

Abstract
We described earlier a novel mode of regulation of Hsp104, a cytosolic chaperone directly involved in the refolding of heat-denatured proteins, and designated it delayed upregulation, or DUR. When Saccharomyces cerevisiae cells grown at the physiological temperature of 24°C, preconditioned at 37°C, and treated briefly at 50°C were shifted back to 24°C, Hsp104 expression was strongly induced after 2.5 h of recovery and returned back to normal after 5 h. Here we show that the endoplasmic reticulum (ER) chaperones BiP/Kar2p and Lhs1p and the mitochondrial chaperone Hsp78 were also upregulated at the physiological temperature during recovery from thermal insult. The heat shock element (HSE) in the KAR2 promoter was found to be sufficient to drive DUR. The unfolded protein element could also evoke DUR, albeit weakly, in the absence of a functional HSE. BiP/Kar2p functions in ER translocation and assists protein folding. Here we found that the synthesis of new BiP/Kar2p molecules was negligible for more than an hour after the shift of the cells from 50°C to 24°C. Concomitantly, ER translocation was blocked, suggesting that preexisting BiP/Kar2p molecules or other necessary proteins were not functioning. Translocation resumed concomitantly with enhanced synthesis of BiP/Kar2p after 3 h of recovery, after which ER exit and protein secretion also resumed. For a unicellular organism like S. cerevisiae, conformational repair of denatured proteins is the sole survival strategy. Chaperones that refold proteins in the cytosol, ER, and mitochondria of S. cerevisiae appear to be subject to DUR to ensure survival after thermal insults.
In their natural habitat, yeast cells experience greatly varying conditions such as rapid changes in temperature, nutrient balance, and hydration. This is why molecular mechanisms to cope with cellular stress have evolved. Molecular chaperones play a key role in survival mechanisms by preventing the denaturation of proteins or by refolding denatured proteins. In the yeast Saccharomyces cerevisiae, like in all organisms, mild heat treatments induce tolerance to otherwise lethal high temperatures by increasing the expression of heat shock proteins (23). Chaperones that repair heat-damaged proteins have been found in the cytosol, the endoplasmic reticulum (ER), and mitochondria (17, 31, 36, 38, 44).
In the yeast cytoplasm, the Hsp100/ClB homologue Hsp104, assisted by the disaccharide trehalose, prevents the denaturation of proteins and directly refolds damaged proteins (31, 44). The expression of Hsp104 is increased upon a shift of cells from the physiological temperature 24°C to the heat shock temperature 37°C. The induction is regulated by the Msn2/4p/STRE regulon and the Hsf1p/HSE regulon. Both regulons are capable of acting alone in the absence of the other (2, 16, 47). When cells grown at 24°C are preconditioned at 37°C, they survive a brief incubation at 48 to 50°C, whereas in the absence of preconditioning they die. Hsp104 is essential for the acquisition of thermotolerance (35, 44).
We found recently that Hsp104 is also subject to another, novel type of regulatory mechanism, which we designated delayed upregulation, or DUR. When cells grown at 24°C, preconditioned for an hour at 37°C, and subjected for 20 min to thermal insult at 50°C were shifted back to 24°C to recover, the expression of Hsp104 was slowly upregulated. After 2 to 4 h at 24°C, the level was many times higher than that at 37°C and returned after 5 h back to that prevailing at 24°C. We found that DUR was mediated via the heat shock element (HSE) and not the stress response elements (STREs) in the HSP104 promoter. However, Msn2/4p transcription factors driving STRE-dependent gene expression were required for DUR, suggesting that Msn2/4p-regulated gene products were involved in DUR of the HSP104 gene (40).
In the lumen of the ER, conformational repair of heat-denatured proteins requires the Hsp70 homologue Lhs1p (36, 37). Unexpectedly, Hsp104 and trehalose were also found to be necessary for the refolding events occurring in the ER lumen, suggesting that cross talk occurs between cytosolic and luminal chaperones over the ER membrane (17, 41). In the mitochondria, the reactivation of protein synthesis after thermal insult involves the Hsp100/ClB homologue Hsp78, which cooperates with the mitochondrial Hsp70 machinery (22, 38).
BiP/Kar2p is a member of the 70-kDa heat shock protein (Hsp70) family, which is resident in the ER. Yeast BiP/Kar2p is an essential protein (30) with multiple roles. It functions in the translocation of newly synthesized polypeptides through the Sec61 translocon into the ER (6, 25). BiP/Kar2p interacts with the luminal J domain of the cochaperone Sec63p, a member of the Hsp40 family, and provides the driving force for translocation by hydrolyzing ATP (4, 10, 39). Like other Hsp70 proteins, BiP/Kar2p consists of two domains, an N-terminal domain harboring an ATPase catalytic site and a C-terminal domain responsible for substrate binding (3, 12, 26). Two models have been proposed for the function of BiP/Kar2p in protein translocation (for a review, see reference 20). In the ratchet or trapping model, BiP/Kar2p binds to the polypeptide emerging from the translocon, preventing it from sliding backwards (24). In the translocation motor model, BiP/Kar2p binds to the emerging polypeptide, and an ATP hydrolysis-dependent conformational change in BiP/Kar2p pulls the protein through the translocon (15). Moreover, BiP/Kar2p assists the folding of polypeptides in the ER by shielding hydrophobic amino acid side chains from the aqueous environment of the ER lumen (43). BiP/Kar2p functions in quality control by binding to misfolded proteins and preventing their exit from the ER (14, 18).
The KAR2 promoter harbors three elements that control basal and stress-induced transcription. The HSE is responsible for the upregulation of expression upon a shift of cells from 24°C to 37°C (21). The unfolded protein response element (UPRE) is activated when unfolded proteins accumulate in the ER, e.g., when N-glycosylation or disulfide bond formation is inhibited (21). The GC region between these stress-inducible elements is responsible for basal expression (21). In this study, we show that delayed upregulation after a thermal insult, or DUR, is not specific for Hsp104. The ER chaperones BiP/Kar2p and Lhs1p are also subject to DUR, as is mitochondrial Hsp78. We found that either the HSE or UPRE in the KAR2 promoter was sufficient for DUR in the absence of the other, although the HSE evoked a stronger response than the UPRE. The biological functions that depend on BiP/Kar2p, namely, the translocation and folding of polypeptides, were abolished after thermal insult. Concomitant with the upregulation of BiP/Kar2p synthesis during recovery, protein translocation and ER exit resumed. The delayed upregulation of chaperones in different cellular compartments appears to ensure the refolding of heat-denatured proteins, and thus survival, after severe heat stress.
MATERIALS AND METHODS
Strains and media.
The yeast strains used for this study are listed in Table 1. They were grown at 24°C in shake flasks in yeast extract-peptone-dextrose medium or in appropriate selection medium overnight to early logarithmic phase or in synthetic complete medium lacking methionine and cysteine for metabolic labeling experiments. Escherichia coli strain DH5α was used for cloning and was grown in Luria-Bertani medium supplemented with ampicillin (100 μg/ml).
TABLE 1.
