

Abstract
The classical interferon (IFN)-dependent antiviral response to viral infection involves the regulation of IFN-stimulated genes (ISGs), one being the gene encoding cellular endoribonuclease RNase L, which arrests protein synthesis and induces apoptosis by nonspecifically cleaving rRNA. Recently, the herpes simplex virus type 1 (HSV-1) protein ICP0 has been shown to block the induction of ISGs by subverting the IFN pathway upstream of the 2′-5′-oligoadenylate synthetase (OAS)/RNase L pathway. We report that ICP0 also prevents rRNA degradation at late stages of HSV-1 infection, independent of its E3 ubiquitin ligase activity, and that the resultant rRNA degradation is independent of the classical RNase L antiviral pathway. Moreover, the degradation is independent of the viral RNase vhs and is independent of IFN response factor 3. These studies indicate the existence of another, previously unidentified, RNase that is part of the host antiviral response to viral infection.
Viral infection of mammalian cells induces a robust antiviral response intended to restrict viral replication and propagation. A predominant characteristic of this innate immune response is the expression and secretion of interferons (IFNs), a family of immunomodulatory cytokines with antiviral and antiproliferative activities (43). Once secreted from infected cells, the type I IFNs (alpha IFN [IFN-α] and IFN-β) bind to their cognate class II cytokine receptors in both paracrine and autocrine fashions and induce the phosphorylation of Jak/Stat molecules, leading to the nuclear translocation of activated Stat1-Stat2-IRF9 heterotrimers. These complexes bind to the IFN-stimulated response elements (ISRE) in promoters of IFN-stimulated genes (ISGs), resulting in the robust induction of these antiviral effector molecules. Among the ISGs most intently studied are the double-stranded RNA (dsRNA)-dependent protein kinase R (PKR), which induces protein synthesis arrest (52), and the ubiquitous 2′-5′-oligoadenylate synthetase (OAS) family of proteins, which function to promote RNA degradation (44). Once upregulated, PKR and OAS become activated by binding to dsRNA, a by-product of viral replication.
OAS catalyzes the synthesis of variable short 2′-5′-linked oligoadenylates (2′-5′A) from ATP, which in turn activate the latent endoribonuclease RNase L to cleave cellular and viral RNA (36). Infection with several viruses, including vaccinia virus and encephalomyocarditis virus, leads to RNase L-dependent rRNA degradation (44). Ultimately, rRNA cleavage results in protein synthesis inhibition and apoptosis and constitutes a significant cellular antiviral event to prevent viral propagation (7). Indeed, several viruses have evolved specific mechanisms to counteract the 2′-5′A pathway, including human immunodeficiency virus type 1, vaccinia virus, and the alphaherpesvirus herpes simplex virus type 1 (HSV-1) (8, 27, 53). HSV-1 infection of conjunctival cells induces the synthesis of 2′-5′A derivatives, which antagonize RNase L activation by competing with genuine 2′-5′A for binding to ankyrin repeats 7 and 8 on RNase L. In addition, HSV-1, like many other viruses, has been shown to inhibit IFN signaling pathways upstream of RNase L activation at multiple points (25, 31, 54, 55).
On the basis of the above findings, it appears that preventing cellular rRNA degradation is of great importance to viral propagation, and viral interference in the IFN-regulated OAS/RNase L pathway is paramount for viral replication and spread. Despite these observations, there is conflicting evidence for the specific contribution of RNase L to an antiviral state in HSV-1-infected cells. Studies with wild-type (wt) and RNase L knockout cells illustrated that HSV-1 infection does not significantly induce RNase L activity in vitro (47) and that the absence of RNase L does not significantly affect viral growth and virulence in an in vivo ocular model of HSV-1 infection (24). This observation is in contrast to another study that concluded that HSV-1 infection of RNase L knockout mice induces a significantly higher mortality rate and heightened susceptibility to herpetic disease and stromal keratitis compared to HSV-1 infection of wt mice (57). These data suggest that either RNase L does not significantly contribute to host defense against HSV-1 infection or that RNase L, as a component of the IFN signaling antiviral pathway, is inhibited during the course of HSV-1 infection by a viral factor.
Expressed early in infection, the HSV-1 immediate early (IE) protein infected-cell protein 0 (ICP0) is a multifunctional transcriptional activator of viral and cellular genes that synergistically functions with another IE protein, ICP4, for several of its transcriptional functions (14). In the absence of ICP0, initiation of lytic replication is diminished and latent genomes reactivate with decreased kinetics. In addition, ICP0 is responsible for surmounting a variety of cellular antiviral responses (14, 30-32). Upon translocating to the nucleus early in infection, ICP0 promotes the proteasome-dependent degradation of an array of cellular antiviral ISGs, including those encoding the nuclear body-associated proteins promyelocytic leukemia protein (PML) and Sp100 (9, 15, 34). To date, ICP0's biological effects have been found to require the N-terminal RING finger domain, which mediates E3 ubiquitin ligase activity (3). The resultant disruption of ND10 nuclear bodies, in addition to other IFN-induced pathways, diminishes cellular antiviral capacity. Recently, ICP0 has been shown to block ISG expression by inhibiting the key transcriptional activators IFN regulatory factor 3 (IRF3) and IRF7 (12, 25, 29). Moreover, ICP0 functions to counteract an IFN-induced barrier to virus replication (32, 33).
Since ICP0 is involved in subverting IFN signaling during the innate immune response to HSV-1 infection and RNase L-mediated rRNA degradation is a component of the cellular antiviral response, we set out to determine if ICP0 prevents cellular rRNA degradation during HSV-1 infection. We report that, in the absence of ICP0 expression, HSV infection results in RNase L- and IRF3-independent rRNA degradation in a variety of cell types at late times postinfection. The resultant rRNA degradation is independent of both the virion host shutoff (vhs) RNase and the E3 ubiquitin ligase activity of ICP0. These studies provide further evidence for the existence of another, previously unidentified cellular endoribonuclease that is part of a host antiviral response to viral infection.
MATERIALS AND METHODS
Cells and viruses.
