

Abstract
The origin of asymmetric charge and mass partitioning observed for gas-phase dissociation of multiply charged macromolecular complexes has been hotly debated. These experiments hold the potential to provide detailed information about the interactions between the macromolecules within the complex. Here, this unusual phenomenon of asymmetric charge partitioning is investigated for several protein homodimers. Asymmetric charge partitioning in these ions depends on a number of factors, including the internal energy, charge state, and gas-phase conformation of the complex, as well as the conformational flexibility of the protein monomer in the complex. High charge states of both cytochrome c and disulfide-reduced α-lactalbumin homodimers dissociate by a symmetrical charge partitioning process in which both fragment monomers carry away roughly an equal number of charges. In contrast, highly asymmetric charge partitioning dominates for the lower charge states. Cytochrome c dimer ions with eleven charges formed by electrospray ionization from two solutions in which the solution-phase conformation differs dissociate with dramatically different charge partitioning. These results demonstrate that these gas-phase complexes retain a clear “memory” of the solution from which they are formed, and that information about their solution-phase conformation can be obtained from these gas-phase dissociation experiments. Cytochrome c dimer ions formed from solutions in which the conformation of the protein is native show greater asymmetric charge partitioning with increasing ion internal energy. Cytochrome c dimers that are conformationally constrained with intramolecular cross-linkers undergo predominantly symmetric charge partitioning under conditions where asymmetric charge partitioning is observed for cytochrome c dimers without cross-links. Similar results are observed for α-lactalbumin homodimers. These results provide convincing evidence that the origin of asymmetric charge partitioning in these homodimers is the result of one of the protein monomers unfolding in the dissociation transition state. A mechanism that accounts for these observations is proposed.
Introduction
Proteins often act in large assemblies in the cellular environment to complete tasks necessary to life. With electrospray ionization,1 it is possible to liberate intact, noncovalent assemblies of biomolecules from their native solution environment into the gas phase. In recent years, great strides have been made in the size of assemblies accessible to analysis by mass spectrometry (MS). Complexes that have molecular masses in excess of 1 MDa, including whole ribosomes2–5 and virus particles,4,5 have been studied using MS. A key advantage of mass spectrometry for the analysis of noncovalent biomolecular complexes is that the stoichiometry of the complex can be determined from a simple mass measurement.6
Once in the gas phase, information about the structures of the biomolecules can be obtained using two-dimensional mass spectrometry, in which an ion of interest is mass selected and reacted and structural information inferred from the resulting mass-analyzed product ions. Information about the sequence of peptides,7 proteins,8,9 DNA,10 and oligosaccharides11 can be obtained from dissociation experiments. Such dissociation methods have also been used to determine the location of posttranslational modifications in proteins.7,12,13 Extensive work has been done to understand fragmentation mechanisms of biomolecules to obtain the maximum amount of structural information from a dissociation spectrum. More subtle probes of structure, such as ion mobility,14–17 gas-phase H/D exchange,18,19 and chemical20 and proton-transfer reactivity,21–23 have been used to obtain information about intramolecular noncovalent interactions in biomolecules. Proteins and peptides can adopt different conformations in the gas phase. Information about the intrinsic structure of biomolecules obtained from these experiments provides insights into the role of water in biomolecule structure.24,25
Limited structural information about noncovalent complexes has also been obtained using two-dimensional mass spectrometry. Several studies have indicated that some specific interactions that occur in solution can remain intact during the process of electrospray ionization.26–29 For example, one study showed that the gas-phase dissociation activation energy of a complimentary DNA duplex is higher than those of the noncomplementary DNA homodimers.26 The gas-phase dissociation activation energies and the solution-phase binding energies of several complementary DNA duplexes were found to be correlated. These and other gas-phase measurements, supported by molecular modeling calculations, indicate that the Watson–Crick base pairing that exists for complementary DNA duplexes in solution can be preserved in the gas phase. Recent work by Gabelica et al. shows that the gas-phase kinetic stability of DNA duplexes as a function of collision energy parallels the calculated solution-phase dissociation enthalpies.27 This observation provides additional support that Watson–Crick base pairing and specific DNA base stacking interactions are preserved in the gas phase. Although some specific interactions that exist in solution can be preserved in the gas phase, this is not always the case.30
Other correlations between gas-phase and solution-phase properties of noncovalent complexes have been reported. For example, myoglobin has a noncovalently bound heme ligand. Dissociation kinetics of apomyoglobin ions formed from solutions of different composition are different.31 To determine the contributions that hydrogen bonds provide in the binding of heme to myoglobin, Douglas and co-workers mutated the amino acids responsible for heme binding and systematically reduced the number of hydrogen bonds in the complex.29 A correlation was observed between the solution-phase Arrhenius activation energy of heme loss for the various mutants and their gas-phase kinetic stabilities. An exception occurs for some mutations of residue 92 that decrease the solution-phase activation energy but increase the gas-phase kinetic stability. These experiments suggest that the same hydrogen bonds responsible for heme binding in solution are responsible for binding in the gas phase. These experiments clearly show that these ions can retain a “memory” of their solution-phase structure and that these structural differences can be probed in gas-phase dissociation experiments.
Many dissociation experiments of whole multimeric noncovalent complexes of multiple proteins have been done with the goal of obtaining structural information from these large complexes. Robinson and co-workers used ESI to form ions of the intact E. coli 50S ribosomal subunit which is a noncovalent complex consisting of 33 proteins and 2 strands of RNA.3 Upon collisional activation of the complex in the gas phase, several identifiable proteins were ejected. These proteins contained a disproportionately high degree of charging for their mass compared to that of the original complex. The authors concluded that it is feasible to begin mapping subunit interactions with mass spectrometry.