Yeast strains used for this study
| Strain | Genotype | Reference or source |
|---|---|---|
| H245 | MATaade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 (W303-1A) | 46 |
| H7 | MATα leu2-3,112 his3-11,15 ura3-251,373 (OL1) | J. Knowles |
| H335 | MATaade2-101 ura3-52 leu2-3 leu2-112 suc2-9 gal2 URA3::HSP150Δ-β-lactamase | 42 |
| H720 | MATα leu2-3,112 his3-11,15 ura3-251,373 ire1::HIS3 | This study |
| H1765 | MATaade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 (pKTH5157)a | This study |
| H1766 | MATaade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 (pKTH5158)a | This study |
| H1767 | MATaade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 (pKTH5159)a | This study |
| H1768 | MATaade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 (pKTH5160)a | This study |
| H1769 | MATaade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 (pKTH5161)a | This study |
| H1770 | MATaade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 (pKTH5162)a | This study |
| H1777 | MATahis3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 hac1::kanMX4 (BY4742) | Euroscarf |
| H1806 | MATaade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 (pKTH5163)a | This study |
| H1807 | MATα leu2-3,112 his3-11,15 ura3-251,373 ire1::HIS3 (pKTH5163)a | This study |
| H1808 | MATahis3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 hac1::kanMX4 (pKTH5163)a | This study |
| H2004 | MATaade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 (pKTH5222)a | This study |
| H2010 | MATα leu2-3,112 his3-11,15 ura3-251,373 ire1::HIS3 (pKTH5222)a | This study |
The plasmid numbers in parentheses designate KAR2 promoter-lacZ fusion variants transformed into the strains (see Fig. 3 and Materials and Methods).
Plasmid construction.
See Fig. 3 for a schematic presentation of the KAR2 promoter-lacZ fusion constructs and Table 1 for the constructed strains. Plasmids pKTH5157 (pJS-U), pKTH5158 (pJS-5HG), pKTH5159 (pJS-5ΔHG), pKTH5160 (pJS-HG), pKTH5161 (pJS-H), and pKTH5162 (pJS401) (49) are based on plasmid pKTH5162 (pJS401) (5), which contains the 2μm DNA origin, the URA3 marker gene, and a transcriptionally silent ICL1 promoter upstream of lacZ, which can be switched on by the insertion of transcriptionally active promoter sequences. To construct plasmid pKTH5163, an XhoI/BamHI-digested KAR2 promoter fragment (nucleotides −280 to −109) from a genome-based PCR with the upper primer 5′ AGC CGC TTC TCG AGC AAA GTG 3′ and the lower primer 5′ CTT CCA GAG GAT CCT TTT CGT G 3′ (restriction sites are underlined) was ligated to the XhoI/BamHI sites of vector pKTH5162 (pJS401). Plasmid pKTH5222, where the wild-type KAR2 promoter HSE (GAACCTTCTGGAAATTTC) was replaced with a disrupted one (AGGCCTATTGTTTATTAA) (GAA repeats are underlined), was constructed using a three-step PCR procedure, with plasmid pKTH5163 as the template. In the first step, the 5′ end of the KAR2 promoter sequence, upstream of the HSE, was amplified with the above-mentioned upper primer and the mutagenizing primer 5′ CCG GGT TTA ATA AAC AAT AGG CCT TAT TAA AAT AGA AAG TTG C 3′. In the second step, the 3′ fragment of the KAR2 promoter, downstream of the HSE, was amplified with the mutagenizing primer 5′ CCT ATT GTT TAT TAA ACC CGG CGC G 3′ and the above-mentioned lower primer. In the third step, the two partially overlapping PCR fragments were annealed, and thereafter the KAR2 promoter fragment (−280 to −109) was amplified with the above-mentioned upper and lower primers, digested with XhoI/BamHI, and ligated to the XhoI/BamHI sites of vector pKTH5162 (pJS401), resulting in plasmid pKTH5222. The IRE1 disruption plasmid pKOHS (28) was transformed into strain H7, resulting in strain H720.
FIG. 3.

Contribution of different promoter elements of KAR2 to DUR. (A) Metabolic labeling of β-galactosidase. The figure shows a schematic presentation of the thermal treatments. Cells (strain H1806) harboring a β-galactosidase reporter construct regulated by the KAR2 promoter were grown at 24°C, divided into 10 parallel samples, and labeled with [35S]methionine-cysteine for successive 45-min periods, as indicated by the bars (a to j). After the termination of labeling, the cells were lysed for immunoprecipitation with β-galactosidase antiserum, and the immunoprecipitates were resolved by SDS-PAGE, followed by autoradiography. (B) Phosphorimager quantification of [35S]β-galactosidase. Normalization was done against the level of [35S]β-galactosidase in untreated cells (sample a). Data are means of six experiments. (C to L) Summary of a series of metabolic labeling experiments and β-galactosidase assays. Metabolic labeling and immunoprecipitation of β-galactosidase were performed with strains harboring different KAR2 promoter-lacZ variants, as shown in panel A. The basal expression of β-galactosidase in the strains was quantified by enzyme activity measurements. The magnitude of β-galactosidase expression in strain H1806, harboring a KAR2 promoter (−280 to −109) directly upstream of the lacZ reporter, was used as a reference for β-galactosidase expression at 24°C (basal), after 1 h at 37°C (HS), and at 24°C 3 h after thermal insult (DUR). 0, no expression; =, same expression as that in H1806; <, expression weaker than that in H1806.
Metabolic labeling, immunoprecipitation, and SDS-PAGE analysis.
Metabolic labeling of cells (2.5 × 107/0.5 ml) was done with [35S]methionine-cysteine (1,000 Ci/mmol; Amersham Biosciences, Buckinghamshire, United Kingdom) in synthetic complete medium lacking methionine and cysteine. Labeling was terminated with NaN3 (final concentration, 10 mM), and immunoprecipitation of lysed cell samples was performed with antiserum against Kar2p (1:100) (45), β-lactamase (1:100), or β-galactosidase (1:100; Biodesign International, Saco, ME) and protein A-Sepharose (Amersham Pharmacia Biotech AB, Uppsala, Sweden), as described previously (36). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was done with 8% (Kar2p and Hsp150Δ-β-lactamase) or 6% (β-galactosidase) gels. Relative radioactivities were quantified from gels, using phosphorimager technology (BAS-1000 instrument with TINA 2.09c software; Fuji).
Real-time quantitative PCR.
