still present when CAT, a potent inhibitor of ANT, is added. They are therefore distinct from the CAT-sensitive inducible proton conductance that is catalysed by ANT in the presence of fatty acids [10–12], AMP [13] or alkenals [14]. The changes in basal proton conductance are not caused by changes in mitochondrial inner-membrane bilayer surface area, or by changes in fatty-acyl-chain composition of membrane phospholipids, since these factors are unchanged when ANT content is altered in flies.
A previous study measured the effects of doubling the content of yeast ANT in yeast mitochondria by genetic manipulation, and found no obvious change in ANT-inhibited state-4 respiration rate [6]. The conclusion that an unchanged state-4 rate means an unchanged basal proton conductance is commonly drawn, but is not always correct. In Drosophila mitochondria, state-4 rates are unaffected by ANT-expression level (Figure 5B), but proton conductance is clearly altered (Figures 6 and 8). Secondary changes in substrate oxidation capacity masked the effects of the proton-conductance change in Figure 5(B), and the same may have been true in the experiments using yeast mitochondria [6]. Consistent with this interpretation, we have found that expression of yeast ANT1 to twice the normal level in S. cerevisiae caused an increase in the basal proton conductance of isolated yeast mitochondria [41]. In the same way, expression of a hybrid ANT (consisting of the N-terminus of yeast ANT2 with the remainder of the protein derived from bovine ANT2) in ANT1−/− ANT2−/− yeast [42] led to less functional ANT than in wild-type yeast, and a lower mitochondrial proton conductance [41]. These results show that ANT concentration affects mitochondrial proton conductance in yeast as it does in higher organisms, contrary to the conclusion in [6].
Other mitochondrial anion carriers might also contribute to the basal proton leak of mitochondria, but their concentrations are low and therefore their contributions would be small. Figure 10 confirms previous studies showing that ANT contributes 1–10% of total mitochondrial protein, depending on the tissue and species. The amount of phosphate carrier may also be significant [43], but most other carriers contribute much less: only 0.3% of total mitochondrial protein is contributed by the pyruvate carrier [44,45], 0.03% by UCP2 [46] and 0.01% by UCP3 [47]. The exception is UCP1, which comprises 1–5% of mitochondrial protein in brown adipose tissue of mice kept below their thermoneutral temperature of 28 °C (see [48]). However, the proton conductance of brown-adipose-tissue mitochondria from UCP1 knock-out mice is not different from that of mitochondria from wild-type mice (kept at 23 °C) in the presence of GDP to inhibit UCP1 function [49]. This suggests that the presence of GDP-inhibited UCP1 does not make a measurable contribution to basal proton conductance, despite its abundance in the membrane, so the effect of ANT on basal proton conductance may be a specific one.
Since the pathway of proton conductance is not dependent on the function or turnover of ANT, it may occur at the protein–phospholipid interface. If so, a partial explanation of the observation that the fatty acyl composition of membrane phospholipids correlates with proton conductance [17,18,20–22] might be that the fatty acyl chains alter the properties of the proton-conductance pathway at the ANT–phospholipid interface. Alternatively (or additionally), the correlation may be indirect: mitochondria with high metabolic requirements may have more polyunsaturated membranes to allow greater diffusion and catalytic activity of membrane proteins [50], and more ANT to service the ATP demand (with a corresponding increase in basal proton conductance catalysed by ANT), independent of any causal relationship between fatty acyl composition and proton conductance. Similarly, the high proton conductance of mitochondria from more active tissues in rat [15] and other species (Figure 10), and from the same tissue in smaller, more metabolically active mammals and birds [16–18], may be a side effect of the greater ANT content needed to service the higher ATP demand. The same argument may explain the high basal proton conductance of mitochondria from mammals compared with those from lizards [19–21], and from rats with high thyroid hormone levels [22,23], where ANT expression is high [51].
Thus changes in phospholipid fatty acyl composition and proton conductance per unit of mitochondrial protein may be secondary results of the changes in the surface area of the inner membrane and in the activity and concentration of ANT that are needed in mitochondria with a higher capacity for ATP production. Much of the increased proton conductance of such mitochondria would be caused by changes in ANT content. This hypothesis would explain the observed differences in mitochondrial proton conductance under many different conditions. The basal-proton-conductance pathway of the ANT might be an unavoidable consequence of the structure and abundance of this carrier protein. Alternatively, it might be an evolved function that ensures that mitochondria with high ANT content and a high capacity for oxidative phosphorylation also have a higher degree of mild uncoupling by the proton leak. The higher leak would protect the mitochondria against the enhanced risk of radical production and oxidative damage that is associated with a greater capacity for oxidative phosphorylation [52].
