

Abstract
Human embryonic stem cells are pluripotent entities, theoretically capable of generating a whole-body spectrum of distinct cell types. However, differentiation of these cells has been observed only in culture or during teratoma formation. Our results show that human embryonic stem cells implanted in the brain ventricles of embryonic mice can differentiate into functional neural lineages and generate mature, active human neurons that successfully integrate into the adult mouse forebrain. Moreover, this study reveals the conservation and recognition of common signals for neural differentiation throughout mammalian evolution. The chimeric model will permit the study of human neural development in a live environment, paving the way for the generation of new models of human neurodegenerative and psychiatric diseases. The model also has the potential to speed up the screening process for therapeutic drugs.
Keywords: chimeric model, human ES cells, neuronal differentiation
Human embryonic stem cells (hESC) are known for their ability to propagate indefinitely in culture as undifferentiated cells; furthermore, they can be induced to differentiate in vitro and in vivo into various cell types (1). However, it is currently unknown whether hESC can differentiate into authentic human neurons in vivo. The transplantation of hESC into the developing mouse brain is a valuable model for studying the differentiation and migration potential of hESC in vivo. Detailed knowledge about human neural differentiation during development will certainly prove valuable in the regenerative medicine field and in drug discovery. We have investigated this process both in vitro and in vivo.
Methods
Culture and Differentiation of hESC. The Cyth25 cell line (Cythera, San Diego) was cultured on mitotically inactivated (mitomycin C-treated) mouse embryonic fibroblasts (Specialty Media, Lavellette, NJ) in DMEM/F12 Glutamax (GIBCO), 20% knockout serum replacement (GIBCO), 0.1 mM nonessential amino acids (GIBCO), 0.1 mM 2-mercaptoethanol (GIBCO), and 4 ng/ml bFGF-2 (R & D Systems). hESC neuronal differentiation was obtained by coculture with PA6 cells for 3–5 weeks under the following differentiation conditions: DMEM/F12 Glutamax (GIBCO), 10% knockout serum replacement (GIBCO), 0.1 mM nonessential amino acids (GIBCO), and 0.1 mM 2-mercaptoethanol (GIBCO). Alkaline phosphatase activity was measured by using the Vector Red Alkaline Phosphatase substrate kit I from Vector Laboratories.
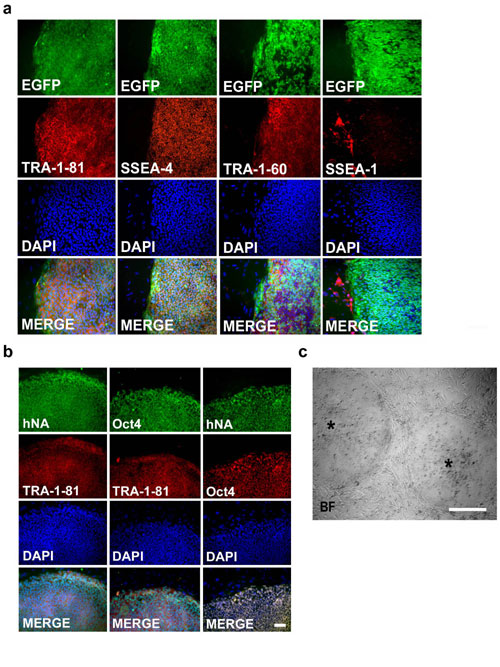
hESC Transfection. hESC were stably transfected to express enhanced GFP (EGFP) by CAG-EGFP self-inactivating lentivirus infection. The self-inactivating lentiviral vector expressing EGFP under control of the CAG promoter was derived from a multiply attenuated HIV vector system but included a U3 deletion and introduction of a central polypurine tract element. Vectors were produced by triple transfection of HEK293 cells followed by ultracentrifugation and titration as described in ref. 2. Undifferentiated cells were exposed to the virus at a titer of 0.5 × 1010 gene transfer units/ml for 1 h followed by a 2-day recovery period. EGFP was detected by native fluorescence at day 3 after transduction in ≈1% of the cells. Cells expressing EGFP were manually selected for stable and uniform EGFP expression. Only subtle loss in EGFP expression was observed during propagation or in vitro differentiation for up to a year after transduction. The EGFP+ cells derived from these colonies are thus polyclonal in origin and are all positive for human nuclear antigen (hNA). The EGFP+ hESC maintain a phenotype similar to the wild-type cells [stage-specific embryonic antigen (SSEA)-4, terato-related antigen (TRA)-1-60, TRA-1-81, and Pit1-Oct1-Unc86 transcription factor octamer-4 (OCT4)-positive].