A key obstacle to using mass spectrometry for the general analysis of protein complexes is a poor understanding of how these complexes dissociate in the gas phase. Many groups have reported a seemingly odd dissociation behavior for large multimeric protein complexes in which a small subunit, typically a protein monomer, is ejected from the complex, with the monomer carrying away a disproportionate amount of charge for its mass relative to the mass of the remaining complex.32–36,38,39 For example, streptavidin exists in solution as a tetramer in which each straptavidin molecule is identical to the others. Gas-phase dissociation of the 14+ charge state of the tetrameric complex results primarily in the formation of the 7+ monomer and the 7+ trimer.36 No dimer ions are observed. Thus, the monomer, which is only one-fourth of the mass of the overall complex, carries off half of the charge!
To explain such highly asymmetric charge partitioning, Smith and co-workers suggested a liquid drop model in which the protein complex is treated as a sphere.36 Upon activation, a fragment “drop” is produced that has a higher surface area-to-mass ratio than the original complex and thus is able to remove a disproportionate amount of charge. This model could not account for the extent of asymmetry, so they speculated that the dissociation of the tetramer may occur by a Coulombically driven process in which a monomer species becomes “unraveled” and is ejected from the aggregate with a disproportionately large share of the charge.34 Some support for this hypothesis was presented by Felytsyn et al., who measured the blackbody infrared radiative dissociation (BIRD) Arrhenius activation parameters for the dissociation of homopentameric Shiga-like toxin I.32 Arrhenius A factors as high as 1039 s−1 were reported, indicating an unusually high transition-state entropy consistent with a large structural change taking place during the dissociation process. The authors suggested that the charge enrichment of the leaving subunit is energetically unfavorable, but a transfer of charge destabilizes the complex sufficiently that charge enrichment is entropically favorable. Heck and co-workers studied the dissociation of four protein homodimers and found that, even for such relatively simple complexes, asymmetric charge partitioning can occur.38 Heck and co-workers concluded that the liquid drop model as originally proposed by Smith and co-workers36 could not quantitatively account for this result.38
Here, we show that the charge partitioning between the dissociation products of protein homodimers is a function of several parameters. These include the charge state of the complex, the dissociation energy, the solution composition from which these complexes are formed, and the conformational flexibility of the proteins in the complex. These results are consistent with those of Felitsyn et al., where charge asymmetry in the dissociation products is energetically unfavorable, but is driven by high entropy.32 These results also demonstrate that the conformational flexibility of the proteins in the complex is a key part of the asymmetric charge partitioning process.
Experimental Section
Mass Spectrometry.
All experiments were conducted using a 110 mm bore 9.4 T Fourier-transform mass spectrometry (FTMS) instrument that has been recently constructed in collaboration with Bruker Daltonics (Billerica, MA) (Figure 1). The ion optics and introduction system are a standard Bruker design. Ions are generated by ESI and accumulated in a storage hexapole prior to injection through multiple ion lenses and two stages of differential pumping into the ultra-high-vacuum (UHV) chamber of the instrument where the ions are trapped in a cylindrical Bruker Infinity ion cell with an outer diameter and length of 74 and 80 mm, respectively. A pulse of nitrogen gas is introduced into the vacuum chamber at a pressure of ~2 × 10−6 Torr to enhance trapping and damp the motion of the ions in the cell. Pressures in the UHV chamber are measured using an uncalibrated ion gauge situated above one of the turbomolecular pumps. During ion excitation and detection, the pressure is typically ~3 × 10−10 Torr. The vacuum chamber of a standard Bruker FTMS instrument is designed to fit into a 160 mm bore magnet. To interface the ion optics to a 110 mm bore magnet, a custom vacuum chamber that fits in the bore of the magnet and extends several centimeters on each side was constructed. To increase the pumping speed of the system, a 500 L/s turbomolecular pump was added to the vacuum chamber opposite the ion source.
Figure 1.

Schematic diagram of (a) the entire Berkeley-Bruker FTMS instrument with a 110 mm bore magnet and (b) a cross section of the vacuum chamber and ion cell.
The heater for the vacuum chamber consists of an oxygen-free copper tube that has 30 grooves cut lengthwise on its outside surface. Two independent heating circuits of 1.2 mm diameter titanium wire are placed in the grooves of the copper tube and are insulated from the copper by alumina tubing. The copper tube is slit lengthwise and compressed to fit tightly inside the vacuum chamber of the instrument. The inner diameter of the copper tube is slightly larger than the outer diameter of the ion cell, making it necessary to stabilize the ion cell in the center of the heater tube with a spring-loaded device. The vacuum chamber inside the magnet consists of two concentric titanium tubes with a 3.2 mm gap between them (Figure 1b). These tubes are sealed at either end, and the space between the titanium tubes is evacuated with a mechanical pump to create a vacuum insulation layer. This arrangement provides even heating of the vacuum chamber for BIRD experiments. The gap between the concentric tubes can be vented to atmospheric pressure to increase the cooling rate of the vacuum chamber. To protect the magnet from high temperatures, a layer of water (~0.4 mm thick, 1 L/min flow rate) is passed between the outer titanium tube and the shim tube of the magnet.