Human embryonic lung (HEL) fibroblast, HepG2 hepatoma, U20S osteosarcoma, and Vero monkey kidney epithelial cells were obtained from the American Type Culture Collection and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% (Vero) or 10% (HEL, HepG2, and U2OS) fetal bovine serum (FBS) and 2 mM l-glutamine. RNase L−/− and RNase L+/+ murine embryo fibroblasts (MEFs; genetic background, C57BL/6) (58) and IRF3−/− and IRF3+/+ MEFs (genetic background, C57BL/6) (41) were maintained in α minimum essential medium supplemented with 10% FBS and 2 mM l-glutamine. A listing of all HSV-1 viruses used in this study is provided in Table 1. AdMLP0 is a type 5 adenovirus expressing ICP0 under the control of the major late promoter, while AdE1E3 is a control adenovirus containing deletions of the E1 and E3 transcriptional units (59). Wild-type and IE mutant HSV-1 infections were completed at multiplicities of infection (MOI) of 1 and 5 PFU per cell, respectively, in serum-free DMEM for 1 h. Adenovirus infections were completed at a MOI of 100 PFU/cell in phosphate-buffered saline (PBS) supplemented with 0.01% MgCl2 and 0.01% CaCl2 for 45 min. UV inactivation of viruses was performed with a UV Stratalinker 2400 (Stratagene) for the length of time required to drop infectious titers by greater than 5 orders of magnitude. Unless otherwise indicated, total cellular RNA was extracted 3 days after wt or mutant HSV infection and 24 h after adenovirus infection.
TABLE 1.
Properties of HSV-1 viruses used in this study
| Virus | Parent strain | Mutation | Reference |
|---|---|---|---|
| KOS | KOS | None (wt) | 46 |
| 7134 | KOS | ICP0 null | 6 |
| dlX3.1 | KOS | ICP0 null | 38 |
| n212 | KOS | ICP0 null | 5 |
| Δsma | KOS | vhs null | 37 |
| 7134Δsma | KOS | ICP0/vhs null | 25 |
| 17syn+ | 17 | None (wt) | 4 |
| d11403 | 17 | ICP0 null | 48 |
| FXE | 17 | RING finger deletion | 13 |
| d22lacZ | KOS | ICP22 null | 26 |
| N38 | KOS | ICP47 null | 50 |
| ΔICP6 | KOS | ICP6 null | 17 |
| d120 | KOS | ICP4 null | 10 |
| 5dl1.2 | KOS | ICP27 null | 28 |
Poly(IC) transfections and treatments.
Poly(IC) (Amersham-Pharmacia) was reconstituted in PBS at 5 mg/ml, denatured at 55°C for 15 min, and left to anneal at room temperature prior to transfection. Unless otherwise indicated, all cell lines were transfected with 1 μg poly(IC) per ml or 500 ng 2′-5′A (R. Silverman) using the Lipofectamine (LF) transfection reagent (Invitrogen) according to the manufacturer's protocol.
RNA extraction and analysis.
RNA was harvested using Trizol (Life Technologies) according to the manufacturer's recommendations. Five micrograms of RNA was diluted into a 3-N-morpholinopropanesulfonic acid (MOPS) solution containing 20% formaldehyde and 50% formamide. Samples were subjected to agarose-gel electrophoresis (1% agarose, 20% formaldehyde) in MOPS running buffer in the presence of ethidium bromide for 1.5 h at a potential difference of 100 V. RNA gels were scanned using the Typhoon Imager (Amersham Biosciences) using ImageQuant (Amersham Biosciences) software.
Viral titer determination.
HEL fibroblasts were seeded into six-well plates, and monolayers were infected with ICP0-null HSV-1 (7134; MOI, 5) or wt HSV-1 (KOS; MOI, 1). At various times postinfection, total virus from infected cultures was harvested and subsequently titered on U20S cells in the presence of 30 mM N,N′-hexamethylene-bisacetamide (HMBA; Sigma). Following 1 hour of inoculation, cell monolayers were maintained in DMEM supplemented with 1% human serum and 30 mM HMBA. After 3 days of plaque formation, cells were fixed in methanol and stained with Giemsa (Sigma). Each experiment was performed in triplicate, with the averages of three experiments illustrated.
RESULTS
ICP0 prevents HSV-1-induced rRNA degradation.
Given the role of ICP0 in mediating host antiviral responses, we sought to determine if ICP0 prevented rRNA degradation in HSV-1-infected cells by monitoring rRNA degradation following infection with wt and ICP0-null HSV. Although ICP0 mutants are grown under complementing conditions, their titers are reproducibly fivefold lower than that of wt virus (data not shown), due to an increase in the particle-to-PFU ratio (48). Thus in this and subsequent experiments, we utilized MOI of 1 and 5 for wt and ICP0-null HSV infections, respectively. Under these conditions, the overall levels and kinetics of viral protein expression in HEL fibroblasts were similar (data not shown). HEL fibroblasts infected with various ICP0-null HSV-1 mutants (described in Table 1), but not wt HSV-1, consistently demonstrated cellular 28S and 18S rRNA degradation after 3 days of infection (Fig. 1A). Moreover, after prolonged periods of KOS infection, we failed to detect rRNA degradation (data not shown). To determine if the rRNA degradation pattern was consistent with an RNase L-mediated event, we transfected HEL fibroblasts with poly(IC), a synthetic dsRNA polyribonucleotide derivative. rRNA degradation characteristic of RNase L activation was observed upon transfection of poly(IC) with LF, but not following treatment with either component on its own. Identical patterns were observed when 2′-5′A was used in place of poly(IC) (data not shown). The degradation patterns observed following transfection of poly(IC) or infection with ICP0-null HSV-1 were dissimilar, suggesting that RNase L may not mediate rRNA degradation following HSV-1 infection in the absence of ICP0 expression. One possible explanation for the disparate rRNA degradation profiles is a difference in degradation kinetics. To address this possibility, HEL fibroblasts were either transfected with poly(IC) or infected with ICP0-null HSV-1. As shown in Fig. 1B, poly(IC)-induced rRNA degradation appears within the first hour of transfection, with complete 28S rRNA degradation occurring within 3 hours, and involves the formation of a series of complex degradation products. In contrast, rRNA degradation following infection with ICP0-null HSV-1 is a late-stage event, with observable degradation 2 days postinfection, and involves the formation of only two major rRNA degradation products.