In Utero hESC Transplantation. Time-pregnant ICR females were anesthetized, and embryos were removed with intact placenta. Approximately 105 EGFP+ hESC in 1 μl of PBS were injected into the lateral ventricle of each embryo (embryonic day 14) with the help of a micro glass capillary and mouth pipette technique. After injection, embryos were placed back into the female with the addition of 1 ml of saline solution. Females were placed in cages, and pups were born by normal vaginal delivery. Weaned animals were kept in individual cages for future experiments. All experiments were performed in compliance with institutional and U.S. National Academies guidelines and with the protocol approved by The Salk Institute Animal Care and Use Committee.
Immunofluorescence. Injected animals were perfused at different time points with 4% paraformaldehyde, and brains were equilibrated in 30% sucrose. Entire brains were processed in 40-μm microtome sections. Immunofluorescence was performed as described by using the following antibodies at the indicated dilutions (3): rabbit anti-β tubulin-III (TUJ1; 1:1,000, Covance, Richmond, CA), guinea pig anti-glial fibrillary acidic protein (1:1,000; Advanced Immunochemical, Long Beach, CA), mouse anti-RIP (1:250, Chemicon), mouse anti-microtubule-associated protein 2(a+b) (1:100, Sigma), mouse anti-S100-β (1:100, Sigma), mouse anti-glutathione transferase π (1:500, BD Pharmingen), mouse anti-OCT4 (1:500, Santa Cruz Biotechnology), and the other marker antibodies, hNA, SSEA-1, SSEA-4, TRA-1-60, TRA-1-81, and Nestin (1:100, Chemicon). All secondary antibodies were purchased from Jackson ImmunoResearch. Fluorescent signals were detected by using a confocal laser scanning head (Bio-Rad MRC 1000) on a Zeiss inverted microscope. Images were processed with photoshop 7 (Adobe Systems, San Jose, CA).
Laser Capture. Forty-micrometer microtome sections were immunostained with anti-EGFP antibody (1:500, Chemicon), rapidly dehydrated through xylenes, and stored until use in a vacuum dessicator. A PixCell II machine (Arcturus, Mountain View, CA) was used to isolate 30 EGFP+ and EGFP-cells from different regions of the brain. Genomic DNA was isolated and prepared as described in ref. 4.
Microscopy. For confocal microscopy, mice were anesthetized 1 and 2 months after injection of EGFP+ hESC and were perfused with 4% paraformaldehyde. Brains were sectioned at a thickness of 100 μm. Dendrites were imaged with a ×40 oil lens and a z-step of 0.5 μm by using a Bio-Rad radiance 2100 confocal microscope. To restrict our study to perforant path inputs, we analyzed the distal portion of the dendritic tree after the first branching point. Seven cells, 15 dendritic segments, and 2,400 protrusions were analyzed. Protrusions were counted on projection images of labeled dendrites. For electron microscopy, mice were anesthetized and perfused with 4% paraformaldehyde plus 0.2% glutaraldehyde. Sections of a thickness of 100 μm were cut, and hESC-derived EGFP+ cells were microinjected under a fluorescence microscope with 5% aqueous lucifer yellow (Aldrich). Slices were then incubated with 2.8 mM 3,3′-diaminobenzidine tetrahydrochloride (DAB) and 6 mM potassium cyanide and then irradiated under conventional epifluorescence by using a 75-W Hg lamp and a fluorescein filter set to induce photoconversion of DAB. Slices were then postfixed overnight in a solution of 3% glutaraldehyde and processed conventionally for electron microscopy. Sections were cut at a thickness of 60 nm and analyzed with a JEOL 100CXII electron microscope at a magnification of ×19,000.