Total RNA was phenol extracted from yeast cells (strain H245) broken with glass beads. For each sample, 100 μg of RNA was treated with 30 U of RNase-free DNase I (QIAGEN GmbH, Hilden, Germany) and purified by using an RNeasy mini kit (QIAGEN GmbH, Hilden, Germany) according to the manufacturer's instructions. The quality of the RNA specimens was tested with an Agilent Technologies 2100 bioanalyzer according to the manufacturer's instructions. Reverse transcription reactions were performed with 2 μg of RNA by using TaqMan reverse transcription reagents (manufactured for Applied Biosystems by Roche Molecular Systems, Inc., Branchburg, NJ) in a reaction volume of 100 μl, of which 2.5 μl was used for PCR amplification. The KAR2 primers (5′ TGA TAA CTT TGA AAC CGC CAT TG 3′ and 5′ GTA ATT GGA TAA GCG ACC TTG GA 3′), LHS1 primers (5′ AAA TTC CAT TAG CGA AAT AGA GAA GTT C 3′ and 5′ CTT CGC CAA ATA GGT TTT TTG C 3′), and HSP78 primers (5′ TGG TGC AAG GCC ATT GAA TA 3′ and 5′ TTG GTA GCA CGA CAA GCT TAG TAT CT 3′) were designed with Primer Express, version 2.0.0 (Applied Biosystems). Amplification and detection of cDNA were done with the ABI Prism 7000 sequence detection system (Applied Biosystems), using SYBR green PCR master mix (Applied Biosystems, Warrington, United Kingdom) as described by the manufacturer.
Other procedures.
For β-galactosidase assays, cells grown to an optical density at 600 nm of 0.6 to 1.0 were broken with glass beads in Z buffer (0.06 M Na2HPO4, 0.04 M NaH2PO4, 0.01 M KCl, 0.001 M MgSO4, 0.05 M β-mercaptoethanol). The β-galactosidase activity was determined by incubating an aliquot of the cell lysate in the presence of o-nitrophenyl-β-D-galactopyranoside (ONPG; Sigma-Aldrich Chemie GmbH, Steinheim, Germany) and by measuring the absorbance at 420 nm. For bulk protein synthesis experiments, the 35S-labeled cells were lysed, subjected to precipitation with cold 20% trichloroacetic acid, and counted for precipitated radioactivity (41). The glucose consumption of yeast cells was followed by determining the glucose concentration in the growth medium of duplicate samples, using a Gluco-quant kit from Roche Diagnostics as described before (41).
RESULTS
Upregulation of Kar2p during recovery at 24°C after thermal insult.
To study the expression of Kar2p after a thermal insult, S. cerevisiae cells (strain H245; see Table 1 for genotypes) were grown at the physiological temperature 24°C, treated for an hour at 37°C (preconditioning), and then shifted to 50°C for 20 min (thermal insult) and finally back to 24°C (recovery). Parallel cell samples were labeled with [35S]methionine-cysteine for successive periods of 45 min, as indicated by the bars in Fig. 1A. The cells were lysed and subjected to immunoprecipitation with Kar2p antiserum, followed by SDS-PAGE analysis (Fig. 1B) and phosphorimager quantification (Fig. 1C). After preconditioning at 37°C, the level of newly synthesized Kar2p was sevenfold higher than the basal level detected after growth at 24°C (sample b). After thermal insult at 50°C and a shift back to 24°C, Kar2p expression was negligible for the first 1.5 h (samples c and d). After 2.5 h of recovery, it was 27 times (sample f) the level detected after growth at 24°C (sample a) and decreased to close to normal in 6 h (sample j). This experiment was repeated three times with strain H245 and two times with another strain (H7), with similar results (not shown).
FIG. 1.
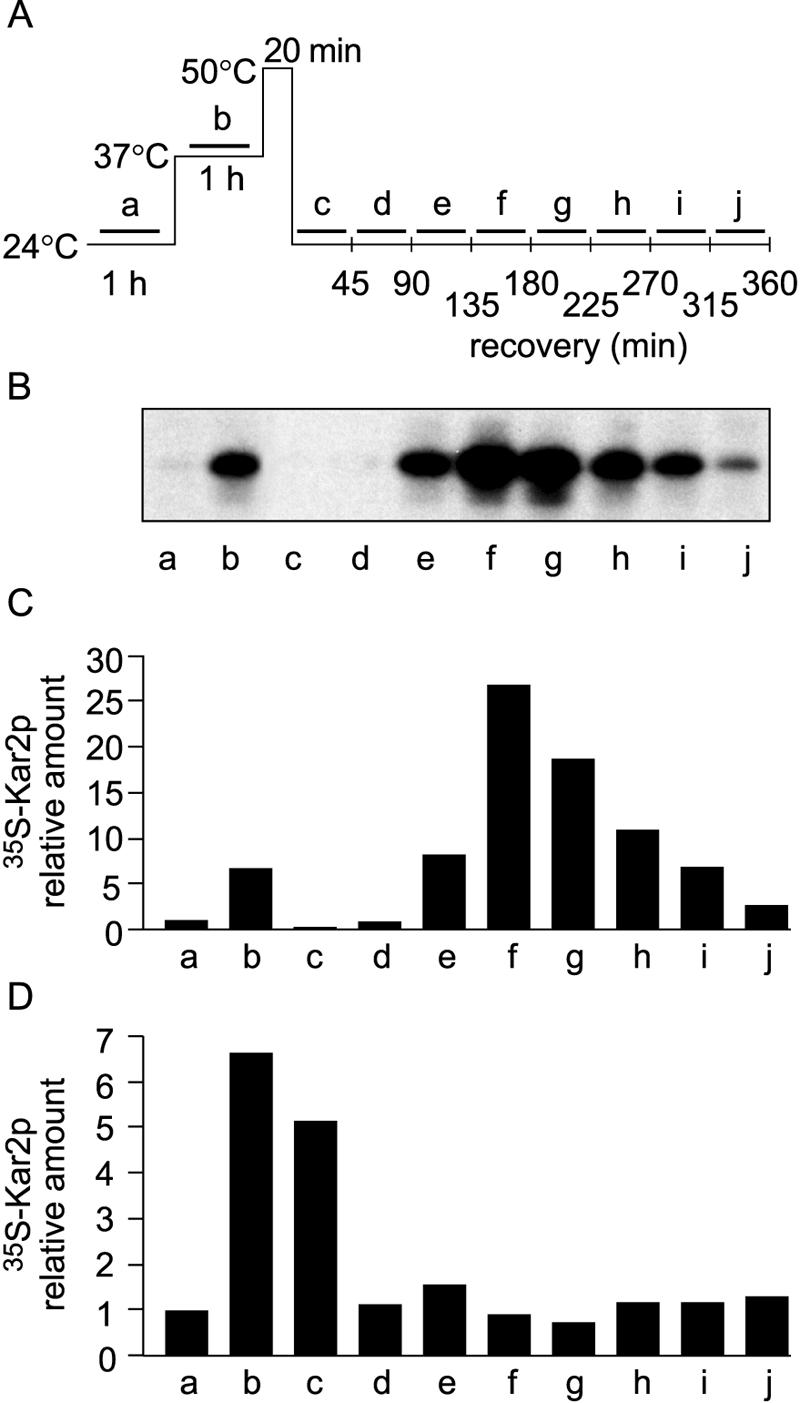
Upregulation of Kar2p expression during recovery from thermal insult at 24°C. (A) Schematic presentation of thermal treatments. (B) Metabolic labeling of Kar2p. Cells (strain H245) grown at 24°C were divided into 10 parallel samples and labeled with [35S]methionine-cysteine for successive 45-min periods, as indicated by the bars in panel A (a to j). After the termination of labeling, the cells were lysed for immunoprecipitation with Kar2p antiserum, and the immunoprecipitates were resolved by SDS-PAGE, followed by autoradiography. (C) Phosphorimager quantification of [35S]Kar2p levels in panel B. Normalization was done against the level of [35S]Kar2p in untreated cells (sample a). (D) The experiment in panel C was repeated, but with the 50°C treatment omitted.