FIG. 1.

ICP0 prevents rRNA degradation induced following infection of HEL fibroblasts with HSV-1. RNA gel electrophoresis was performed in fibroblasts treated with LF, poly(IC), or LF plus poly(IC) or infected with wild-type HSV-1 viruses and mutant HSV-1 viruses derived from strain KOS (A, B, and D) or 17 (C).
In order to determine if the rRNA degradation following HSV-1 infection occurs in the absence of ICP0 with viral strains other than KOS, we infected HEL fibroblasts with wt strain 17 and its ICP0-null derivative dl1403. As shown in Fig. 1C, dl1403 induced an identical degradation pattern to that of 7134, which was absent in 17syn+-infected cells. Infection with the strain 17-derived ICP0 RING finger domain mutant FXE, which has abolished E3 ubiquitin ligase activity, did not elicit rRNA degradation.
Previous studies of HSV-1 replication have shown that ICP0 functions synergistically with other IE proteins, such as ICP4, in mediating some of its biological functions (14). To determine if ICP0 is the sole IE protein required for the prevention of rRNA degradation, HEL fibroblasts were infected with a panel of single IE mutant viruses (Table 1). Aside from 7134, no other IE mutant virus infection induced rRNA degradation (Fig. 1D), illustrating that ICP0 is the only IE protein required for the prevention of rRNA degradation in HSV-1-infected HEL fibroblasts.
Cell type specificity of ICP0-null-HSV-1-induced rRNA degradation.
Since the expression of RNase L is cell type specific (21, 35), we set out to determine if the rRNA degradation observed in ICP0-null-HSV-1-infected HEL fibroblasts can occur in other cell types. Infection of human osteosarcoma (U20S; Fig. 2A) and hepatoma (HepG2; Fig. 2B) cells with ICP0-null HSV-1 mutants consistently induced an rRNA degradation pattern similar to that in HEL fibroblasts, while KOS-infected cells displayed intact rRNA. Infection of green monkey kidney epithelial (Vero) and human alveolar epithelial (A549) cells with ICP0-null HSV-1 consistently induced rRNA degradation comparable to that of U20S cells (data not shown). Notably, the extent of rRNA degradation was reproducibly less in these cell lines compared to HEL fibroblasts (Fig. 1A). As with HEL fibroblasts, poly(IC) transfection induced rRNA degradation in A549, Vero, and U20S cells, indicating the presence of functional RNase L. In contrast, poly(IC) transfection of HepG2 cells did not induce rRNA degradation (Fig. 2B), which is consistent with the lack of functional RNase L in this cell line (49).
FIG. 2.

The extent of rRNA degradation observed following ICP0-null-HSV-1 infection is cell type specific. RNA gel analysis was performed on U20S (A) and HepG2 (B) cells transfected with poly(IC) or infected with the indicated HSVs.
rRNA degradation in ICP0-null-HSV-1-infected cells correlates with a reduction in viral titers.
As shown in Fig. 1 and 2, HSV-1 infection consistently induces rRNA degradation under conditions where ICP0 is absent in a variety of cell lines at later times of infection. In an effort to determine the biological significance of this late rRNA degradation event for HSV replication, HEL fibroblasts were infected with either KOS (MOI, 1) or 7134 (MOI, 5). The efficiency of virus replication was determined in a standard viral-growth assay. As shown in Fig. 3, the growth kinetics of KOS and 7134 were similar until 24 h postinfection, at which point 7134 titers were reduced relative to KOS. By 3 days postinfection, KOS titers were consistently ∼2 logs higher than that of 7134.
FIG. 3.

Reduction in ICP0-null-HSV titers at late times postinfection correlates with rRNA degradation. Total infectious virus was isolated from HEL fibroblasts infected with ICP0-null HSV-1 or wt HSV-1 at various times postinfection and titered on the complementing U20S cell line. Titer values were adjusted to 106 infected cells and plotted on a logarithmic scale. The results represent three separate experiments performed in triplicate, with error bars representing the standard deviations of individual experimental means.
rRNA degradation in ICP0-null-HSV-1-infected cells is independent of RNase L.
Given the difference in rRNA degradation patterns between transfected and infected cells (Fig. 1A and B) and the ability to induce rRNA degradation following infection in RNase L-deficient HepG2 cells (Fig. 2B), we set out to conclusively determine the role of RNase L in mediating rRNA degradation following ICP0-null-HSV-1 infection. We infected wt MEFs (Fig. 4A) and RNase L−/− MEFs (Fig. 4B) with KOS, 7134, dlX3.1, or n212. Both wt and RNase L−/− MEFs infected with ICP0-null HSV-1 displayed an rRNA degradation pattern similar to that in HEL fibroblasts. Consistent with the lack of RNase L, poly(IC)-induced rRNA degradation was observed in wt but not RNase L−/− MEFs. Taken together, these results show that the rRNA degradation observed following HSV-1 infection in the absence of ICP0 expression occurs independently of RNase L.
FIG. 4.

The rRNA degradation observed following ICP0-null-HSV-1 infection is independent of RNase L. RNA gel analysis of wt MEFs (A) and RNase L−/− MEFs (B) transfected with poly(IC) or infected with wt HSV-1 or ICP0-null HSV-1 (derived from strain KOS).
rRNA degradation in ICP0-null-HSV-1-infected cells is independent of the viral RNase vhs.