Electrophysiology. Brain slices were prepared as described in refs. 5 and 6. In brief, 18-month-old adult mice were anesthetized with halothane (Ayerst Laboratories) and rapidly decapitated. The brains were immediately harvested and placed into ice-cold cutting solution that consisted of 120 mM choline-Cl, 3 mM KCl, 8 mM MgCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 10–20 mM glucose. A vibratome apparatus was used to cut 300-mm-thick slices. After they were cut, slices were kept at room temperature (≈25°C) in artificial cerebrospinal fluid, which was composed of 124 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 10 mM glucose, bubbled with a mixture of 95% O2/5% CO2, making the final pH 7.4 (300–315 mOsm/kg). Experiments were performed at room temperature. EGFP-expressing neurons in acute brain slices were identified by using wide-field illumination at 488 nm and emission at 520 nm. Pipettes were sealed on cell bodies by using video-enhanced differential interference contrast optics (7). Intracellular solution contained 130 mM K-methanesulfonate, 4 mM NaCl, 2 mM Mg-ATP, 0.3 mM Na-GTP, and 10 mM Hepes; pH was adjusted to 7.3 with CH3SO4 (300 mOsm/kg). Pipette resistance was 5–7 MΩ. Neuronal recordings were made in voltage-clamp mode by using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). Signals were filtered at 10 kHz. Holding currents were adjusted to 0 pA at the beginning of recording to identify the membrane potential of a neuron. No correction was made for the junction potential between the bath and the pipette. Data are presented as means ± SD. Recorded cells were filled with 5 μM Alexa Fluor 594 (Molecular Probes) through the recording pipette to ascertain that the recorded cell was indeed EGFP+. Alexa Fluor 594 was detected by using wide-field illumination at 595 nm and emission at 615 nm. After recording, slices were fixed in 4% paraformaldehyde for >24 h and then imaged by using a SPOT RT charge-coupled device camera (Diagnostic Instruments, Sterling Heights, MI) to visualize recorded neurons.
Results
Cyth25 hESC were cocultured with mouse embryonic fibroblasts in a defined medium and were immunoreactive for undifferentiated markers, including OCT4, TRA-1-60, TRA-1-81, and SSEA-4, but not for the murine embryonic marker SSEA-1 (see Fig. 5 a and b, which is published as supporting information on the PNAS web site). In addition, >90% of the colonies showed alkaline phosphatase activity, and most of the cells kept a stable karyotype even at higher passages (>50) (data not shown). Undifferentiated colonies were also positive for a specific hNA (Fig. 5c). Moreover, cells were immunonegative for Nestin, a neural precursor marker; TUJ1 and microtubule-associated protein 2(a+b), immature neuronal markers; NeuN, a mature neuronal marker; glial fibrillary acidic protein and S100-β, astrocyte markers; and oligodendrocyte marker 4, glutathione transferase π, and RIP, oligodendrocyte markers (data not shown), suggesting that these cells remained undifferentiated in culture.
The ability of Cyth25 hESC to differentiate into neurons was examined by coculture with the mouse skull bone marrow-derived stromal cell line PA6 (8). After 3–4 weeks, most of the colonies became positive for Nestin, and, after 5 weeks, >95% of the colonies formed extensive processes and expressed neuronal markers, such as microtubule-associated protein 2(a+b) (Fig. 1 a–e). Neurons generated by this process were immunopositive for hNA (Fig. 1f). For transplantation purposes, undifferentiated Cyth25 hESC were infected with the self-inactivating lentivirus carrying the reporter gene EGFP (9). Colonies with undifferentiated morphology (round and compact colonies with well defined borders containing cells with a high ratio of nucleus to cytoplasm and prominent nucleoli; see Fig. 5c) were selected, isolated by mechanical dissociation, and briefly trypsinized to produce small clumps of cells in suspension. By the time of the transfection, the vast majority of the cells highly expressed the EGFP reporter gene. We estimated that ≈105 hESC were grafted into the lateral ventricle of embryonic-day-14 ICR mice. The experiment was done in four different pregnant females, and results are summarized in Table 1. Neither teratomas nor tumors were observed at any time. Furthermore, no rejection or immunological reaction was observed, indicating the immunotolerance of the embryonic brain.
Fig. 1.

In vitro neuronal differentiation of Cyth25 hESC. (a) Example of a differentiated hESC colony after 5 months in PA6 feeder coculture. (b–d) DAPI nuclear staining (b), Nestin (c), and microtubule-associated protein 2(a+b) (d). (e) Merged image of c and d.(f) Differentiated cells that are positive for TUJ1 colocalize with hNA. (Scale bars, 50 μm.)