Chemicals.
Equine cytochrome c, bovine ubiquitin, α-lactalbumin from bovine milk, and phospholipase A2 from Naja mossambica were purchased from Sigma-Aldrich Co. (St. Louis, MO), and were used without further purification except for the chemical modifications described below. Ammonium acetate, potassium phosphate, sodium hydroxide, and sodium chloride were purchased from Fisher Scientific (Pittsburgh, PA). BS3 was purchased from Pierce Chemical (Rockford, IL).
Intramolecular cross-linking of cytochrome c with BS3 is done by modifying a procedure used by Fales and co-workers.40 To avoid intermolecular cross-linkages between cytochrome c monomers, cross-linker reactions are done in dilute protein solution. The cross-linked protein is subsequently concentrated to assist in dimer formation. A 150 μL sample of 100 μM equine cytochrome c is added to 1350 μL of 100 mM phosphate-buffered saline (potassium phosphate buffer at pH 7.6 with 0.3 M sodium chloride). To this solution is added 30 μL of a freshly prepared 5 mM solution of BS3 in dry DMSO. The reaction is allowed to proceed at room temperature for 30 min before being quenched with 100 μL of an aqueous solution of 100 mM ammonium acetate. Buffer exchange and solution concentration are accomplished using a Microcon centrifuge tube with a 10 kDa cutoff membrane filter from Millipore Co. (Bedford, MA). Three 500 μL aliquots of the reaction solution are concentrated in a single centrifuge tube, and the buffer is then exchanged four times with 1.0 M aqueous ammonium acetate, pH 7, and three times with 100 mM ammonium acetate, pH 7. The final recovered volume of 15 μL had an estimated final concentration of 150–200 μM.
Acetamidation of α-lactalbumin from bovine milk was done by Dr. David King (University of California, Berkeley). All four intramolecular disulfide bonds of α-lactalbumin are reduced with tris(carboxyethyl)-phosphine, acetamidated in the dark with iodoacetamide for 1 h, and purified using HPLC to remove buffer and unreacted reagents. Mass measurements confirm that all disulfide bonds are broken and acetamidated.
Dimer Formation.
Formation of nonspecific protein dimers is enhanced by using protein concentrations of 100–200 μM and adjusting interface voltages for “gentle” ion introduction. The dimer signal is improved by tuning for selective ion accumulation in the external hexapole of the instrument. This is done by accumulating the ions in the hexapole for 3–4 s (vs ≤1.0 s typically), increasing the hexapole dc offset from ~2.7 to 3.5–4.5 V, and injecting multiple hexapole accumulations into the ion cell prior to detection.
Proteins are electrosprayed from solution conditions in which the native conformation of the protein is intact (100 mM ammonium acetate in water, pH ~7)41,42 or denatured (1:1 water/methanol and 2% acetic acid). Solutions containing methanol are electrosprayed with a syringe pump flow rate of 1–2 μL/min and nitrogen-assisted nebulization. Ions formed from buffered aqueous solutions are generated using nanoelectrospray using borosilicate capillaries that are pulled to a ~4 μm tip with a model P-87 capillary puller (Sutter Instruments, Navato, CA). A small volume of solution (2–6 μL) is injected into the borosilicate capillary, and a platinum wire is inserted into the solution at the end of the capillary. The capillary is positioned ~2 mm from the source inlet, and a potential of ~1100 V is applied to the platinum wire. An unmodified Bruker Apollo source with a heated glass capillary is used for these experiments.
Gas-Phase Deprotonation and Collisional Activation.
To compare the dissociation of the same charge states of protein ions produced from solutions in which their conformations are native or denatured, it is necessary to reduce the charge state of some complexes. Gas-phase charge state reduction is done by introducing triethylamine at a pressure of (1–5) × 10−8 Torr into the UHV chamber through a variable leak valve (Varian, Lexington, MA). Gas-phase deprotonation is continued for 30–70 s prior to ion excitation and detection. For dissociation experiments, the variable leak valve is closed prior to the introduction of N2 collision gas and ion excitation and dissociation to reduce the pressure of triethylamine.
To dissociate the complexes, the charge state of interest is isolated using correlated sweeps prior to sustained off-resonance irradiation–collisionally activated dissociation (SORI–CAD)43 in which an excitation waveform is applied +600 Hz off-resonance for 250 ms with an applied peak-to-peak potential of 3–18 V. A pulse of nitrogen is introduced for 90 ms, briefly raising the pressure to ~2 × 10−6 Torr.
Results and Discussion
Gas-phase homodimer ions of cytochrome c can be readily formed from concentrated solutions (10−4 M) using electrospray ionization. The gas-phase dissociation pathways and energetics of these nonspecific homodimer ions were investigated. Unlike protonated homodimers of small molecules, the dissociation behavior of protein homodimers can be asymmetric (with respect to charge) and much more complex. Effects of the charge state, the dissociation energy of the complex, the gas-phase conformation, and the conformational flexibility of the protein in the complex are shown for cytochrome c dimers in the sections below.
Effects of Charge State.
A typical electrospray solution of a 100 μM cytochrome c solution in 1:1 water/methanol with 2% acetic acid is shown in Figure 2a. The charge state distribution of the monomer ions formed from this denaturing solution is centered around the 10+ charge state (M10). The abundance of the dimer ions is much lower and is centered around the 15+ charge state (D15). Note that the m/z ratios for the even charge states of the dimer overlaps with those of some monomer charge states, but the relative abundances of the dimers and the monomers for these charge states can be easily determined from the isotope patterns for these peaks (Figure 2a, inset). The charge state distribution can be readily shifted to higher m/z (lower charge state) by gas-phase proton-transfer reactions with triethylamine introduced into the ion cell after ion introduction but prior to detection (Figure 2b). Dimer ions with as few as nine charges can be readily formed.