Acknowledgments
We thank Mike Ashburner and John Roote for mutant Drosophila stocks, Linda Partridge for the Dahomey Drosophila stock, Dr H. Terada for mutant yeast strains, Linda Partridge and Kevin Brindle for helpful advice, and Stephen Roebuck and Julie Buckingham for invaluable technical assistance. This work was supported by the Medical Research Council (M. D. B., J. L. P., A. O. and E. J. C.), the Cambridge Commonwealth Trust (A. O. and E. J. C.) and National Institutes of Health grant NS41850 awarded to D. C. W.
References
- 1.Brand M. D., Chien L. F., Ainscow E. K., Rolfe D. F. S., Porter R. K. The causes and functions of mitochondrial proton leak. Biochim. Biophys. Acta. 1994;1187:132–139. doi: 10.1016/0005-2728(94)90099-x. [DOI] [PubMed] [Google Scholar]
- 2.Brand M. D. The proton leak across the mitochondrial inner membrane. Biochim. Biophys. Acta. 1990;1018:128–133. doi: 10.1016/0005-2728(90)90232-s. [DOI] [PubMed] [Google Scholar]
- 3.Rolfe D. F. S., Brand M. D. Contribution of mitochondrial proton leak to skeletal muscle respiration and to standard metabolic rate. Am. J. Physiol. 1996;271:C1380–C1389. doi: 10.1152/ajpcell.1996.271.4.C1380. [DOI] [PubMed] [Google Scholar]
- 4.Rolfe D. F. S., Brand M. D. The physiological significance of mitochondrial proton leak in animal cells and tissues. Biosci. Rep. 1997;17:9–16. doi: 10.1023/a:1027327015957. [DOI] [PubMed] [Google Scholar]
- 5.Rolfe D. F. S., Newman J. M., Buckingham J. A., Clark M. G., Brand M. D. Contribution of mitochondrial proton leak to respiration rate in working skeletal muscle and liver and to SMR. Am. J. Physiol. 1999;276:C692–C699. doi: 10.1152/ajpcell.1999.276.3.C692. [DOI] [PubMed] [Google Scholar]
- 6.Sheldon J. G., Williams S. P., Fulton A. M., Brindle K. M. 31P NMR magnetization transfer study of the control of ATP turnover in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 1996;93:6399–6404. doi: 10.1073/pnas.93.13.6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marcinek D. J., Schenkman K. A., Ciesielski W. A., Conley K. E. Mitochondrial coupling in vivo in mouse skeletal muscle. Am. J. Physiol. Cell Physiol. 2004;286:C457–C463. doi: 10.1152/ajpcell.00237.2003. [DOI] [PubMed] [Google Scholar]
- 8.Brand M. D., Affourtit C., Esteves T. C., Green K., Lambert A. J., Miwa S., Pakay J. L., Parker N. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radical Biol. Med. 2004;37:755–767. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 9.Esteves T. C., Brand M. D. The reactions catalyzed by the mitochondrial uncoupling proteins UCP2 and UCP3. Biochim. Biophys. Acta. 2005;1709:35–44. doi: 10.1016/j.bbabio.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 10.Andreyev A., Bondareva T. O., Dedukhova V. I., Mokhova E. N., Skulachev V. P., Volkov N. I. Carboxyatractylate inhibits the uncoupling effect of free fatty acids. FEBS Lett. 1988;226:265–269. doi: 10.1016/0014-5793(88)81436-4. [DOI] [PubMed] [Google Scholar]
- 11.Andreyev A., Bondareva T. O., Dedukhova V. I., Mokhova E. N., Skulachev V. P., Tsofina L. M., Volkov N. I., Vygodina T. V. The ATP/ADP-antiporter is involved in the uncoupling effect of fatty acids on mitochondria. Eur. J. Biochem. 1989;182:585–592. doi: 10.1111/j.1432-1033.1989.tb14867.x. [DOI] [PubMed] [Google Scholar]
- 12.Skulachev V. P. Uncoupling: new approaches to an old problem of bioenergetics. Biochim. Biophys. Acta. 1998;1363:100–124. doi: 10.1016/s0005-2728(97)00091-1. [DOI] [PubMed] [Google Scholar]