Table 1. Summary of experimental results obtained with hESC transplantation in mouse embryos.
| Experiment | Cell passage | No. of embryos that died | No. of embryos with good integration | No. of embryos with poor integration | No. of embryos with no integration |
|---|---|---|---|---|---|
| 1 | 30 | 0 | 7 | 0 | 2 |
| 2 | 40 | 1 | 4 | 4 | 0 |
| 3 | 45 | 4 | 1 | 0 | 3 |
| 4 | 52 | 0 | 1 | 5 | 0 |
Good integration means six or more EGFP+ cells per 40-μm brain section. Poor integration refers to fewer than six EGFP+ cells per 40-μm brain section.
Transplanted cells were identified in brain slices by EGFP fluorescence. In the animals with detected incorporated cells, an average of six EGFP+ cells were found in each 40-μm brain microtome section. Despite the high variability observed in different animals, we estimate that <0.1% of the brain cells are of human origin. Most EGFP+ cells colocalized with hNA, and serial 3D reconstruction of confocal sections revealed only one nucleus (Fig. 2a). However, we did observe hNA+ cells that were not EGFP+, suggesting that some cells may have their EGFP expression silenced upon differentiation, contrary to what was observed in vitro (Fig. 1f). A few EGFP+ cells were observed that did not overlap with the hNA marker and may represent differential antibody accessibility in some differentiated cells. However, laser-captured EGFP+ cells were PCR-positive for the human-specific, single-copy TATA-binding protein (TBP) gene but not for the mouse gene, using specific primers set for each species (10) (Fig. 2b). If all of the EGFP+ were fused with mouse cells, the intensities of the mouse TBP band in EGFP+ and EGFP-cells should have been the same; this was clearly not the case. Although we cannot completely exclude the possibility, our results show no evidence of hESC fusion with host cells. Hence, it seems unlikely that fusion plays a dominant role in the observed differentiation of hESC in mouse brain. Two months after transplantation, recipients' brains showed widespread incorporation of hESC in a variety of regions, such as cortex, hippocampus, thalamus, cerebellum, striatum, and corpus callosum, indicating that transplanted cells migrated out from the ventricle into various host brain regions, both ipsilateral and contralateral to the injection site, engrafting at various levels of the neuraxis (Fig. 2 c and d). A small fraction of the transplanted cells integrated individually or in small clusters into the host tissue with morphometric dimensions similar to those of adjacent host cells, including shape, size, and orientation, and adjusted to the preexisting cellular architecture.
Fig. 2.

Widespread chimerism of hESC in the mouse developing brain visualized by EGFP fluorescence. (a) 3D reconstruction of 1-μm confocal sections of a hNA+/EGFP+-colabeled cell in the subgranular layer of the ventricular zone (SVZ). ve, ventricle. (b) An enriched population (≈200 cells) of laser-captured EGFP (+) cells was PCR-positive for the human-specific TBP sequence (hTBP). The same number of non-EGFP cells (-) was positive only for the mouse sequence (mTBP). (c and d) Morphological aspect of hESC in different areas of the host brain, such as cortex (c) and thalamus (d). (e–h) In vivo neural differentiation of hESC; EGFP+ cells are negative for proliferative markers (such as Ki67; SVZ region in e) and express astrocyte (S100-β in the cortex; region in f), oligodendrocyte (glutathione transferase π in the hypothalamus; region in g) and neuron (Tuj-1; SVZ region in h) markers. (Scale bars, 10 μm.)
The remaining cells in the host ventricle formed clusters attached to the walls but without apparent consequences for the animals for up to 18 months of age (Fig. 6, which is published as supporting information on the PNAS web site, and Table 1). It is important to note that the integration was not uniform in all animals. Some animals had no or few detectable cells. We hypothesize that this variation is likely due to a variety of reasons including cell number, cell passage, and manipulation or embryonic handling before transplantation. The ventricle-associated cells were weakly immunoreactive for Nestin but negative for cleaved Caspase-3 and Ki67 (Fig. 2e). In addition, they did not incorporate BrdUrd by either 7 or 30 days after administration, indicating that they were not in an active proliferative state but remained quiescent (data not shown).