Figure 2.

Electrospray ionization mass spectra of cytochrome c from a 50:50 water/methanol solution with 2% acetic acid and a solution concentration of 10−4 M detected (a) directly and (b) after charge reduction with triethylamine. The inset is an expansion around m/z 1766 showing the isotopic distributions from the M7 (monomer) and D14 (dimer) ions.
Dissociation spectra of the odd charge states of the cytochrome c dimer ions, from D19 to D11, are shown in Figure 3. Dissociation of even charge states of the dimer ions is more complicated in that a symmetric dissociation pathway results in two monomers with the same m/z as the dimer being activated. Although these ions can be resolved on the basis of their isotope patterns, these fragment monomer ions are subsequently activated by the SORI excitation waveform. To avoid this complication, only odd charge states of the dimers are investigated.
Figure 3.
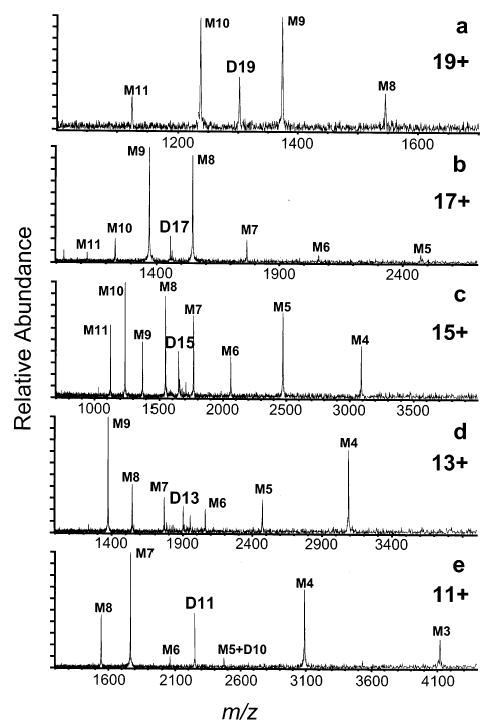
SORI–CAD spectra of odd charge states of cytochrome c dimer ions electrosprayed from a 50:50 water/methanol solution with 2% acetic acid: (a) 19+, (b) 17+, (c) 15+, (d) 13+, and (e) 11+ dimer charge states. The dimer ions in (e) are produced by gas-phase deprotonation of higher charge state dimers.
D19 dissociates primarily by a symmetric pathway, generating complementary M10/M9 and M11/M8 pairs. Similarly, D17 dissociation is also largely symmetric, but the spread in product ion charge states is larger than that for D19. The spectrum for D15 clearly shows evidence for two fragmentation pathways: a symmetric process to give the M8/M7 complementary pair, and an asymmetric charge partitioning process to give the distribution of product monomers centered around the M10/M5 complementary pair. The asymmetric pathway becomes the dominant process for D13 and D11 under all dissociation conditions investigated. For D11, very little symmetric dissociation is observed.
These results clearly show that the relative extent of symmetric vs asymmetric charge partitioning in the dissociation process for these protein dimers is a strong function of the charge state of the complex. Higher charge states dissociate predominantly via a symmetric process, whereas lower charge states dissociate almost exclusively through an asymmetric pathway.
Effects of Conformation.
D11 ions can also be readily formed by electrospray of 10−4 M aqueous cytochrome c solutions with 100 mM ammonium acetate buffer. In this solution, cytochrome c should have a native conformation.41,42 The electrospray mass spectrum of this solution is dominated by the M7 ion. D11 is produced in low abundance (Figure 4). Dissociation of this ion under conditions similar to those for D19 to D11 formed from denaturing solutions results in a charge distribution that is broad, but highly symmetric, producing M6/M5, M7/M4, and M8/M3 complementary pairs in decreasing abundance (Figure 5a).
Figure 4.

Electrospray ionization mass spectrum of cytochrome c formed from 100 mM ammonium acetate in water at a solution concentration of 10−4 M.
Figure 5.
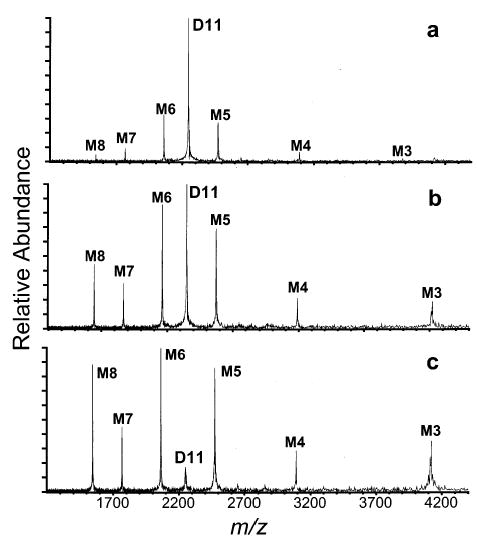
SORI–CAD spectra of cytochrome c D11 ions formed from 100 mM ammonium acetate in water shown as a function of maximum kinetic energy: (a) 120 eV, (b) 210 eV, (c) 360 eV.