Next, we performed the above experiment with strain H245, omitting the 50°C incubation step (Fig. 1D), for reference. Without the thermal insult, Kar2p expression was not induced during recovery at 24°C (samples c to j) but declined to the basal level within 90 min. Thus, the delayed upregulation of Kar2p expression resulted specifically from the thermal insult. We established earlier that bulk protein synthesis is negligible right after a thermal insult and recovers gradually to normal in 5 h (19, 40). Thus, the delayed upregulation of Kar2p expression was not due to a general increase in protein synthesis, but to a specific mechanism.
Delayed upregulation detected at the mRNA level.
Real-time quantitative PCR (qPCR) using KAR2 primers on total RNA from H245 cells demonstrated upregulation of the KAR2 gene at the mRNA level. Upon a shift from 24°C to 37°C, the level of KAR2 transcripts increased about sevenfold (Fig. 2B; results are means of three experiments with independently isolated RNA samples). After the thermal insult, the level abruptly decreased but then increased within the next 90 min to about sixfold the basal level, after which it declined. Thus, the mRNA level reached the maximum an hour before de novo BiP/Kar2p synthesis was maximal. To study whether DUR is specific for one ER chaperone only, i.e., BiP/Kar2p, we repeated the qPCR experiment with another ER chaperone, Lhs1p. The Hsp70 homologue Lhs1p is involved in the refolding of heat-denatured proteins (36, 37). The level of LHS1 mRNA increased 2.5-fold after 2 hours of recovery (Fig. 2C). Hsp78 is a mitochondrial chaperone required for mitochondrial thermotolerance and the resumption of respiratory function after a thermal insult (38). To study whether DUR is a phenomenon which extends even beyond the cytosol and the ER, we performed the qPCR experiment with HSP78. The level increased 15-fold after 2 hours of recovery (Fig. 2D). These results show that delayed upregulation is a mechanism controlling chaperone expression in the cytosol, the ER, and mitochondria during recovery from thermal insult.
FIG. 2.
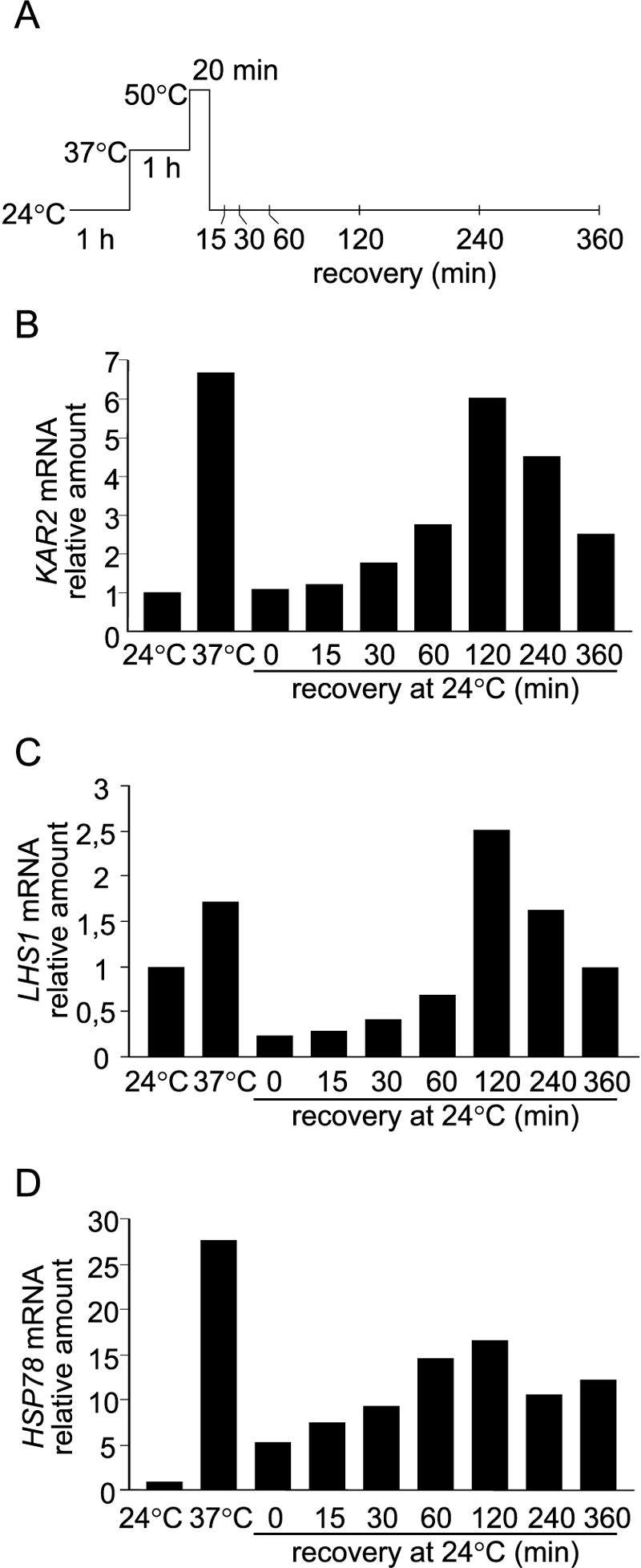
Real-time quantitative PCR analysis of mRNA levels. (A) Cells (H245) were exposed to heat treatments and harvested at the indicated time points. Total RNA was extracted, and 2-μg samples were subjected to reverse transcription-PCR analysis with KAR2 (B), LHS1 (C), or HSP78 primers (D) (see Materials and Methods). The value for untreated cells at 24°C was set to 1.
Promoter elements of KAR2 responsible for DUR.
KAR2 is an essential gene, and mutation of its upstream sequences is potentially lethal. Thus, to study which regulatory elements of the KAR2 promoter are responsible for DUR, we left the genomic KAR2 gene intact and used ΔICL1-lacZ fusion constructs, where the E. coli β-galactosidase gene was placed under the control of different KAR2 promoter variants (49). Strain H1806 (Table 1) contains the KAR2 promoter fragment known to be sufficient for KAR2 expression (nucleotides −280 to −109) (29) directly upstream of the lacZ reporter (Fig. 3 shows a schematic presentation of KAR2 promoter constructs). H1806 cells were exposed to the heat treatments and metabolic labeling, and β-galactosidase expression was monitored by immunoprecipitation, like the BiP/Kar2p expression shown in Fig. 1A to C. Enzymatic assays were used only for measuring basal activity at 24°C due to eventual thermal inactivation and reactivation of β-galactosidase molecules synthesized prior to the thermal insult. After the thermal insult at 50°C, the expression of β-galactosidase under the control of the KAR2 promoter was regulated similarly to that of authentic genomic Kar2p (Fig. 3B; data are means of six experiments). Thus, this assay system was found to be functional for investigating the role of KAR2 promoter elements in DUR.