The identity of the incorporated cells was analyzed by immunostaining with specific antibodies for neurons, astrocytes, and oligodendrocytes. Immature neurons (TUJ1+) were found in the subventricular zone (Fig. 2h). In the dentate gyrus, NeuN+ cells showed the mature morphological characteristics of granule cells, including dendritic trees extending into the molecular layer and axonal projections into the hilar area, characteristics of native mature neurons in this region (Fig. 3c). In addition, transplanted cells that integrated into the hippocampus expressed calbindin, a calcium-binding protein typically found in mature granule neurons (Fig. 7, which is published as supporting information on the PNAS web site). Intriguingly, the diameter of EGFP+/calbindin+ granular cell bodies (11.1 ± 0.6 μm; n = 5 in two different animals) was similar to that of mouse cells in the same region (11.1 ± 3.3 μm; n = 1,000 in two different animals), indicating the ability of the human transplanted cells to adjust their size in relation to adjacent neurons (human granular cell body diameter is, on average, 16.9 ± 3.4 μm; n = 1,000 in several samples analyzed). Furthermore, mature neurons in the cortex frequently displayed the morphologies of projection neurons, with pyramidal cell bodies, long apical dendrites reaching into the superficial cortical layers, and basal axons extending into the corpus callosum (Fig. 2c).
Fig. 3.

Maturation of neuron-derived hESC. (a and c) Visualization of spines in EGFP+/NeuN+ cells in the cortex (a) or in the dentate gyrus (c) 2 months after transplantation. The boxed areas correspond to the enlarged images of the dendrites in b and d, respectively. (Scale bars, 10 μm.) (e and f) Electron micrographs of synaptic terminals (arrowheads) on the soma of EGFP+ neurons (*) in the granule cell layer of the dentate gyrus. (Scale bars, 0.75 μm.)
Evidence of synaptic inputs was apparent in the presence of arborized dendrites with spines, suggesting that glutamate-containing terminals contacted these dendrites (Fig. 3 b and d). We used confocal microscopy to examine the density of dendritic protrusions on EGFP+ neurons in the granule cell layer. Protrusions at least 0.4 μm in length were included in the counts, and their numbers were divided by the dendritic length to obtain linear density. We obtained a density of 2.5 ± 0.08 protrusions per micrometer of dendrite, a value comparable to that obtained in adult mouse dentate gyrus (11), indicating that the morphology of EGFP+ neurons is similar to mature neurons and suggesting the presence of glutamatergic inputs. Ultrastructural analysis also confirmed that EGFP+ cells in the granular zone of the dentate gyrus received synaptic input and exhibited mature features, such as pools of presynaptic vesicles adjacent to a postsynaptic density (Fig. 3 e and f). In humans, granule cells in the hippocampus are formed mainly in the first 4–6 months after birth (12). Interestingly, the transplanted cells could regulate their maturation speed and size within the recipient granular layer, and they established contact with host neurons, revealing remarkable conservation of signaling across these two species.
hESC-derived astrocytes expressed S100-β, showing no difference in size or branching pattern when compared with host cells in the same region (Fig. 2f). In addition to neurons and astrocytes, EGFP+ oligodendrocytes were found in the mouse brains with hESC transplants. The identity of these oligodendrocytes was confirmed by glutathione transferase π immunofluorescence. Their size and orientation were indistinguishable from those of their host counterparts (Fig. 2g).
To investigate whether mature, hESC-derived neurons are functional even a long time (18 months) after transplantation, electrophysiological recordings of EGFP+ cells from adult animals were made in acute brain slices. Recorded EGFP+ cells (n = 5, from two different animals) showed neuronal properties similar to mature pyramidal cortical neurons under comparable recording conditions (13). The membrane potential of the recorded neurons was -64.8 ± 3.1 mV. Membrane resistance was 38.1 ± 3.2 MΩ. The amplitude of the first action potential in a train was 1,572 ± 286 pA. Two neurons generated a burst of three to four action potentials in response to depolarization (Fig. 4).
Fig. 4.

hESC differentiate into functionally mature neurons in the mouse brain. (a) A neuron filled with Alexa Fluor 594 and expressing EGFP. (Scale bar, 20 μm.) (b) Low-magnification merge of EGFP, Alexa Fluor 594, and bright-field images shows the position of a recorded neuron in the mouse cortex. (Scale bar, 1 mm.) (c) Whole-cell recording from the neuron shown in a. The neuron generates a train of action potentials (Top) in response to depolarization according to the voltage protocol shown in Middle. Time expansion of the first two action potentials is labeled by an asterisk.