When D11 ions are produced by charge stripping ions formed from a denaturing solution, the dissociation process is almost completely asymmetric (Figure 3e), yet it is predominantly symmetric when these D11 ions are formed from a solution in which cytochrome c has a native conformation. In addition, the charge distribution for the latter ions shows an energy dependence (vide infra), whereas very little energy dependence is observed for the former ions. These D11 ions clearly “remember” the solution from which they were formed. Thus, the gas-phase conformation of these ions clearly plays a role in their dissociation behavior. Ultimately, if we can understand how conformation affects gas-phase dissociation pathways and energetics, it should be possible to deduce elements of solution-phase structure from these gas-phase experiments.
Effects of Dissociation Energy.
SORI–CAD is a “slow” heating method in which an ion is activated incrementally by many low-energy collisions.43 By increasing the magnitude of the excitation waveform, the maximum kinetic energy of the ion can be increased, resulting in increased energy deposition into the ion.44 Spectra of D11 ions formed from “native” solution conditions and activated with increasing amounts of maximum kinetic energy are shown in Figure 5. At the lowest energy investigated, the dissociation is largely symmetric (Figure 5a). At the highest energy investigated, the asymmetric and symmetric dissociation processes become comparable in magnitude (Figure 5c). Increasing the energy deposition results in an increasing extent of asymmetric dissociation. Thus, energy deposition can clearly play a role in asymmetric dissociation. Interestingly, D11 formed directly from denaturing solutions dissociates predominantly asymmetrically over the same range of collision energies, with the dominant products being the M7/M4 pair. A slight enhancement in the abundance of the M8/M3 ion pair is observed at higher collision energies.
These results are consistent with those of Smith and Heck and their co-workers.37,38 Smith and co-workers found that even charge states of cytochrome c homodimers, D18–D24, dissociated with highly symmetric charge partitioning.37 Heck and coworkers observed asymmetric charge partitioning for D11 ions.38 The results presented here clearly show a gradual change from symmetric to asymmetric charge partitioning with decreasing charge state. Heck and co-workers measured asymmetric charge partitioning for D11 ions formed from solutions in which the protein is in its native conformation. The results presented here show a symmetric charge partitioning of D11 ions formed from similar solutions when the ions are activated using gentle conditions and increasingly asymmetric charge partitioning at higher dissociation energies. This apparent discrepancy is explained by noting that Heck and co-workers used a time-of-flight mass spectrometer, an instrument that needs to make measurements within a very short time frame. To observe dissociation on the time frame of their experiment, much more energy must be deposited into the ions (a kinetic shift). Because there is clearly an energy dependence in the degree of asymmetric charge partitioning, care must be taken when results from experiments conducted on instruments with different kinetic windows are interpreted.
Effects of Conformational Flexibility.
Cytochrome c monomers can adopt many different conformations in the gas phase.19,45 The conformation can depend on the solution-phase structure as well as the overall charge state of the ion. Both folding and unfolding of cytochrome c monomer ions in the gas phase have been reported.46,47 To obtain a better understanding of how a conformational change of a protein might influence the dissociation behavior of the protein complex, the conformational flexibility of cytochrome c was reduced by reacting the protein with the nonspecific cross-linker BS3, which is capable of spanning 11.4 Å and irreversibly linking two lysine residues residing on the surface of the protein as illustrated in Scheme 1. The linking reactions are performed in 100 mM potassium phosphate buffer, pH 7.6, to ensure that the protein is in its native conformation during the linking procedure.41,42 Mass spectra collected under typical conditions reveal that each monomer receives between zero and three linkers and that each linker is covalently attached to two lysines without any partially reacted linker groups (Figure 6). To avoid intermolecular cross-linking, linking reactions are performed in dilute (10 μM) cytochrome c solution and the reaction is quenched prior to the sample being concentrated to enhance dimer formation (~150 μM). Mass spectra of cross-linked cytochrome c obtained using electrospray interface conditions that disrupt noncovalent protein dimer formation show no cytochrome c dimer signal, indicating that essentially all of the linkages are intramolecular.
Scheme 1.

Figure 6.
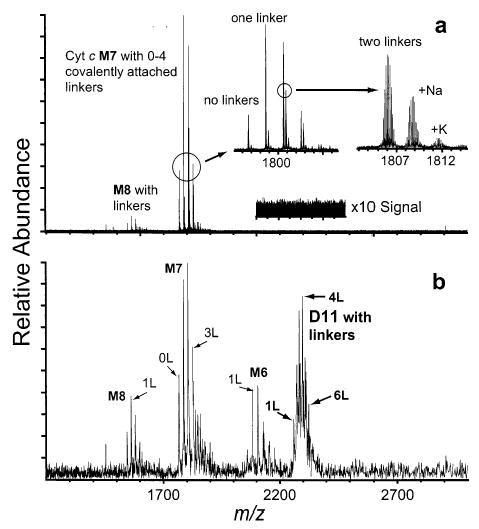
Electrospray ionization mass spectra of cytochrome c that has been reacted with cross-linking reagents. The spectra are tuned with interface conditions optimizing the (a) monomer signal and (b) dimer signal. The absence of dimer ions in (a) indicates intramolecular cross-linking rather than cross-linking between monomer species.