Next, we investigated the contribution to DUR of the different elements of the KAR2 promoter by using a series of KAR2 promoter-lacZ fusion construct variants (Fig. 3). The KAR2 promoter harbors the following three elements that control basal and stress-induced transcription: the HSE, a GC-rich region, and the UPRE (29). To investigate the significance of the HSE for DUR, we used a KAR2 promoter-lacZ fusion construct containing only the HSE of the KAR2 promoter. This construct was transformed into yeast strain H245, resulting in strain H1769 (Table 1 and Fig. 3D). Metabolic labeling experiments and immunoprecipitation with a β-galactosidase antibody showed that in strain H1769, DUR occurred as in strain H1806, which contained an unmodified KAR2 promoter (Fig. 3D). This suggested that the HSE alone could evoke DUR. The basal expression of β-galactosidase was abolished in strain H1769. However, basal expression was maintained when the GC-rich region was present in addition to the HSE, whether the 5′ region was present (strain 1766; Fig. 3E) or not (strain 1768; Fig. 3F). These data are in agreement with previous work showing that the GC-rich region regulates the basal expression of KAR2 (29).
The HSE consensus sequence consists of inverted GAA repeats (1). Next, we mutated the four consecutive GAA repeats in the KAR2 HSE (see Materials and Methods). The mutated KAR2 promoter-lacZ fusion construct was transformed into yeast strain H245, resulting in strain H2004 (Table 1 and Fig. 3G). In the absence of a functional HSE, the upregulation of β-galactosidase at 37°C (HS column in Fig. 3) was reduced compared to that in control strain H1806, containing an unmodified KAR2 promoter. This was expected because the HSE has been reported to regulate the induction of KAR2 at 37°C (21, 29). However, DUR occurred similarly to that in the control strain H1806 (Fig. 3C and G). This shows that some other promoter element can evoke DUR in the absence of a functional HSE. Next, we investigated whether backing up the HSE was accomplished by the UPRE. First, we used a construct with a mutated HSE and a deleted UPRE (strain H1767; Fig. 3H). In strain H1767, β-galactosidase expression was undetectable both by SDS-PAGE analysis and according to enzymatic assays (Fig. 3H). Evidently, the GC-rich region could not drive DUR in the absence of the HSE and the UPRE. This is in agreement with the work of Zimmer and coworkers (49), who reported a lack of β-galactosidase activity under ER stress conditions for this construct. We then used a construct containing only the UPRE in the KAR2 promoter (strain H1765; Fig. 3I). Weak DUR was detected. This suggests that the UPRE is able to drive DUR on its own, albeit with a lower efficiency than that by the HSE.
The induction of BiP/Kar2p expression in response to a perturbation of protein folding in the ER lumen activates the unfolded protein response (UPR) pathway and occurs via the UPRE (21, 29). The UPR signal is transferred from the ER lumen to the cytosol by the transmembrane kinase Ire1p (7, 28). Ire1p causes splicing of HAC1 mRNA. Hac1p is the transcription factor that recognizes the UPRE in the target genes (8, 27). To study the involvement of these key proteins of the unfolded protein response in DUR, we performed the above experiments with strains H1807 and H1808, which harbor the β-galactosidase reporter construct under the control of an unmodified KAR2 promoter in strains lacking the IRE1 or HAC1 gene, respectively (Fig. 3J and K). Both deletions block the activation of the UPR pathway (7, 27). In these strains, DUR occurred to the same extent as in the reference strain H1806 (Fig. 3J and K). Both strains survived the thermal treatments, as their bulk protein synthesis and rate of glucose consumption after thermal insult were similar to those in normal cells (not shown). These data show that in the absence of the unfolded protein response, the HSE alone was responsible for DUR. Next, we transformed the strain lacking IRE1 with the KAR2 promoter-lacZ construct with a mutated HSE, resulting in strain H2010. For this strain, the induction of β-galactosidase expression at 37°C was abolished, as expected, due to the absence of a functional HSE (Fig. 3L). During recovery, β-galactosidase expression did not peak before 3 h of recovery, as with DUR, but started to resume only gradually. A maximum which was 1.5 times the basal expression level at 24°C before the heat treatments was reached only after 4.5 h of recovery. This level did not decrease later, but instead persisted. We have previously shown with numerous strains from different genetic backgrounds that total protein synthesis is abolished by thermal insult and resumes after 4 to 5 h of recovery to levels between one and two times the basal expression level (19, 40, 41; unpublished data). Thus, the slow recovery of β-galactosidase expression to a steady level in strain H2010 must have been due to the resumption of protein synthesis and not to DUR. Thus, a functional UPRE could not drive DUR in the absence of a functional unfolded protein response pathway. In conclusion, the HSE was sufficient to drive DUR. The UPRE could back up the HSE to some extent and worked via the unfolded protein response pathway.
Biological functions of BiP/Kar2p after thermal insult.
Kar2p functions in the translocation of polypeptides into the ER, in the folding of newly synthesized polypeptides emerging into the ER through the translocon, and in the retention of incorrectly folded or assembled proteins in the ER (for a review, see reference 13). After a thermal insult, BiP/Kar2p synthesis resumes after 2 hours of recovery (Fig. 1). To study the recovery of the biological functions of BiP/Kar2p after thermal insult, we followed the translocation of a reporter protein, Hsp150Δ-β-lactamase. The Hsp150Δ fragment is a signal peptide-containing, 321-amino-acid N-terminal fragment of the natural yeast secretory glycoprotein Hsp150 (34) which helps the β-lactamase portion fused to its C terminus to fold properly in the yeast ER (42). The Hsp150 signal peptide confers posttranslational translocation (32). The cytosolic, ER, and mature forms of the reporter can be distinguished by their different extents of O-glycosylation, as shown earlier (32). In SDS-PAGE, the unglycosylated cytoplasmic form migrates like a 66-kDa protein, the ER-resident form migrates like a 110-kDa protein, and the fully glycosylated, mature form of Hsp150Δ-β-lactamase migrates like a 145-kDa protein (11, 32). Hsp150Δ-β-lactamase was expressed under the control of the heat-activated HSP150 promoter (34). Cells (strain H335) were pulse labeled with [35S]methionine-cysteine for 5 min (Fig. 4A, bars). After different chase periods, aliquots were removed and the media were separated from the cells, which were then lysed. Immunoprecipitation with β-lactamase antiserum was performed with the cell lysates and the media, and the immunoprecipitates were resolved by SDS-PAGE, followed by autoradiography and phosphorimager quantification.
FIG. 4.