Discussion
Our results show that hESC, transplanted into the ventricle of the developing mammalian brain, can give rise to neuronal and glial lineages, suggesting that they are responsive to environmental cues that regulate cell fate determination and differential migration. Access to large areas of the neuroepithelium, the lack of immunological response, and the neurogenically enriched environment of the embryonic brain appear to be responsible for the first steps of hESC-grafted cell differentiation. Donor-derived neurons seem to respond to local differentiation signals, acquiring morphological and functional features similar to host neurons and displaying remarkable regional specificity. Such a contribution to the host tissue formation probably requires a close physical interaction between the transplanted and intrinsic cells.
Depending on the injection site, murine ES cells and teratocarcinoma NT2 cells transplanted to the adult mouse brain frequently develop tumors (14, 15). hESC have been transplanted into immunocompromised adult mice to form teratomas that contain derivatives of the three embryonic germ layers, demonstrating the potency of these cells (1). Interestingly, our results suggest that transplanted hESC in the embryonic-day-14 mouse ventricle stop dividing and do not form teratomas. Tumor formation seems to be caused by different factors, including the number of viable cells and the age and site of host tissue. In contrast, both murine ES and teratocarcinoma cells have been shown to participate in normal development upon introduction into early embryos at the blastocyst stage (16).
Human undifferentiated embryonic cells were also transplanted into somites of chick embryos of 1.5–2 days of development (17). Surprisingly, transplanted cells could migrate, differentiate, and integrate with host tissues, indicating that the host chick embryonic environment may modulate their differentiation. Recent data suggest that, after transplantation, undifferentiated hESC can survive in the striatum of a rat model of Parkinson's disease (18). This finding indicates that hESC can be used as long-term carriers of therapeutic gene product(s). However, there is no evidence that these cells can functionally integrate into the existing nervous system of the host and contribute to behavioral recovery. Transplantation of human fetal, adult neuronal and hematopoietic stem cells into mouse embryos, and their subsequent contribution to a variety of organs, has been reported (19–23). Moreover, the transplantation of human neural stem cells into fetal monkey brain showed an extensive incorporation and neuronal differentiation (24). hESC-derived neural progenitors were also transplanted into the ventricles of newborn mice and adult rats and showed neural differentiation (25–27). Functional neuronal differentiation, detected by synaptic formation, was obtained from human neural stem cells transplantation into gerbil ischemic brain and mouse spinal-cord-injured models, revealing the potential of human cells to form synaptic connections with other species (28, 29). Together with these studies, the data presented here point to conserved neural differentiation signals from mouse to man.
It will be of interest to extend this approach to other tissues of the embryo, allowing the evaluation of functional differentiation of hESC in specific fetal environments, without the use of ethically controversial blastocyst transplantation. In addition, it will be important to standardize these procedures to assure reliable, reproducible, and, hopefully, quantitative assays in the future. The introduction of hESC-derived neurons and glia into the developing nervous system is a new approach for the study of neurological disorders, mainly in those cases when the animal model does not entirely recapitulate the neurodegenerative process observed in humans, such as in the ataxia telangiectasia syndrome (30). Moreover, the system can be applied to study the neural phenotype of certain human diseases in which individuals die before the development of a functional nervous system. Mouse chimeric models have already produced insights into neurodegenerative diseases such as amyotrophic lateral sclerosis (31). Genetic manipulation of hESC (by recombination or delivery of small interfering RNA) (32, 33) or somatic nuclear transfer (34–36) will allow the generation of a large spectrum of modified donor hESC-derived precursor cells that can then be assayed in vivo in a wild-type or mutant recipient brain, producing a mouse–human chimeric nervous system. Such a strategy might allow the direct examination of the effect of potential therapeutic molecules on hESC in a live and functioning nervous system, even in long-term paradigms.
Supplementary Material
Acknowledgments
We are in debt to Kevin D'Amour for technical advice with Cyth25 cells and Mark Ellisman at the National Center for Microscopy and Imaging Research at the University of California at San Diego (La Jolla). We thank The Salk Institute administration for the establishment of a non-National Institutes of Health core in the Stem Cell Facility through the support of the Lookout Fund and Mathers Foundation. A.R.M. is supported by the Pew Latin America Fellows, K.N. is supported by the Japan Society for the Promotion of Science Postdoctoral Fellowship for Research Abroad, and N.T. is supported by the International Human Frontier Science Program Organization.