Using gentle electrospray interface conditions and selective ion accumulation in the external hexapole of the instrument, it is possible to form a distribution of cytochrome c D11 ions with one to six linkers. These ions are isolated and activated with a SORI pulse applied +600 Hz from the cytochrome c D11 ion with a total of four attached linkers (Figure 7). Under the same conditions as the Figure 5 data, predominantly symmetric dissociation is observed at all energies. The asymmetric dissociation of cytochrome c dimer ions formed from native solution conditions is not observed for the cross-linked protein dimers. The most asymmetric fragment charge states correlate with the degree of conformational flexibility remaining in the monomer units. For example, in the dissociation process D11 → M8 + M3, the M8 ion predominantly has only one or no linkers, and the M3 ion has predominantly three linkers. Thus, the monomer species that has the greatest conformational flexibility is the one that carries away the majority of the charge. In contrast, for the D11 → M6 + M5 symmetric dissociation process, the M6 and M5 ions have similar numbers of attached linkers. Clearly, the conformational flexibility of the protein in the complex plays an important role in the appearance of asymmetric dissociation.
Figure 7.

SORI–CAD spectra of cytochrome c dimers with one to six intramolecular cross-linkers attached. Protein ions were formed by electrospray ionization using a 100 mM ammonium acetate solution. Results are shown as a function of maximum kinetic energy: (a) 120 eV, (b) 210 eV, (c) 360 eV.
Because of the nonspecific nature of BS3 attachment, it is expected that the degree of conformational stability conferred by the linking reaction will vary depending on which lysine pairs are linked. Fales and co-workers examined cytochrome c reacted with cross-linkers of differing lengths.40 Equine cytochrome c contains 19 lysine residues, and many of these are located close enough for intramolecular linking reactions to occur.40 However, only five or six primary linkage sites were identified from ESI mass spectra of tryptic digests of cytochrome c after linking reactions.40 The most reactive lysine pair reported is K25/K27, the dominant cross-link obtained using reaction conditions optimized for a single linker attachment. Under more vigorous reaction conditions, four or five additional linking sites were identified with apparently similar reactivities. Two of these linking sites occur within a single α-helix, while the remainder cross-link lysine pairs in different regions of the molecule brought together by the tertiary structure of the protein. Thus, BS3 linking reagents have the potential to stabilize the secondary structure of the protein, and a strong probability of stabilizing the tertiary structure, particularly in the case where two or more linkers are attached.
An additional effect of reacting protein molecules with linkers is a decrease in the number of basic sites available for protonation; a cytochrome c molecule with three attached linkers will have the number of potentially protonated lysine residues reduced by six. The reduction in the number of basic sites alone is not expected to significantly contribute to the measured variation in dissociation asymmetry of cytochrome c dimers. Equine cytochrome c has 24 basic residues, and residues other than the standard basic residues can be protonated.48 An individual cytochrome c monomer can hold 24 protons,49 indicating that there is a large excess in the number of sites available for protonation even after the modification of six lysine side chains.
Conformational Flexibility of α-Lactalbumin.
The non-specific cross-linking of cytochrome c has the effect of increasing gas-phase conformational rigidity. A complementary experiment is the irreversible reduction of intramolecular disulfide bonds that has the effect of decreasing the gas-phase conformational rigidity of a protein. The protein α-lactalbumin has four intramolecular disulfide bonds. A measurable abundance of the 13+ homodimer (D13) of the protein can be formed from denaturing solutions in which the protein concentration is 125 μM (Figure 8a). Dissociation of α-lactalbumin D13 results in a symmetric and narrow charge distribution of product monomers for all dissociation energies investigated (Figure 9a,b).
Figure 8.

Electrospray ionization mass spectra of α-lactalbumin using a 50:50 water/methanol solution with 2% acetic acid and a protein concentration of 125 μM with all four intramolecular disulfide bonds (a) intact, (b) broken, (c) and broken and charged stripped with triethylamine. The inset shows isotopic resolution of the α-lactalbumin 13+ dimer with intact disulfide bonds.
Figure 9.
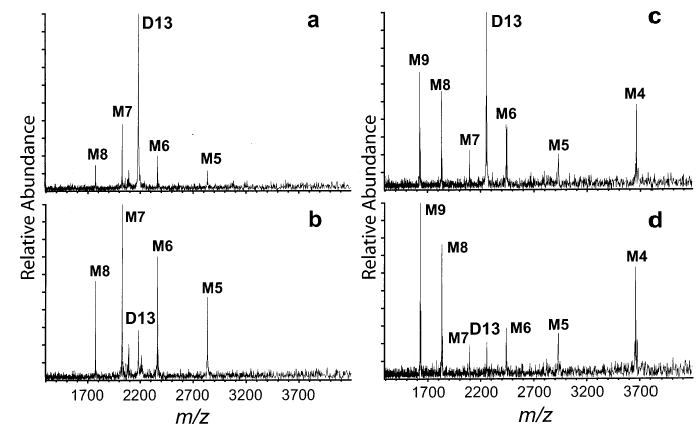
SORI–CAD spectra of D13 of α-lactalbumin using different maximum kinetic energies with disulfide bonds intact [(a) 50 eV and (b) 170 eV] and with disulfide bonds broken and all eight cysteines in α-lactalbumin acetamidated to prevent the disulfide bonds from reforming [(c) 50 eV and (d) 170 eV].
The ESI mass spectrum of the same protein after disulfide bond reduction and acetamidation is shown in Figure 8b. Higher charge state ions of the dimer are observed. For the dimer, the 15–19+ charge states are dominant. To compare equivalent charge states of the disulfide-broken and disulfide-intact α-lactalbumin, all disulfide-reduced ions, including D15–D19, are charge stripped in the gas phase using triethylamine as the deprotonating agent (Figure 8c). Dissociation spectra of D13 formed by charge stripping are shown in Figure 9c,d.