Translocation and secretion of a reporter glycoprotein during recovery from thermal insult. (A) Schematic presentation of the experiment. Cells (strain H335) were divided into six parallel samples, subjected to thermal treatments, labeled with [35S]methionine-cysteine for 5 min, as indicated by the bars, and chased for different periods, as indicated in panel B. After the termination of labeling, the growth media were separated from the cells, which were then lysed. The cell lysates and growth media were subjected to immunoprecipitation with β-lactamase antiserum, and the immunoprecipitates were resolved by SDS-PAGE, followed by autoradiography. (B) Cytoplasmic (66 kDa; triangles), ER-specific (110 kDa; squares), and mature (145 kDa; circles) forms of β-lactamase were quantified by phosphorimaging and plotted against the chase time.
After the pulse at 24°C, 60% of the β-lactamase was found in the unglycosylated 66-kDa cytoplasmic form, 13% was in the 110-kDa ER form, and 26% was in the fully glycosylated 145-kDa form (Fig. 4B, panel a). During the chase, the fraction of mature molecules increased and that of the cytosolic form decreased. The portion of ER-resident β-lactamase (Fig. 4B, panel a, squares) remained low, which probably reflects an equal flow of translocation into and exit from the ER. Finally, after 30 min of chase, practically all of the β-lactamase was in the mature form (Fig. 4B, panel a, circles), meaning that it had exited the ER and become glycosylated in the Golgi. After pretreatment at 37°C and a thermal insult at 50°C, a 5-minute pulse failed to label β-lactamase molecules (not shown) due to a halt of protein synthesis (19, 40). After 1 h of recovery at 24°C, a 5-minute pulse followed by different chase times showed that sufficient β-lactamase was produced that the experiment could be performed (Fig. 4B, panel b). About 80% of the β-lactamase was in the cytoplasm after the pulse and was not translocated after 40 min of chase (Fig. 4B, panel b, triangles). When the cells were pulse labeled after 2 h of recovery at 24°C, <20% was mature and 80% was cytoplasmic after the 40-min chase (Fig. 4B, panel c). After 3 h of recovery, the cytoplasmic pool decreased from 90% to 40% (Fig. 4B, panel d, triangles), while the mature pool only increased from 3% to 35% (circles) during the chase. Concomitantly, the ER-resident pool increased from 5% to 20% (squares), suggesting that molecules did not efficiently exit the ER. After 4 h of recovery, the translocation kinetics was only slightly slower than before the thermal insult (Fig. 4B, panel e). Finally, after 6 h of recovery, the translocation and maturation kinetics had recovered to the same level as that at 24°C before the heat treatments (Fig. 4B, panel f). In conclusion, ER translocation resumed concomitantly with the enhanced synthesis of BiP/Kar2p after 3 h of recovery, after which ER exit and protein secretion also resumed.
DISCUSSION
We showed earlier that exocytic proteins located in the ER at the time of thermal insult can be denatured and thereafter renatured during recovery at a physiological temperature by an ATP-dependent mechanism (19). At 50°C, a reporter enzyme lost its activity and formed secretion-incompetent aggregates. During the recovery period at 24°C, the aggregates slowly disassembled, and the enzyme thereafter regained its activity, finally exited the ER, and was secreted into the culture medium (36). While the denatured molecules were undergoing repair, their native counterparts synthesized during the recovery period bypassed the damaged ones and were secreted normally (37). Lhs1p belongs to the GRP170 subgroup of the Hsp70 family, shares 24% identical amino acids with BiP/Kar2p, and resides in the ER (9, 33). We found that Lhs1p was necessary for the conformational repair of denatured protein aggregates. Both Lhs1p and BiP/Kar2p were found to be associated with the aggregates (19, 36). Unlike BiP/Kar2p, Lhs1p is not essential under normal conditions but is required for the efficient acquisition of thermotolerance. Whether BiP/Kar2p also has a role in conformational repair of the damaged proteins is not known.
Here we showed that the expression of BiP/Kar2p, Lhs1p, and Hsp78 was subject to DUR during recovery at a physiological temperature following a thermal insult. When cells which had been preconditioned at 37°C and treated for 20 min at 50°C were shifted back to 24°C, the level of newly synthesized Kar2p/BiP was first negligible. It then started to increase slowly, reaching after 2.5 h of recovery a maximum level which was many times higher than that at 37°C. Thereafter, the rate of synthesis started to decline and was normal after 6 h. The expression of Lhs1p and Hsp78 also underwent delayed upregulation after a thermal insult.
The KAR2 promoter harbors two stress-responsive elements. The HSE is responsible for the upregulation of KAR2 expression when the cells are shifted from a physiological temperature to the heat shock temperature 37°C. The UPRE drives upregulation when unfolded proteins accumulate in the ER. Both promoter elements act independently (21, 29). We found that the HSE in the KAR2 promoter alone was able to drive DUR. The HSE has been thought to serve the upregulation of Kar2p expression upon a shift of the cells from a physiological temperature to 37°C (21). However, its regulatory role appears to be much more prominent in DUR. Also the UPRE alone could evoke DUR, albeit less efficiently than the HSE. Thus, the HSE and the UPRE could act independently and were able to back each other up.
The expression of BiP/Kar2p is rapidly elevated when newly synthesized and translocated polypeptides fail to acquire the proper conformation in the ER. The accumulation of misfolded proteins is signaled from the ER lumen to the cytosol via the transmembrane kinase Ire1p, leading to activation of the UPRE by the transcription factor Hac1p (7, 21, 28, 29). We have shown before that correctly folded proteins residing in the ER at the time of thermal insult are instantly denatured and aggregated and then slowly renatured while the cells are recovering at a physiological temperature (19, 37). The functioning of the UPRE in DUR was also dependent on Ire1p. Thus, the heat-denatured proteins in the ER did elicit the activation of the unfolded protein response, but only hours after the cells had been recovering at a physiological temperature.
We found that after a thermal insult, BiP/Kar2p synthesis was abolished. This prompted us to ask whether the BiP/Kar2p molecules that existed prior to the thermal insult were damaged at 50°C. We found that for some hours after the thermal insult, newly synthesized reporter polypeptides were not translocated, as they accumulated in the cell in a cytosolic unglycosylated form. Thus, the heat-treated BiP/Kar2p molecules were not able to perform the translocation function, or other vital components required for ER translocation were inactivated at 50°C. After 4 h of recovery at 24°C, when BiP/Kar2p expression was upregulated due to DUR, the reporter protein was translocated and glycosylated, exited the ER, and was secreted into the medium. Earlier, we found that the intracellular transport of vacuolar carboxypeptidase Y and cell wall invertase also resumed after 3 to 4 h of recovery (36, 37). Thus, the enhanced synthesis of new Kar2p/BiP molecules accomplished by DUR may serve to ensure the resumption of translocation and ER exit functions. Under ER stress conditions, elevated amounts of BiP/Kar2p have been reported to restore a cell growth defect caused by overproduction of Δpro, a mutated fungal secretory proteinase, which accumulates as misfolded aggregates which bind directly to BiP/Kar2p in the yeast ER lumen (48).