Author contributions: A.R.M., K.N., and F.H.G. designed research; A.R.M., K.N., N.T., and V.M.S. performed research; K.N., N.T., and V.M.S. contributed new reagents/analytic tools; A.R.M., K.N., N.T., V.M.S., and F.H.G. analyzed data; and A.R.M. wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: hESC, human ES cells; EGFP, enhanced GFP; hNA, human nuclear antigen; SSEA, stage-specific embryonic antigen; TBP, TATA-binding protein.
References
- 1.Thomson, J. A., Itskovitz-Eldor, J., Shapiro, S. S., Waknitz, M. A., Swiergiel, J. J., Marshall, V. S. & Jones, J. M. (1998) Science 282, 1145-1147. [DOI] [PubMed] [Google Scholar]
- 2.Zufferey, R., Nagy, D., Mandel, R. J., Naldini, L. & Trono, D. (1997) Nat. Biotechnol. 15, 871-875. [DOI] [PubMed] [Google Scholar]
- 3.Gage, F. H., Coates, P. W., Palmer, T. D., Kuhn, H. G., Fisher, L. J., Suhonen, J. O., Peterson, D. A., Suhr, S. T. & Ray, J. (1995) Proc. Natl. Acad. Sci. USA 92, 11879-11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Millar, D. S., Warnecke, P. M., Melki, J. R. & Clark, S. J. (2002) Methods 27, 108-113. [DOI] [PubMed] [Google Scholar]
- 5.Sandler, V. M. & Barbara, J. G. (1999) J. Neurosci. 19, 4325-4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sandler, V. M. & Ross, W. N. (1999) J. Neurophysiol. 81, 216-224. [DOI] [PubMed] [Google Scholar]
- 7.Stuart, G. J. & Sakmann, B. (1994) Nature 367, 69-72. [DOI] [PubMed] [Google Scholar]
- 8.Kawasaki, H., Suemori, H., Mizuseki, K., Watanabe, K., Urano, F., Ichinose, H., Haruta, M., Takahashi, M., Yoshikawa, K., Nishikawa, S., et al. (2002) Proc. Natl. Acad. Sci. USA 99, 1580-1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyoshi, H., Blomer, U., Takahashi, M., Gage, F. H. & Verma, I. M. (1998) J. Virol. 72, 8150-8157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Imbert, G., Trottier, Y., Beckmann, J. & Mandel, J. L. (1994) Genomics 21, 667-668. [DOI] [PubMed] [Google Scholar]
- 11.Williams, R. S. & Matthysse, S. (1983) J. Comp. Neurol. 215, 154-164. [DOI] [PubMed] [Google Scholar]
- 12.Seress, L. (1992) Epilepsy Res. Suppl. 7, 3-28. [PubMed] [Google Scholar]
- 13.Stuart, G., Schiller, J. & Sakmann, B. (1997) J. Physiol. 505, 617-632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deacon, T., Dinsmore, J., Costantini, L. C., Ratliff, J. & Isacson, O. (1998) Exp. Neurol. 149, 28-41. [DOI] [PubMed] [Google Scholar]
- 15.Miyazono, M., Lee, V. M. & Trojanowski, J. Q. (1995) Lab. Invest. 73, 273-283. [PubMed] [Google Scholar]
- 16.Kleppner, S. R., Robinson, K. A., Trojanowski, J. Q. & Lee, V. M. (1995) J. Comp. Neurol. 357, 618-632. [DOI] [PubMed] [Google Scholar]
- 17.Goldstein, R. S., Drukker, M., Reubinoff, B. E. & Benvenisty, N. (2002) Dev. Dyn. 225, 80-86. [DOI] [PubMed] [Google Scholar]
- 18.Park, S., Kim, E. Y., Ghil, G. S., Joo, W. S., Wang, K. C., Kim, Y. S., Lee, Y. J. & Lim, J. (2003) Neurosci. Lett. 353, 91-94. [DOI] [PubMed] [Google Scholar]
- 19.Brustle, O., Choudhary, K., Karram, K., Huttner, A., Murray, K., Dubois-Dalcq, M. & McKay, R. D. (1998) Nat. Biotechnol. 16, 1040-1044. [DOI] [PubMed] [Google Scholar]