Since the rRNA degradation observed following ICP0-null-HSV-1 infection occurs in the absence of RNase L, then either a viral or an alternative cellular RNase is responsible for the observed degradation. Previously, HSV-1 vhs was shown to induce a rapid arrest of macromolecular biosynthesis by associating with the eukaryotic initiation factor 4H (eIF4H) and inducing nonspecific destabilization of cellular and viral mRNA (45). Due to its nuclease activity, we sought to determine if vhs contributes to the rRNA degradation observed following HSV-1 infection in the absence of ICP0 expression by infecting HEL fibroblasts with wt HSV-1 or mutants deficient in ICP0 and/or vhs. While ICP0-null-HSV (7134 and 7134Δsma)-infected cells displayed rRNA degradation, KOS- and vhs-null-HSV-infected cells possessed intact rRNA (Fig. 5), illustrating that the rRNA degradation observed following HSV-1 infection in the absence of ICP0 expression is independent of vhs.
FIG. 5.
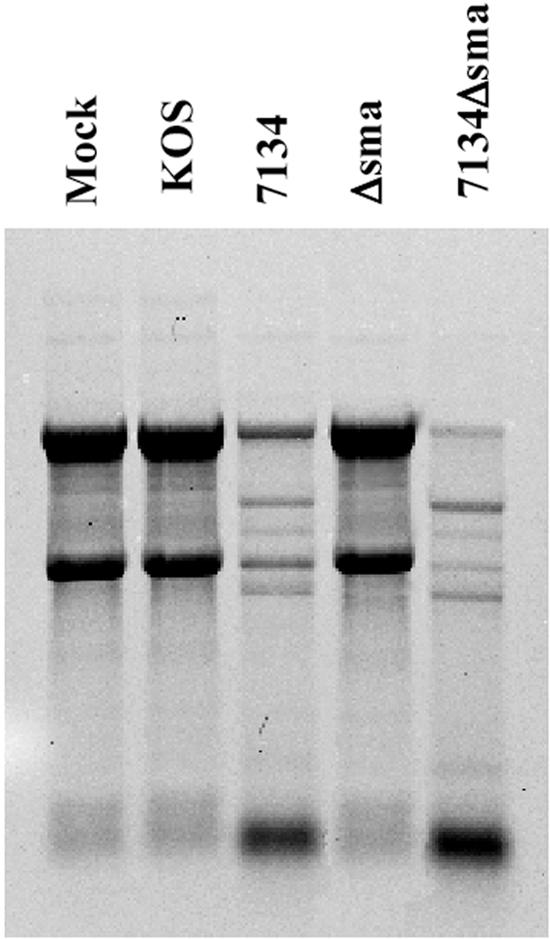
The rRNA degradation observed following ICP0-null-HSV-1 infection is independent of the viral RNase vhs. Shown is an RNA gel analysis of HEL fibroblasts infected with the indicated ICP0-null, vhs-null, or ICP0/vhs-null HSV-1 mutants.
ICP0 overexpression does not prevent RNase L-mediated rRNA degradation.
We have shown that infection with HSV-1 leads to RNase L-independent rRNA degradation that is blocked upon expression of ICP0. However, it remains unclear if ICP0 is capable of also counteracting the IFN-mediated RNase L pathway. To determine if ICP0 expression is sufficient to block RNase L-mediated rRNA degradation, we infected Vero cells with an adenovirus encoding ICP0 under the transcriptional control of the major late promoter (AdMLP0) and subsequently challenged the infected-cell cultures with poly(IC) transfection or ICP0-null-HSV infection (7134). Immunofluorescence microscopy illustrated that approximately 80% of cells expressed ICP0 following AdMLP0 infection (data not shown). In addition, Western blot analysis of AdMLP0- and KOS-infected-cell lysates indicated that both viruses expressed similar amounts of ICP0 (data not shown). While ICP0 preexpression prevented rRNA degradation in 7134-infected cells, thus restoring a wt phenotype, it did not prevent poly(IC)-induced, RNase L-mediated rRNA degradation (Fig. 6), illustrating that ICP0 does not inhibit RNase L-mediated rRNA degradation. Infection with a control adenovirus (AdE1E3) did not complement the ICP0-null phenotype. Moreover, poly(IC) transfection following KOS infection induced rRNA degradation, indicating that wt HSV does not prevent RNase L-mediated rRNA degradation (data not shown). Similar results were observed in AdMLP0-infected A549 cells (data not shown). To corroborate these observations, we utilized 0-28 cells, a stable Vero-derived cell line that expresses ICP0 under the control of its endogenous promoter. Under these conditions, similar results to Fig. 6 were observed (data not shown). Therefore, whether expressed by a recombinant adenovirus or wt HSV or within a stable cell line, ICP0 does not prevent RNase L-mediated rRNA degradation.
FIG. 6.
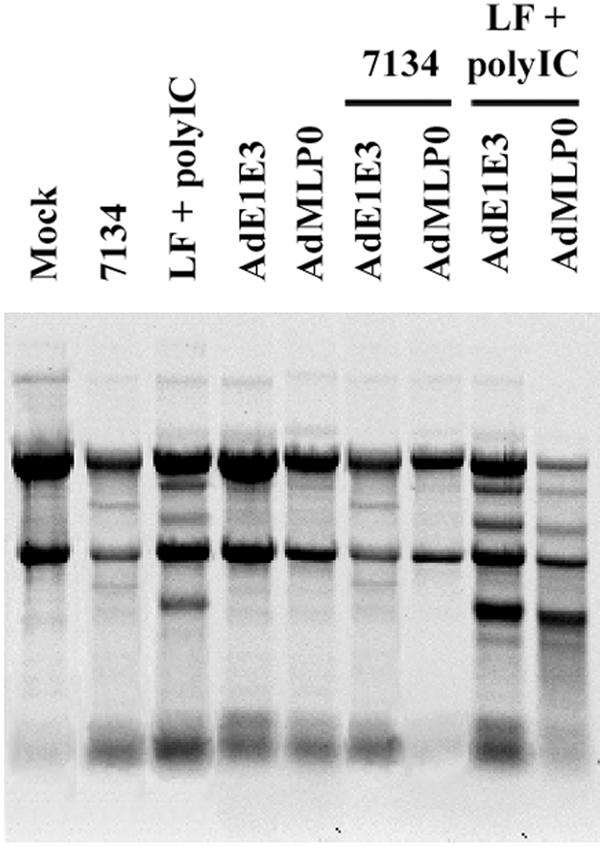
ICP0 overexpression does not prevent RNase L-specific rRNA degradation. Vero cells were infected with control adenovirus (AdE1E3) or adenovirus encoding ICP0 (AdMLP0) and subsequently challenged with poly(IC) transfection (10 ng) or HSV-1 infection.
rRNA degradation in ICP0-null-HSV-1-infected cells is independent of IRF3.