We have shown before that major cellular functions are abolished by severe heat stress at 48 to 50°C. After a shift of the cells to a physiological temperature, these functions do resume, slowly but efficiently. ATP production resumes in about 4 h. Protein synthesis is normal after 4 to 6 h, and cell division is normal after 20 h (17, 19, 40, 41). The inhibition of these functions must be due to protein denaturation, followed by reactivation due to conformational repair performed by chaperones. Here we showed that the expression of chaperones in the cytosol, the ER, and the mitochondria is upregulated during recovery at a physiological temperature from thermal insult. DUR of chaperones must be part of the molecular mechanisms ensuring the survival of cells after severe heat stress. We propose that the production of excessive levels of chaperones during recovery from thermal insult serves to ensure efficient conformational repair of heat-denatured proteins and thus cell survival under extreme environmental conditions. Because unicellular organisms cannot rely on apoptosis to remove damaged cells, survival is dependent on the conformational repair of denatured proteins.
Acknowledgments
This work was supported by the Academy of Finland (grant 1780444) and the Sigrid Jusélius Foundation. M.M. is a Biocentrum Helsinki Fellow, and L.S. is a Ph.D. student of the Helsinki Graduate School in Biotechnology and Molecular Biology.
We thank Ricardo Nunes Bastos for constructing plasmid pKTH5222, Heidi Holkeri for creating strain H720, and Anna Liisa Nyfors for excellent technical assistance. We thank Thomas Zimmer, Friedrich Schiller University, Jena, Germany, for the KAR2 promoter-lacZ constructs, Colin J. Stirling, University of Manchester, Manchester, United Kingdom, for the Kar2p antibody, Mary-Jane Gething, University of Melbourne, Melbourne, Australia, for the IRE1 disruption plasmid, and the groups named in Table 1, who generously donated yeast strains.
REFERENCES
- 1.Amin, J., J. Ananthan, and R. Voellmy. 1988. Key features of heat shock regulatory elements. Mol. Cell. Biol. 8:3761-3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amorós, M., and F. Estruch. 2001. Hsf1p and Msn2/4p cooperate in the expression of Saccharomyces cerevisiae genes HSP26 and HSP104 in a gene- and stress type-dependent manner. Mol. Microbiol. 39:1523-1532. [DOI] [PubMed] [Google Scholar]
- 3.Blond-Elguindi, S., S. E. Cwirla, W. J. Dower, R. J. Lipshutz, S. R. Sprang, J. F. Sambrook, and M. J. Gething. 1993. Affinity panning of a library of peptides displayed on bacteriophages reveals the binding specificity of BiP. Cell 75:717-728. [DOI] [PubMed] [Google Scholar]
- 4.Brodsky, J. L., and R. Schekman. 1993. A Sec63p-BiP complex from yeast is required for protein translocation in a reconstituted proteoliposome. J. Cell Biol. 123:1355-1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caspary, F., A. Hartig, and H.-J. Schüller. 1997. Constitutive and carbon source-responsive promoter elements are involved in the regulated expression of the Saccharomyces cerevisiae malate synthase gene MLS1. Mol. Gen. Genet. 255:619-627. [DOI] [PubMed] [Google Scholar]
- 6.Corsi, A. K., and R. Schekman. 1997. The lumenal domain of Sec63p stimulates the ATPase activity of BiP and mediates BiP recruitment to the translocon in Saccharomyces cerevisiae. J. Cell Biol. 137:1483-1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cox, J. S., C. E. Shamu, and P. Walter. 1993. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73:1197-1206. [DOI] [PubMed] [Google Scholar]
- 8.Cox, J. S., and P. Walter. 1996. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 87:391-404. [DOI] [PubMed] [Google Scholar]
- 9.Craven, R. A., J. R. Tyson, and C. J. Stirling. 1997. A novel subfamily of Hsp70s in the endoplasmic reticulum. Trends Cell Biol. 7:277-282. [DOI] [PubMed] [Google Scholar]
- 10.Cyr, D. M., T. Langer, and M. G. Douglas. 1994. DnaJ-like proteins: molecular chaperones and specific regulators of Hsp70. Trends Biochem. Sci. 19:176-181. [DOI] [PubMed] [Google Scholar]
- 11.Fatal, N., L. Karhinen, E. Jokitalo, and M. Makarow. 2004. Active and specific recruitment of a soluble cargo protein for endoplasmic reticulum exit in the absence of functional COPII component Sec24p. J. Cell Sci. 117:1665-1673. [DOI] [PubMed] [Google Scholar]
- 12.Flynn, G. C., T. G. Chappell, and J. E. Rothman. 1989. Peptide binding and release by proteins implicated as catalysts of protein assembly. Science 245:385-390. [DOI] [PubMed] [Google Scholar]
- 13.Gething, M. J. 1999. Role and regulation of the ER chaperone BiP. Semin. Cell Dev. Biol. 10:465-472. [DOI] [PubMed] [Google Scholar]
- 14.Gething, M. J., K. McCammon, and J. Sambrook. 1986. Expression of wild-type and mutant forms of influenza hemagglutinin: the role of folding in intracellular transport. Cell 46:939-950. [DOI] [PubMed] [Google Scholar]
- 15.Glick, B. S. 1995. Can Hsp70 proteins act as force-generating motors? Cell 80:11-14. [DOI] [PubMed] [Google Scholar]
- 16.Grably, M. R., A. Stanhill, O. Tell, and D. Engelberg. 2002. HSF and Msn2/4p can exclusively or cooperatively activate the yeast HSP104 gene. Mol. Microbiol. 44:21-35. [DOI] [PubMed] [Google Scholar]
- 17.Hänninen, A. L., M. Simola, N. Saris, and M. Makarow. 1999. The cytoplasmic chaperone Hsp104 is required for conformational repair of heat-denatured proteins in the yeast endoplasmic reticulum. Mol. Biol. Cell 10:3623-3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hurtley, S. M., D. G. Bole, H. Hoover-Litty, A. Helenius, and C. S. Copeland. 1989. Interactions of misfolded influenza virus hemagglutinin with binding protein (BiP). J. Cell Biol. 108:2117-2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jämsä, E., N. Vakula, A. Arffman, I. Kilpeläinen, and M. Makarow. 1995. In vivo reactivation of heat-denatured protein in the endoplasmic reticulum of yeast. EMBO J. 14:6028-6033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jensen, R. E., and A. E. Johnson. 1999. Protein translocation: is Hsp70 pulling my chain? Curr. Biol. 9:R779-R782. [DOI] [PubMed] [Google Scholar]
- 21.Kohno, K., K. Normington, J. Sambrook, M. J. Gething, and K. Mori. 1993. The promoter region of the yeast KAR2 (BiP) gene contains a regulatory domain that responds to the presence of unfolded proteins in the endoplasmic reticulum. Mol. Cell. Biol. 13:877-890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krzewska, J., T. Langer, and K. Liberek. 2001. Mitochondrial Hsp78, a member of the Clp/Hsp100 family in Saccharomyces cerevisiae, cooperates with Hsp70 in protein refolding. FEBS Lett. 489:92-96. [DOI] [PubMed] [Google Scholar]