A dramatic difference is observed in the degree of asymmetry of charge partitioning between the monomer fragments produced from the disulfide-broken and disulfide-intact protein homodimers. Without the disulfide bonds, the charge partitioning of α-lactalbumin is highly asymmetric (Figure 9c,d). The most prominent charge partitioning pathway is formation of the M9/M4 complementary pair. This pathway is not observed for the disulfide-intact counterpart. These results are entirely consistent with the cross-linking experiments with cytochrome c. Conformational flexibility of the protein in the homodimer plays an important role in the observation of asymmetric charge partitioning. By making a protein less conformationally flexible, by either cross-linking or disulfide bond formation, the asymmetric dissociation pathway is shut down or greatly reduced.
A different result is obtained for higher charge states of disulfide-broken α-lactalbumin. Disulfide-broken α-lactalbumin D17 dissociates with very broad, but overall symmetric, charge partitioning under all dissociation energies studied (Figure 10). This is consistent with the results reported for cytochrome c in which the higher charge states undergo symmetric charge partitioning.
Figure 10.
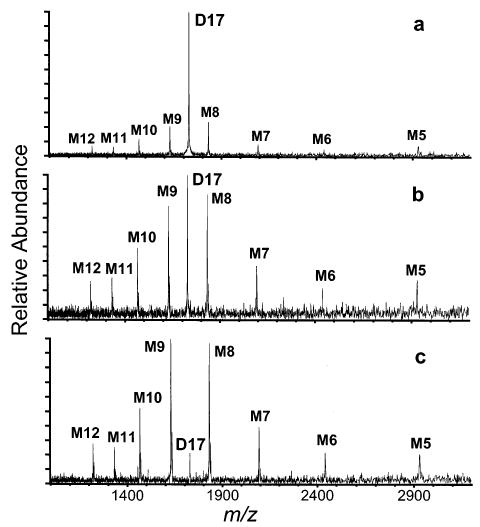
SORI–CAD spectra of the 17+ dimers of α-lactalbumin, with all four intramolecular disulfide bonds broken and all of the cysteines acetamidated, as a function of maximum kinetic energy: (a) residual excitation after isolation, (b) 70 eV, (c) 90 eV.
Charge Partitioning and Disulfide Bonds.
Other protein homodimers with varying numbers of disulfide bonds were also examined to determine the relationship between gas-phase conformational flexibility and asymmetric charge partitioning. One measure of asymmetric charge partitioning is the width of the distribution of product monomer charge states; a narrow distribution corresponds to a more symmetric process. In this experiment, similar charge states of ubiquitin, α-lactalbumin, and phospholipase A2 are isolated and dissociated, and the resulting charge distributions of the monomers are compared (Figure 11). Bovine ubiquitin has no disulfide bonds at all. Dissociation of ubiquitin D11 results in a symmetric but broad distribution of product ions (Figure 11c). Dissociation of bovine α-lactalbumin D13, which has four disulfide bonds, produces a narrower distribution even though the protein dimer is larger and more highly charged than ubiquitin (Figure 11b). Phospholipase A2 (PLA2) from Mozambique cobra venom has seven disulfide bonds, an exceptionally large number for a 13.2 kDa protein. D11 of PLA2 dissociates exclusively to the M6/M5 complementary pair at all dissociation energies investigated (Figure 11a). The charge partitioning between the monomers is the most symmetric of all the protein dimers investigated. As the number of disulfide bonds increases, the gas-phase conformational flexibility of the proteins decreases, and the resulting charge state distributions of the product monomers narrow.
Figure 11.

SORI–CAD spectra of (a) phospholipase A2 (seven disulfide bonds, 13.2 kDa), (b) α-lactalbumin (four disulfide bonds, 14.2 kDa), and (c) ubiquitin (no disulfide bonds, 8.6 kDa).
Dissociation Mechanism.
Symmetric dissociation of a homodimer to produce identically charged monomers is the expected result and is readily understood. To observe asymmetric charge partitioning upon dissociation of a homodimer, it is necessary to break the symmetry of the system. The only way to do this under gentle dissociation conditions is to change the conformation of one of the monomers in the dimer. The results we present here indicate that, in the dissociation transition state, one of the proteins in the homodimer unfolds, and in doing so, it carries away the vast majority of charge.
The apparent gas-phase basicity of unfolded or elongated proteins is greater than that of a folded protein of the same charge state,21,23,48 making it energetically favorable to transfer protons from a folded to an unfolded or unfolding protein. This is primarily due to lower Coulomb repulsion between charges for more elongated conformations. For this proton transfer to take place, the protons on the complex must be able to sample the most energetically favorable positions. The work of McLafferty and co-workers, who have done extensive studies on the gas-phase H/D exchange reactivity of cytochrome c ions, indicates that protons are quite mobile along the surface of a protein.19,46,50 Nearly complete H/D exchange of high charge states of cytochrome c can take place (a maximum of 193 out of 198 solution-exchangeable hydrogens for the 15+ charge state) with D2O.19 This occurs even though H/D exchange with D2O is only favorable at the protonated sites.51,52 Very recently, Wells et al. formed gas-phase cytochrome c dimer ions by reacting M15+ with M6− to form D9+.53 Upon activation, the complex dissociates to form predominantly the M6+/M3+ complementary pair, indicating that at least nine protons are transferred to the originally negatively charged monomer upon complex formation and ion activation. The asymmetric dissociation observed in these experiments is most likely the result of different conformations for the two monomers.