Given that rRNA degradation is a hallmark of apoptosis (19) and that ICP0 blocks IRF3 (25), a transcription factor implicated in apoptosis (20), we set out to determine the role of IRF3 in RNase L-dependent and -independent rRNA degradation. wt and IRF3−/− MEFs (Fig. 7A and B, respectively) were transfected with poly(IC) or infected with wt or ICP0-null HSV-1. Both wt and IRF3−/− MEFs infected with ICP0-null HSV-1 displayed rRNA degradation similar to that observed in HEL fibroblasts. Notably, poly(IC)-mediated rRNA degradation was not observed in IRF3−/− MEFs, suggesting that IRF3 is essential for RNase L-mediated rRNA degradation.
FIG. 7.

The rRNA degradation observed following ICP0-null-HSV-1 infection is independent of IRF3. RNA gel analysis was performed on wt MEFs (A) or IRF3−/− MEFs (B) transfected with poly(IC) or infected with the indicated wt and ICP0-null mutant (derived from strain KOS) HSVs.
To confirm these results, we investigated rRNA profiles following transfection of poly(IC) or infection with ICP0-null HSV-1 in Jak1-deficient parental cells (U4C) or a derivative, P2.1, that expresses only low levels of IRF3 (23). While poly(IC)-induced, RNase L-mediated rRNA degradation was markedly reduced in P2.1 cells compared to U4C cells, levels of HSV-1-mediated rRNA degradation were similar in both (data not shown). Taken together, these data indicate that, while ICP0-null-HSV-1-induced rRNA degradation is IRF3 independent, poly(IC)-induced RNase L-mediated rRNA degradation requires IRF3.
DISCUSSION
The innate antiviral response to viral replication involves the combined activities of ISGs in an effort to suppress viral replication and to induce apoptosis of infected cells to limit viral spread. Once activated by dsRNA, the endoribonuclease RNase L mediates both of these responses following infection with viruses such as vaccinia virus and encephalomyocarditis virus (7). Although HSV-1 has been shown to block RNase L-mediated RNA degradation by synthesizing decoy 2′-5′A derivatives that antagonize RNase L activity (8), the specific contribution of RNase L to host antiviral capacity remains controversial. However, HSV-1 countermeasures to other IFN pathways are well documented (30).
Since the HSV-1 IE protein ICP0 has been shown to be instrumental in surmounting several of these IFN-dependent antiviral pathways, we investigated the possibility that ICP0 prevents cellular rRNA degradation following HSV-1 infection by blocking RNase L activity. We observed that, in the absence of ICP0 expression, HSV-1 strains 17 and KOS induced rRNA degradation at late stages postinfection. Individual deletion of the remaining IE gene products failed to demonstrate the hallmarks of rRNA degradation. While this observation suggests that ICP0 is necessary to block rRNA degradation, it does not discount the possibility that an additional viral protein(s) is also involved. It is well established by Aubert and Blaho and Zachos et al. that ICP27-null-HSV infection results in many of the hallmarks associated with apoptosis through the destabilization of cellular Bcl-2 protein and a reduction in Bcl-2 RNA levels, including DNA fragmentation (1, 56). However, evidence exists which illustrates that rRNA degradation is a cell type-dependent occurrence that is independent of DNA fragmentation (39).
Intracellular antiviral pathways are predominantly mediated by immediate early response factors and are thus activated at early times of infection. Here, however, we detected rRNA degradation at late times of infection. While we currently do not fully understand the biological implications of a delayed cellular immune response, a number of factors could impact on the delayed kinetics we observed. As mentioned above, it is likely that viral proteins other than ICP0 assist in blocking this cellular antiviral response. Viruses routinely encode multiple proteins to disable cellular antiviral pathways (40, 42). Under conditions where only ICP0 activity is absent, rRNA degradation would remain partially inhibited, with subsequent degradation products requiring sufficient accumulation to become visible. Furthermore, these experiments were performed in vitro using relatively high multiplicities of infection, conditions that may not reflect in vivo infections. Notwithstanding these arguments, in a standard viral-growth assay viral titers in KOS- and 7134-infected cultures were similar at 24 h postinfection, when rRNA appears completely intact. However, by 2 days postinfection, when rRNA degradation becomes detectable in 7134-infected cultures, the production of 7134 decreases relative to KOS. Finally, by 3 days postinfection, when rRNA degradation is nearly complete in 7134-infected cultures, 7134 titers have dropped by approximately 2 logs compared to wild-type HSV-1. Thus the decrease in 7134 virus production correlates with rRNA degradation. This, along with our observations that ICP0-null mutants induce rRNA degradation, leads us to believe that an important function of ICP0 is the prevention of rRNA degradation. Of interest, rRNA degradation in the absence of ICP0 was consistently more pronounced in nonimmortalized, nontransformed cells. Given that IFN is both antiviral and antiproliferative in nature, it is likely that IFN-mediated immune responses are more intact in “primary” cells as opposed to immortalized or transformed cells.
Of particular interest, the ICP0 RING finger mutant was capable of blocking rRNA degradation, illustrating that the E3 ubiquitin ligase activity of ICP0 does not contribute to preventing rRNA degradation. Although ICP0 contains multiple functional domains, including a nuclear localization signal, a herpesvirus-associated ubiquitin-specific protease binding domain, and an ND10 localization domain, the biological functions of ICP0 have to date been found to rely on its E3 ubiquitin ligase activity (14, 25). Therefore, this is the first ICP0-regulated biological phenomenon that is independent of ICP0's E3 ubiquitin ligase activity. Studies are under way to determine the mechanism(s) whereby ICP0 blocks rRNA degradation following HSV-1 infection.