- 23.Lindquist, S., and G. Kim. 1996. Heat-shock protein 104 expression is sufficient for thermotolerance in yeast. Proc. Natl. Acad. Sci. USA 93:5301-5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matlack, K. E., B. Misselwitz, K. Plath, and T. A. Rapoport. 1999. BiP acts as a molecular ratchet during posttranslational transport of prepro-alpha factor across the ER membrane. Cell 97:553-564. [DOI] [PubMed] [Google Scholar]
- 25.McClellan, A. J., J. B. Endres, J. P. Vogel, D. Palazzi, M. D. Rose, and J. L. Brodsky. 1998. Specific molecular chaperone interactions and an ATP-dependent conformational change are required during posttranslational protein translocation into the yeast ER. Mol. Biol. Cell 9:3533-3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKay, D. B. 1993. Structure and mechanism of 70-kDa heat-shock-related proteins. Adv. Protein Chem. 44:67-98. [DOI] [PubMed] [Google Scholar]
- 27.Mori, K., T. Kawahara, H. Yoshida, H. Yanagi, and T. Yura. 1996. Signalling from endoplasmic reticulum to nucleus: transcription factor with a basic-leucine zipper motif is required for the unfolded protein-response pathway. Genes Cells 1:803-817. [DOI] [PubMed] [Google Scholar]
- 28.Mori, K., W. Ma, M. J. Gething, and J. Sambrook. 1993. A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 74:743-756. [DOI] [PubMed] [Google Scholar]
- 29.Mori, K., A. Sant, K. Kohno, K. Normington, M. J. Gething, and J. F. Sambrook. 1992. A 22 bp cis-acting element is necessary and sufficient for the induction of the yeast KAR2 (BiP) gene by unfolded proteins. EMBO J. 11:2583-2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Normington, K., K. Kohno, Y. Kozutsumi, M. J. Gething, and J. Sambrook. 1989. S. cerevisiae encodes an essential protein homologous in sequence and function to mammalian BiP. Cell 57:1223-1236. [DOI] [PubMed] [Google Scholar]
- 31.Parsell, D. A., A. S. Kowal, M. A. Singer, and S. Lindquist. 1994. Protein disaggregation mediated by heat-shock protein Hsp104. Nature 372:475-478. [DOI] [PubMed] [Google Scholar]
- 32.Paunola, E., T. Suntio, E. Jämsä, and M. Makarow. 1998. Folding of active beta-lactamase in the yeast cytoplasm before translocation into the endoplasmic reticulum. Mol. Biol. Cell 9:817-827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rasmussen, S. W. 1994. Sequence of a 20.7 kb region of yeast chromosome XI includes the NUP100 gene, an open reading frame (ORF) possibly representing a nucleoside diphosphate kinase gene, tRNAs for His, Val and Trp in addition to seven ORFs with weak or no significant similarity to known proteins. Yeast 10(Suppl. A):S69-S74. [DOI] [PubMed] [Google Scholar]
- 34.Russo, P., M. Simonen, A. Uimari, T. Teesalu, and M. Makarow. 1993. Dual regulation by heat and nutrient stress of the yeast HSP150 gene encoding a secretory glycoprotein. Mol. Gen. Genet. 239:273-280. [DOI] [PubMed] [Google Scholar]
- 35.Sanchez, Y., and S. L. Lindquist. 1990. HSP104 required for induced thermotolerance. Science 248:1112-1115. [DOI] [PubMed] [Google Scholar]
- 36.Saris, N., H. Holkeri, R. A. Craven, C. J. Stirling, and M. Makarow. 1997. The Hsp70 homologue Lhs1p is involved in a novel function of the yeast endoplasmic reticulum, refolding and stabilization of heat-denatured protein aggregates. J. Cell Biol. 137:813-824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saris, N., and M. Makarow. 1998. Transient ER retention as stress response: conformational repair of heat-damaged proteins to secretion-competent structures. J. Cell Sci. 111:1575-1582. [DOI] [PubMed] [Google Scholar]
- 38.Schmitt, M., W. Neupert, and T. Langer. 1996. The molecular chaperone Hsp78 confers compartment-specific thermotolerance to mitochondria. J. Cell Biol. 134:1375-1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scidmore, M. A., H. H. Okamura, and M. D. Rose. 1993. Genetic interactions between KAR2 and SEC63, encoding eukaryotic homologues of DnaK and DnaJ in the endoplasmic reticulum. Mol. Biol. Cell 4:1145-1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seppä, L., A. L. Hänninen, and M. Makarow. 2004. Upregulation of the Hsp104 chaperone at physiological temperature during recovery from thermal insult. Mol. Microbiol. 52:217-225. [DOI] [PubMed] [Google Scholar]
- 41.Simola, M., A. L. Hänninen, S. M. Stranius, and M. Makarow. 2000. Trehalose is required for conformational repair of heat-denatured proteins in the yeast endoplasmic reticulum but not for maintenance of membrane traffic functions after severe heat stress. Mol. Microbiol. 37:42-53. [DOI] [PubMed] [Google Scholar]
- 42.Simonen, M., E. Jämsä, and M. Makarow. 1994. The role of the carrier protein and disulfide formation in the folding of beta-lactamase fusion proteins in the endoplasmic reticulum of yeast. J. Biol. Chem. 269:13887-13892. [PubMed] [Google Scholar]
- 43.Simons, J. F., S. Ferro-Novick, M. D. Rose, and A. Helenius. 1995. BiP/Kar2p serves as a molecular chaperone during carboxypeptidase Y folding in yeast. J. Cell Biol. 130:41-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singer, M. A., and S. Lindquist. 1998. Multiple effects of trehalose on protein folding in vitro and in vivo. Mol. Cell 1:639-648. [DOI] [PubMed] [Google Scholar]
- 45.Steel, G. J., D. M. Fullerton, J. R. Tyson, and C. J. Stirling. 2004. Coordinated activation of Hsp70 chaperones. Science 303:98-101. [DOI] [PubMed] [Google Scholar]
- 46.Thomas, B. J., and R. Rothstein. 1989. Elevated recombination rates in transcriptionally active DNA. Cell 56:619-630. [DOI] [PubMed] [Google Scholar]
- 47.Treger, J. M., A. P. Schmitt, J. R. Simon, and K. McEntee. 1998. Transcriptional factor mutations reveal regulatory complexities of heat shock and newly identified stress genes in Saccharomyces cerevisiae. J. Biol. Chem. 273:26875-26879. [DOI] [PubMed] [Google Scholar]
- 48.Umebayashi, K., A. Hirata, H. Horiuchi, A. Ohta, and M. Takagi. 1999. Unfolded protein response-induced BiP/Kar2p production protects cell growth against accumulation of misfolded protein aggregates in the yeast endoplasmic reticulum. Eur. J. Cell Biol. 78:726-738. [DOI] [PubMed] [Google Scholar]
- 49.Zimmer, T., A. Ogura, A. Ohta, and M. Takagi. 1999. Misfolded membrane-bound cytochrome P450 activates KAR2 induction through two distinct mechanisms. J. Biochem. 126:1080-1089. [DOI] [PubMed] [Google Scholar]