- 13.Cadenas S., Buckingham J. A., St-Pierre J., Dickinson K., Jones R. B., Brand M. D. AMP decreases the efficiency of skeletal-muscle mitochondria. Biochem. J. 2000;351:307–311. [PMC free article] [PubMed] [Google Scholar]
- 14.Echtay K. S., Esteves T. C., Pakay J. L., Jekabsons M. B., Lambert A. J., Portero-Otin M., Pamplona R., Vidal-Puig A., Wang S., Roebuck S. J., Brand M. D. A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J. 2003;22:4103–4110. doi: 10.1093/emboj/cdg412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rolfe D. F. S., Hulbert A. J., Brand M. D. Characteristics of mitochondrial proton leak and control of oxidative phosphorylation in the major oxygen-consuming tissues of the rat. Biochim. Biophys. Acta. 1994;1188:405–416. doi: 10.1016/0005-2728(94)90062-0. [DOI] [PubMed] [Google Scholar]
- 16.Porter R. K., Brand M. D. Body mass dependence of H+ leak in mitochondria and its relevance to metabolic rate. Nature (London) 1993;362:628–630. doi: 10.1038/362628a0. [DOI] [PubMed] [Google Scholar]
- 17.Porter R. K., Hulbert A. J., Brand M. D. Allometry of mitochondrial proton leak: influence of membrane surface area and fatty acid composition. Am. J. Physiol. 1996;271:R1550–R1560. doi: 10.1152/ajpregu.1996.271.6.R1550. [DOI] [PubMed] [Google Scholar]
- 18.Brand M. D., Turner N., Ocloo A., Else P. L., Hulbert A. J. Proton conductance and fatty acyl composition of liver mitochondria correlates with body mass in birds. Biochem. J. 2003;376:741–748. doi: 10.1042/BJ20030984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brand M. D., Couture P., Else P. L., Withers K. W., Hulbert A. J. Evolution of energy metabolism: proton permeability of the inner membrane of liver mitochondria is greater in a mammal than in a reptile. Biochem. J. 1991;275:81–86. doi: 10.1042/bj2750081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brookes P. S., Buckingham J. A., Tenreiro A. M., Hulbert A. J., Brand M. D. The proton permeability of the inner membrane of liver mitochondria from ectothermic and endothermic vertebrates and from obese rats: correlations with standard metabolic rate and phospholipid fatty acid composition. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 1998;119:325–334. doi: 10.1016/s0305-0491(97)00357-x. [DOI] [PubMed] [Google Scholar]
- 21.Hulbert A. J., Else P. L., Manolis S. C., Brand M. D. Proton leak in hepatocytes and liver mitochondria from archosaurs (crocodiles) and allometric relationships for ectotherms. J. Comp. Physiol. B. 2002;172:387–397. doi: 10.1007/s00360-002-0264-1. [DOI] [PubMed] [Google Scholar]
- 22.Hafner R. P., Nobes C. D., McGown A. D., Brand M. D. Altered relationship between protonmotive force and respiration rate in non-phosphorylating liver mitochondria isolated from rats of different thyroid hormone status. Eur. J. Biochem. 1988;178:511–518. doi: 10.1111/j.1432-1033.1988.tb14477.x. [DOI] [PubMed] [Google Scholar]
- 23.Brand M. D., Steverding D., Kadenbach B., Stevenson P. M., Hafner R. P. The mechanism of the increase in mitochondrial proton permeability induced by thyroid hormones. Eur. J. Biochem. 1992;206:775–781. doi: 10.1111/j.1432-1033.1992.tb16984.x. [DOI] [PubMed] [Google Scholar]
- 24.Brookes P. S., Rolfe D. F. S., Brand M. D. The proton permeability of liposomes made from mitochondrial inner membrane phospholipids: comparison with isolated mitochondria. J. Membr. Biol. 1997;155:167–174. doi: 10.1007/s002329900168. [DOI] [PubMed] [Google Scholar]
- 25.Brookes P. S., Hulbert A. J., Brand M. D. The proton permeability of liposomes made from mitochondrial inner membrane phospholipids: no effect of fatty acid composition. Biochim. Biophys. Acta. 1997;1330:157–164. doi: 10.1016/s0005-2736(97)00160-0. [DOI] [PubMed] [Google Scholar]