There are three mutually exclusive mechanisms that could account for the observed rRNA degradation following ICP0-null-HSV-1 infection. In the first mechanism, RNase L mediates cellular rRNA degradation in response to HSV-1 infection and ICP0 prevents this by either directly inhibiting RNase L or indirectly blocking an upstream activator of the OAS-RNase L pathway. We provide evidence, however, that RNase L does not mediate rRNA cleavage following HSV-1 infection. In addition to the RNase L-deficient hepatoma cell line (HepG2), both control MEFs and RNase L knockout MEFs infected with ICP0-null HSV-1 displayed rRNA degradation. Furthermore, RNase L-mediated rRNA degradation exhibits markedly different kinetics from that of ICP0-null-HSV-1-induced rRNA degradation and produces a disparate rRNA degradation profile. In addition, ICP0 overexpression did not prevent RNase L-specific rRNA cleavage, further illustrating that ICP0 does not block the OAS-RNase L pathway during HSV-1 infection. Recently, a poly(IC)-containing liposome complex (NS-9) was shown to induce rRNA degradation in an IRF3-dependent manner (51). In agreement with these data, we report that poly(IC)-induced, RNase L-mediated rRNA degradation is IRF3 dependent and further conclude that ICP0-null-HSV-1-induced rRNA degradation is IRF3 independent. Taken together, these data illustrate that ICP0 prevents the RNase L- and IRF3-independent rRNA degradation event that is induced following HSV-1 infection. These results also parallel those of other studies that determined that RNase L activity does not contribute to the host antiviral response during HSV-1 infection (24, 47). Interestingly, RNase L activity does not contribute to cellular antiviral responses during infection with varicella-zoster virus, a related alphaherpesvirus (11).
In a second putative mechanism, the rRNA degradation observed following ICP0-null-HSV-1 infection is mediated by the viral RNase vhs. However, in the absence of ICP0 and vhs, rRNA degradation was still prominent in HEL fibroblasts, discrediting vhs as the causative RNase. This conclusion is in agreement with previous research illustrating that rRNA is resistant to vhs-mediated degradation (16, 22).
In the last mechanism, HSV-1 induces the activity of a cellular RNase other than RNase L and ICP0 blocks the resultant rRNA degradation. Indeed, certain cytopathic strains of hepatitis A virus and the murine coronavirus mouse hepatitis virus have been shown to induce rRNA degradation independent of RNase L and of other known viral and cellular RNases (2, 18). Several lines of research are currently under way to determine the identity of this causative RNase, its effects on cell viability and apoptosis, and the biological significance of rRNA degradation induced by ICP0-null-HSV-1 infection. In conclusion, ICP0-null-HSV-1 infection induces cellular rRNA degradation in a variety of cell types that is independent of the classical RNase L pathway. Furthermore, the resultant degradation is independent of the viral RNase vhs, and ICP0 prevents this cellular response to infection in a manner independent of its E3 ubiquitin ligase activity. Although the specific mechanism of this rRNA degradation remains unknown, these studies indicate the existence of another ICP0-mediated viral countermeasure to the anti-HSV-1 response and provide evidence for the existence of a previously unidentified RNase that is part of the host antiviral response.
Acknowledgments
We thank R. Silverman, T. Taniguchi, R. Everett, P. Schaffer, S. Read, S. Rice, S. Weller, S. Silverstein, and D. Johnson for viral mutants and cell lines and D. Cummings and C. Whitty for technical assistance.
This work was sponsored by the Canadian Institutes of Health Research and the U.S. Army Medical Research and Materiel Command. K.L.M. holds an Rx&D Health Sciences Career Award.
REFERENCES
- 1.Aubert, M., and J. A. Blaho. 1999. The herpes simplex virus type 1 regulatory protein ICP27 is required for the prevention of apoptosis in infected human cells. J. Virol. 73:2803-2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banerjee, S., S. An, A. Zhou, R. H. Silverman, and S. Makino. 2000. RNase L-independent specific 28S rRNA cleavage in murine coronavirus-infected cells. J. Virol. 74:8793-8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boutell, C., S. Sadis, and R. D. Everett. 2002. Herpes simplex virus type 1 immediate-early protein ICP0 and is isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 76:841-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown, S. M., D. A. Ritchie, and J. H. Subak-Sharpe. 1973. Genetic studies with herpes simplex virus type 1. The isolation of temperature-sensitive mutants, their arrangement into complementation groups and recombination analysis leading to a linkage map. J. Gen. Virol. 18:329-346. [DOI] [PubMed] [Google Scholar]
- 5.Cai, W., T. L. Astor, L. M. Lipak, C. Cho, D. M. Coen, and P. A. Schaffer. 1993. The herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. J. Virol. 67:7501-7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai, W., and P. A. Schaffer. 1989. Herpes simplex virus type 1 ICP0 plays a critical role in the de novo synthesis of infectious virus following transfection of viral DNA. J. Virol. 63:4570-4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castelli, J., K. A. Wood, and R. J. Youle. 1998. The 2-5A system in viral infection and apoptosis. Biomed. Pharmacother. 52:386-390. [DOI] [PubMed] [Google Scholar]
- 8.Cayley, P. J., J. A. Davies, K. G. McCullagh, and I. M. Kerr. 1984. Activation of the ppp(A2′p)nA system in interferon-treated, herpes simplex virus-infected cells and evidence for novel inhibitors of the ppp(A2′p)nA-dependent RNase. Eur. J. Biochem. 143:165-174. [DOI] [PubMed] [Google Scholar]