- 20.Lagasse, E., Shizuru, J. A., Uchida, N., Tsukamoto, A. & Weissman, I. L. (2001) Immunity 14, 425-436. [DOI] [PubMed] [Google Scholar]
- 21.Verfaillie, C. M. (2002) Nat. Immunol. 3, 314-317. [DOI] [PubMed] [Google Scholar]
- 22.Tamaki, S., Eckert, K., He, D., Sutton, R., Doshe, M., Jain, G., Tushinski, R., Reitsma, M., Harris, B., Tsukamoto, A., et al. (2002) J. Neurosci. Res. 69, 976-986. [DOI] [PubMed] [Google Scholar]
- 23.Uchida, N., Buck, D. W., He, D., Reitsma, M. J., Masek, M., Phan, T. V., Tsukamoto, A. S., Gage, F. H. & Weissman, I. L. (2000) Proc. Natl. Acad. Sci. USA 97, 14720-14725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ourednik, V., Ourednik, J., Flax, J. D., Zawada, W. M., Hutt, C., Yang, C., Park, K. I., Kim, S. U., Sidman, R. L., Freed, C. R. & Snyder, E. Y. (2001) Science 293, 1820-1824. [DOI] [PubMed] [Google Scholar]
- 25.Zhang, S. C., Wernig, M., Duncan, I. D., Brustle, O. & Thomson, J. A. (2001) Nat. Biotechnol. 19, 1129-1133. [DOI] [PubMed] [Google Scholar]
- 26.Reubinoff, B. E., Itsykson, P., Turetsky, T., Pera, M. F., Reinhartz, E., Itzik, A. & Ben-Hur, T. (2001) Nat. Biotechnol. 19, 1134-1140. [DOI] [PubMed] [Google Scholar]
- 27.Tabar, V., Panagiotakos, G., Greenberg, E. D., Chan, B. K., Sadelain, M., Gutin, P. H. & Studer, L. (2005) Nat. Biotechnol. 23, 601-606. [DOI] [PubMed] [Google Scholar]
- 28.Ishibashi, S., Sakaguchi, M., Kuroiwa, T., Yamasaki, M., Kanemura, Y., Shizuko, I., Shimazaki, T., Onodera, M., Okano, H. & Mizusawa, H. (2004) J. Neurosci. Res. 78, 215-223. [DOI] [PubMed] [Google Scholar]
- 29.Cummings, B. J., Uchida, N., Tamaki, S. J., Salazar, D. L., Hooshmand, M., Summers, R., Gage, F. H. & Anderson, A. J. (2005) Proc. Natl. Acad. Sci. USA 102, 14069-14074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barlow, C., Hirotsune, S., Paylor, R., Liyanage, M., Eckhaus, M., Collins, F., Shiloh, Y., Crawley, J. N., Ried, T., Tagle, D. & Wynshaw-Boris, A. (1996) Cell 86, 159-171. [DOI] [PubMed] [Google Scholar]
- 31.Clement, A. M., Nguyen, M. D., Roberts, E. A., Garcia, M. L., Boillee, S., Rule, M., McMahon, A. P., Doucette, W., Siwek, D., Ferrante, R. J., et al. (2003) Science 302, 113-117. [DOI] [PubMed] [Google Scholar]
- 32.Zwaka, T. P. & Thomson, J. A. (2003) Nat. Biotechnol. 21, 319-321. [DOI] [PubMed] [Google Scholar]
- 33.Tiscornia, G., Singer, O., Ikawa, M. & Verma, I. M. (2003) Proc. Natl. Acad. Sci. USA 100, 1844-1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hwang, W. S., Ryu, Y. J., Park, J. H., Park, E. S., Lee, E. G., Koo, J. M., Jeon, H. Y., Lee, B. C., Kang, S. K., Kim, S. J., et al. (2004) Science 303, 1669-1674. [DOI] [PubMed] [Google Scholar]
- 35.Hwang, W. S., Lee, B. C., Lee, C. K. & Kang, S. K. (2005) J. Vet. Sci. 6, 87-96. [PubMed] [Google Scholar]
- 36.Simerly, C., Navara, C., Hyun, S. H., Lee, B. C., Kang, S. K., Capuano, S., Gosman, G., Dominko, T., Chong, K. Y., Compton, D., et al. (2004) Dev. Biol. 276, 237-252. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}