- 26.Cadenas S., Echtay K. S., Harper J. A., Jekabsons M. B., Buckingham J. A., Grau E., Abuin A., Chapman H., Clapham J. C., Brand M. D. The basal proton conductance of skeletal muscle mitochondria from transgenic mice overexpressing or lacking uncoupling protein-3. J. Biol. Chem. 2002;277:2773–2778. doi: 10.1074/jbc.M109736200. [DOI] [PubMed] [Google Scholar]
- 27.Couplan E., del Mar Gonzalez-Barroso M., Alves-Guerra M. C., Ricquier D., Goubern M., Bouillaud F. No evidence for a basal, retinoic, or superoxideinduced uncoupling activity of the uncoupling protein 2 present in spleen or lung mitochondria. J. Biol. Chem. 2002;277:26268–26275. doi: 10.1074/jbc.M202535200. [DOI] [PubMed] [Google Scholar]
- 28.Roussel D., Harding M., Runswick M. J., Walker J. E., Brand M. D. Does any yeast mitochondrial carrier have a native uncoupling protein function? J. Bioenerg. Biomembr. 2002;34:165–176. doi: 10.1023/a:1016027302232. [DOI] [PubMed] [Google Scholar]
- 29.Graham B. H., Waymire K. G., Cottrell B., Trounce I. A., MacGregor G. R., Wallace D. C. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat. Genet. 1997;16:226–234. doi: 10.1038/ng0797-226. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y. Q., Roote J., Brogna S., Davis A. W., Barbash D. A., Nash D., Ashburner M. stress sensitive B encodes an adenine nucleotide translocase in Drosophila melanogaster. Genetics. 1999;153:891–903. doi: 10.1093/genetics/153.2.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miwa S., St-Pierre J., Partridge L., Brand M. D. Superoxide and hydrogen peroxide production by Drosophila mitochondria. Free Radical Biol. Med. 2003;35:938–948. doi: 10.1016/s0891-5849(03)00464-7. [DOI] [PubMed] [Google Scholar]
- 32.Bhattacharya S. K., Thakar J. H., Johnson P. L., Shanklin D. R. Isolation of skeletal muscle mitochondria from hamsters using an ionic medium containing ethylenediaminetetraacetic acid and nagarse. Anal. Biochem. 1991;192:344–349. doi: 10.1016/0003-2697(91)90546-6. [DOI] [PubMed] [Google Scholar]
- 33.Pande S. V., Blanchaer M. C. Reversible inhibition of mitochondrial adenosine diphosphate phosphorylation by long chain acyl coenzyme A esters. J. Biol. Chem. 1971;246:402–411. [PubMed] [Google Scholar]
- 34.Gornall A. G., Bardawill C. J., David M. M. Determination of serum proteins by means of the biuret reaction. J. Biol. Chem. 1949;177:751–766. [PubMed] [Google Scholar]
- 35.Reynafarje B., Costa L. E., Lehninger A. L. O2 solubility in aqueous media determined by a kinetic method. Anal. Biochem. 1985;145:406–418. doi: 10.1016/0003-2697(85)90381-1. [DOI] [PubMed] [Google Scholar]
- 36.Streicher-Scott J., Lapidus R., Sokolove P. M. Use of carboxyatractylate and tight-binding inhibitor theory to determine the concentration of functional mitochondrial adenine nucleotide translocators in a reconstituted system. Anal. Biochem. 1993;210:69–76. doi: 10.1006/abio.1993.1152. [DOI] [PubMed] [Google Scholar]
- 37.Brand M. D. Measurement of mitochondrial protonmotive force. In: Brown G. C., Cooper C. E., editors. Bioenergetics: a Practical Approach. Oxford: IRL Press; 1995. pp. 39–62. [Google Scholar]
- 38.Cadenas S., Brand M. D. Effects of magnesium and nucleotides on the proton conductance of rat skeletal-muscle mitochondria. Biochem. J. 2000;348:209–213. [PMC free article] [PubMed] [Google Scholar]