- 9.Chelbi-Alix, M. K., and H. deThe. 1999. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18:935-941. [DOI] [PubMed] [Google Scholar]
- 10.DeLuca, N. A., A. M. McCarthy, and P. A. Schaffer. 1985. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J. Virol. 56:558-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desloges, N., M. Rahaus, and M. H. Wolff. 2004. Varicella-zoster virus does not significantly induce the cell defence mechanism mediated by the 2-5A/RNase L pathway during its replication cycle. Med. Microbiol. Immunol. 194:25-31. [DOI] [PubMed] [Google Scholar]
- 12.Eidson, K. M., W. E. Hobbs, B. J. Manning, P. Carlson, and N. A. DeLuca. 2002. Expression of herpes simplex virus ICP0 inhibits the induction of interferon-stimulated genes by viral infection. J. Virol. 76:2180-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Everett, R. D. 1989. Construction and characterization of herpes simplex virus type 1 mutants with defined lesions in immediate early gene 1. J. Gen. Virol. 70:1185-1202. [DOI] [PubMed] [Google Scholar]
- 14.Everett, R. D. 2000. ICP0, a regulator of herpes simplex virus during lytic and latent infection. Bioessays 22:761-770. [DOI] [PubMed] [Google Scholar]
- 15.Everett, R. D., and G. G. Maul. 1994. HSV-1 IE protein Vmw110 causes redistribution of PML. EMBO J. 13:5062-5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng, P., D. N. Everly, Jr., and G. S. Read. 2001. mRNA decay during herpesvirus infections: interaction between a putative viral nuclease and a cellular translation factor. J. Virol. 75:10272-10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldstein, D. J., and S. K. Weller. 1988. Factor(s) present in herpes simplex virus type 1-infected cells can compensate for the loss of the large subunit of the viral ribonucleotide reductase: characterization of an ICP6 deletion mutant. Virology 166:41-51. [DOI] [PubMed] [Google Scholar]
- 18.Goswami, B. B., M. Kulka, D. Ngo, and T. A. Cebula. 2004. Apoptosis induced by a cytopathic hepatitis A virus is dependent on caspase activation following ribosomal RNA degradation but occurs in the absence of 2′-5′ oligoadenylate synthetase. Antivir. Res. 63:153-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Houge, G., B. Robaye, T. S. Eikhom, J. Golstein, G. Mellgren, B. T. Gjertsen, M. Lanotte, and S. O. Doskeland. 1995. Fine mapping of 28S rRNA sites specifically cleaved in cells undergoing apoptosis. Mol. Cell. Biol. 15:2051-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim, T., T. Y. Kim, Y. H. Song, I. M. Min, J. Yim, and T. K. Kim. 1999. Activation of interferon regulatory factor 3 in response to DNA-damaging agents. J. Biol. Chem. 274:30686-30689. [DOI] [PubMed] [Google Scholar]
- 21.Krause, D., and R. H. Silverman. 1993. Tissue-related and species-specific differences in the 2-5A oligomer size requirement for activation of 2-5A-dependent RNase. J. Interferon Res. 13:13-16. [DOI] [PubMed] [Google Scholar]
- 22.Krikorian, C. R., and G. S. Read. 1991. In vitro mRNA degradation system to study the virion host shutoff function of herpes simplex virus. J. Virol. 65:112-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leaman, D. W., A. Salvekar, R. Patel, G. C. Sen, and G. R. Stark. 1998. A mutant cell line defective in response to double-stranded RNA and in regulating basal expression of interferon-stimulated genes. Proc. Natl. Acad. Sci. USA 95:9442-9447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leib, D. A., M. A. Machalek, B. R. Williams, R. H. Silverman, and H. W. Virgin. 2000. Specific phenotypic restoration of an attenuated virus by knockout of a host resistance gene. Proc. Natl. Acad. Sci. USA 97:6097-6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin, R., R. S. Noyce, S. E. Collins, R. D. Everett, and K. L. Mossman. 2004. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J. Virol. 78:1675-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Long, M. C., V. Leong, P. A. Schaffer, C. A. Spencer, and S. A. Rice. 1999. ICP22 and the UL13 protein kinase are both required for herpes simplex virus-induced modification of the large subunit of RNA polymerase II. J. Virol. 73:5593-5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinand, C., C. Montavon, T. Salehzada, M. Silhol, B. Lebleu, and C. Bisbal. 1999. RNase L inhibitor is induced during human immunodeficiency virus type 1 infection and down regulates the 2-5A/RNase L pathway in human T cells. J. Virol. 73:290-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCarthy, A. M., L. McMahan, and P. A. Schaffer. 1989. Herpes simplex virus type 1 ICP27 deletion mutants exhibit altered patterns of transcription and are DNA deficient. J. Virol. 63:18-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Melroe, G. T., N. A. DeLuca, and D. M. Knipe. 2004. Herpes simplex virus 1 has multiple mechanisms for blocking virus-induced interferon production. J. Virol. 78:8411-8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mossman, K. L., and A. A. Ashkar. 2005. Herpesviruses and the innate immune response. Viral Immunol. 18:267-281. [DOI] [PubMed] [Google Scholar]
- 31.Mossman, K. L., P. F. Macgregor, J. J. Rozmus, A. B. Goryachev, A. M. Edwards, and J. R. Smiley. 2001. Herpes simplex virus triggers and then disarms a host antiviral response. J. Virol. 75:750-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mossman, K. L., H. A. Saffran, and J. R. Smiley. 2000. Herpes simplex virus ICP0 mutants are hypersensitive to interferon. J. Virol. 74:2052-2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mossman, K. L., and J. R. Smiley. 2002. Herpes simplex virus ICP0 and ICP34.5 counteract distinct interferon-induced barriers to virus replication. J. Virol. 76:1995-1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muller, S., and A. Dejean. 1999. Viral immediate-early proteins abrogate the modification by SUMO-1 of PML and Sp100 proteins, correlating with nuclear body disruption. J. Virol. 73:5137-5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nilsen, T. W., D. L. Wood, and C. Baglioni. 1981. 2′,5′-Oligo(A)-activated endoribonuclease. Tissue distribution and characterization with a binding assay. J. Biol. Chem. 256:10751-10754. [PubMed] [Google Scholar]
- 36.Player, M. R., and P. F. Torrence. 1998. The 2-5A system: modulation of viral and cellular processes through acceleration of RNA degradation. Pharmacol. Ther. 78:55-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Read, G. S., B. M. Bradley, and K. Knight. 1993. Isolation of a herpes simplex virus type 1 mutant with a deletion in the virion host shutoff gene and identification of multiple forms of the vhs (UL41) polypeptides. J. Virol. 67:7149-7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sacks, W. R., and P. A. Schaffer. 1987. Deletion mutants in the gene encoding the herpes simplex virus type 1 immediate-early protein ICP0 exhibit impaired growth in cell culture. J. Virol. 61:829-839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Samali, A., B. Gilje, S. O. Doskeland, T. G. Cotter, and G. Houge. 1997. The ability to cleave 28S ribosomal RNA during apoptosis is a cell-type dependent trait unrelated to DNA fragmentation. Cell Death Differ. 4:289-293. [DOI] [PubMed] [Google Scholar]
- 40.Samuel, C. E. 2001. Antiviral actions of interferons. Clin. Microbiol. Rev. 14:778-809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sato, M., H. Suemori, N. Hata, M. Asagiri, K. Ogasawara, K. Nakao, T. Nakay, M. Katsuki, S. Noguchi, N. Tanaka, and T. Taniguchi. 2000. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-α/β gene induction. Immunity 13:539-548. [DOI] [PubMed] [Google Scholar]
- 42.Sen, G. C. 2001. Viruses and interferon. Annu. Rev. Microbiol. 55:255-281. [DOI] [PubMed] [Google Scholar]
- 43.Sen, G. C., and R. M. Ransohoff. 1993. Interferon-induced antiviral actions and their regulation. Adv. Virus Res. 42:57-102. [DOI] [PubMed] [Google Scholar]
- 44.Silverman, R. H., and N. M. Cirino. 1997. RNA decay by the interferon-regulated 2-5A system as a host defense against viruses, p. 295-309. In J. B. Hartford and D. R. Morris (ed.), mRNA metabolism and post-transcriptional gene regulation. Wiley-Liss Inc., New York, N.Y.
- 45.Smiley, J. R. 2004. Herpes simplex virus virion host shutoff protein: immune evasion mediated by a viral RNase? J. Virol. 78:1063-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith, K. O. 1964. Relationship between the envelope and the infectivity of herpes simplex virus. Proc. Soc. Exp. Biol. Med. 115:814-816. [DOI] [PubMed] [Google Scholar]
- 47.Smith, T. J., R. H. Silverman, and D. A. Leib. 2003. RNase L activity does not contribute to host RNA degradation induced by herpes simplex virus infection. J. Gen. Virol. 84:925-928. [DOI] [PubMed] [Google Scholar]
- 48.Stow, N. D., and E. C. Stow. 1986. Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate-early polypeptide Vmw110. J. Gen. Virol. 67:2571-2585. [DOI] [PubMed] [Google Scholar]
- 49.Tnani, M., and B. A. Bayard. 1998. Lack of 2′,5′-oligoadenylate-dependent RNase expression in the human hepatoma line HepG2. Biochim. Biophys. Acta 1402:139-150. [DOI] [PubMed] [Google Scholar]
- 50.Umene, K. 1986. Conversion of a fraction of the unique sequences to part of the inverted repeats in the S component of the herpes simplex virus type 1 genome. J. Gen. Virol. 67:1035-1048. [DOI] [PubMed] [Google Scholar]
- 51.Uno, T., K. Hirabayashi, M. Murai, J. Yano, and K. Ozato. 2005. The role of IFN regulatory factor-3 in the cytotoxic activity of NS-9, a polyinosinic-polycytidylic acid/cationic liposome complex, against tumor cells. Mol. Cancer Ther. 4:799-805. [DOI] [PubMed] [Google Scholar]
- 52.Williams, B. R. 1999. PKR: a sentinel kinase for cellular stress. Oncogene 18:6112-6120. [DOI] [PubMed] [Google Scholar]
- 53.Xiang, Y., R. C. Condit, S. Vijaysri, B. Jacobs, B. R. Williams, and R. H. Silverman. 2002. Blockade of interferon induction and action by the E3L double-stranded RNA binding proteins of vaccinia virus. J. Virol. 76:5251-5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yokota, S., N. Yokosawa, T. Kubota, T. Suzutani, I. Yoshida, S. Miura, K. Jimbow, and N. Fujii. 2001. Herpes simplex virus type 1 suppresses the interferon signaling pathway by inhibiting phosphorylation of STATs and Janus kinases during an early infection stage. Virology 286:119-124. [DOI] [PubMed] [Google Scholar]
- 55.Yokota, S.-I., N. Yokosawa, T. Okabayashi, T. Suzutani, S. Miura, K. Jimbow, and N. Fujii. 2004. Induction of suppressor of cytokine signaling-3 by herpes simplex virus type 1 contributes to inhibition of the interferon signaling pathway. J. Virol. 78:6282-6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zachos, G., M. Koffa, C. M. Preston, J. B. Clements, and J. Conner. 2001. Herpes simplex virus type 1 blocks the apoptotic host cell defense mechanisms that target Bcl-2 and manipulates activation of p38 mitogen-activated protein kinase to improve viral replication. J. Virol. 75:2710-2728. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 57.Zheng, X., R. H. Silverman, A. Zhou, T. Goto, B. S. Kwon, H. E. Kaufman, and J. M. Hill. 2001. Increased severity of HSV-1 keratitis and mortality in mice lacking the 2-5A-dependent RNase L gene. Investig. Ophthalmol. Vis. Sci. 42:120-126. [PubMed] [Google Scholar]
- 58.Zhou, A., J. Paranjape, T. L. Brown, H. Nie, S. Naik, B. Dong, A. Chang, B. Trapp, R. Fairchild, C. Colmenares, and R. H. Silverman. 1997. Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. EMBO J. 16:6355-6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu, X., C. S. H. Young, and S. Silverstein. 1988. Adenovirus vector expressing functional herpes simplex virus ICP0. J. Virol. 62:4544-4553. [DOI] [PMC free article] [PubMed] [Google Scholar]