- 39.Brand M. D., Hafner R. P., Brown G. C. Control of respiration in non-phosphorylating mitochondria is shared between the proton leak and the respiratory chain. Biochem. J. 1988;255:535–539. [PMC free article] [PubMed] [Google Scholar]
- 40.Murdock D. G., Boone B. E., Esposito L. A., Wallace D. C. Up-regulation of nuclear and mitochondrial genes in the skeletal muscle of mice lacking the heart/muscle isoform of the adenine nucleotide translocator. J. Biol. Chem. 1999;274:14429–14433. doi: 10.1074/jbc.274.20.14429. [DOI] [PubMed] [Google Scholar]
- 41.Cornwall E. Cambridge: University of Cambridge; 2004. The role of the adenine nucleotide translocase in mitochondrial proton leak; p. 197. PhD thesis. [Google Scholar]
- 42.Hashimoto M., Shinohara Y., Majima E., Hatanaka T., Yamazaki N., Terada H. Expression of the bovine heart mitochondrial ADP/ATP carrier in yeast mitochondria: significantly enhanced expression by replacement of the N-terminal region of the bovine carrier by the corresponding regions of the yeast carriers. Biochim. Biophys. Acta. 1999;1409:113–124. doi: 10.1016/s0005-2728(98)00155-8. [DOI] [PubMed] [Google Scholar]
- 43.Palmieri F. Mitochondrial carrier proteins. FEBS Lett. 1994;346:48–54. doi: 10.1016/0014-5793(94)00329-7. [DOI] [PubMed] [Google Scholar]
- 44.Shearman M. S., Halestrap A. P. The concentration of the mitochondrial pyruvate carrier in rat liver and heart mitochondria determined with α-cyano-β-(1-phenylindol-3-yl)acrylate. Biochem. J. 1984;223:673–676. doi: 10.1042/bj2230673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paradies G. Interaction of α-cyano[14C]cinnamate with the mitochondrial pyruvate translocator. Biochim. Biophys. Acta. 1984;766:446–450. doi: 10.1016/0005-2728(84)90260-3. [DOI] [PubMed] [Google Scholar]
- 46.Pecqueur C., Alves-Guerra M. C., Gelly C., Levi-Meyrueis C., Couplan E., Collins S., Ricquier D., Bouillaud F., Miroux B. Uncoupling protein 2, in vivo distribution, induction upon oxidative stress, and evidence for translational regulation. J. Biol. Chem. 2001;276:8705–8712. doi: 10.1074/jbc.M006938200. [DOI] [PubMed] [Google Scholar]
- 47.Harper J. A., Stuart J. A., Jekabsons M. B., Roussel D., Brindle K. M., Dickinson K., Jones R. B., Brand M. D. Artifactual uncoupling by uncoupling protein 3 in yeast mitochondria at the concentrations found in mouse and rat skeletal-muscle mitochondria. Biochem. J. 2002;361:49–56. doi: 10.1042/0264-6021:3610049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stuart J. A., Harper J. A., Brindle K. M., Jekabsons M. B., Brand M. D. A mitochondrial uncoupling artifact can be caused by expression of uncoupling protein 1 in yeast. Biochem. J. 2001;356:779–789. doi: 10.1042/0264-6021:3560779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Monemdjou S., Kozak L. P., Harper M. E. Mitochondrial proton leak in brown adipose tissue mitochondria of Ucp1-deficient mice is GDP insensitive. Am. J. Physiol. 1999;276:E1073–E1082. doi: 10.1152/ajpendo.1999.276.6.E1073. [DOI] [PubMed] [Google Scholar]
- 50.Hulbert A. J., Else P. L. Membranes as possible pacemakers of metabolism. J. Theor. Biol. 1999;199:257–274. doi: 10.1006/jtbi.1999.0955. [DOI] [PubMed] [Google Scholar]
- 51.Dummler K., Muller S., Seitz H. J. Regulation of adenine nucleotide translocase and glycerol 3-phosphate dehydrogenase expression by thyroid hormones in different rat tissues. Biochem. J. 1996;317:913–918. doi: 10.1042/bj3170913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Skulachev V. P. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q. Rev. Biophys. 1996;29:169–202. doi: 10.1017/s0033583500005795. [DOI] [PubMed] [Google Scholar]