The results with the intramolecular cross-linked cytochrome c dimers as well as disulfide-intact/reduced α-lactalbumin clearly indicate that, by reducing the conformational flexibility of the protein subunit, the extent of asymmetric dissociation of the dimer complex decreases. By forming intramolecular bonds between different regions of the protein, the maximum extent of unfolding that is possible is reduced. These experiments clearly show that protein unfolding is required for asymmetric dissociation to occur. The high transition-state entropies measured by Klassen and co-workers for dissociation of a homopentameric complex indicate that the unfolding must take place during the transition state.32 This is also indicated by our results, showing that the complexes can remember their solution-phase structure. Dissociation of D11 ions of cytochrome c from native solution conditions produces largely symmetric charge partitioning at low energy, with increasing asymmetry at higher energy. These results suggest that the lowest energy process is symmetric dissociation. This is also consistent with the observation that high charge state dimers, which are much more fragile than lower charge state dimers, dissociate symmetrically. The increasingly asymmetric charge partitioning with increasing dissociation energy observed for D11 of cytochrome c formed from native solution conditions is consistent with the unfolding of cytochrome c in the transition state. Unfolding low charge states of a protein in the gas phase requires energy,23,46,47 but the high entropy of this process makes it competitive at high dissociation energies. Higher charge state complexes dissociate before unfolding, resulting in symmetric dissociation.
Conclusions
Mass spectrometry is becoming a powerful tool to determine accurate information about the stoichiometry of large biomolecule complexes in solution. Two-dimensional mass spectrometry is being used to try to obtain structural information from such large macromolecular complexes, e.g., to determine how proteins in a complex are organized and how they interact. However, results from dissociation experiments can be notoriously difficult to interpret without a good understanding of dissociation mechanisms and energetics.
The origin of asymmetric dissociation in macromolecular complexes has been hotly debated. For protein homodimers, we demonstrate that the unusual asymmetric charge partitioning that is typically observed when dissociating macromolecular complexes depends on the internal energy, charge state, and conformation of the complex as well as the conformational flexibility of the monomers in the complex. We show that the charge asymmetry in the dissociation of these protein homodimers is due to a protein unfolding in the dissociative transition state. This unfolding requires more energy, but the process is entropically favored.
While these results and the proposed mechanism are for protein homodimers, it is expected that this same mechanism should apply to larger macromolecular complexes consisting of many subunits. By understanding in detail how these complexes dissociate, we hope that it will be possible to extract useful structural information about the organization of the subunits in large complexes. It should also be possible to obtain information about solution-phase structure from these gas-phase experiments. Gas-phase complexes can clearly retain a memory of the solution from which they are formed; the dissociation behavior of a complex can be dramatically different when the ions are formed from solutions in which the solution-phase structure differs. A better understanding of dissociation mechanisms and energetics of these large complexes should greatly enhance our ability to deduce information about these complexes in solution from these gas-phase experiments. We are currently extending these measurements to larger macromolecular complexes.
Acknowledgments
We acknowledge Dr. David King (University of California, Berkeley) for generously preparing α-lactalbumin, Mr. Anthony Iavarone for helpful discussions, and Mr. Russell Cooper for preliminary work on cross-linking cytochrome c. Funding for this work has been provided by the National Institutes of Health (Grant R01-GM64712-01).
References
- 1.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 2.Benjamin DR, Robinson CV, Hendrick JP, Hartl FU, Dobson CM. Proc Natl Acad Sci USA. 1998;95:7391–7395. doi: 10.1073/pnas.95.13.7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rostom AA, Fucini P, Benjamin DR, Juenemann R, Nierhaus KH, Hartl FU, Dobson CM, Robinson CV. Proc Natl Acad Sci USA. 2000;97:5185–5190. doi: 10.1073/pnas.97.10.5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siuzdak G, Bothner B, Yeager M, Brugidou C, Fauquet CM, Hoey K, Chang CM. Chem Biol. 1996;3:45–48. doi: 10.1016/s1074-5521(96)90083-6. [DOI] [PubMed] [Google Scholar]
- 5.Tito MA, Tars K, Valegard K, Hajdu J, Robinson CV. J Am Chem Soc. 2000;122:3550–3551. [Google Scholar]
- 6.Loo JA. Int J Mass Spectrom. 2000;200:175–186. [Google Scholar]
- 7.Mirgorodskaya E, Roepstorff P, Zubarev RA. Anal Chem. 1999;71:4431–4436. doi: 10.1021/ac990578v. [DOI] [PubMed] [Google Scholar]
- 8.Kelleher NL, Lin HY, Valaskovic GA, Aaserud DJ, Fridriksson EK, McLafferty FW. J Am Chem Soc. 1999;121:806–812. [Google Scholar]
- 9.O’Connor PB, Speir JP, Senko MW, Little DP, McLafferty FW. J Mass Spectrom. 1995;30:88–93. [Google Scholar]
- 10.Little DP, Aaserud DJ, Valaskovic GA, McLafferty FW. J Am Chem Soc. 1996;118:9352–9359. [Google Scholar]
- 11.Desaire H, Sirich TL, Leary JA. Anal Chem. 2001;73:3513–3520. doi: 10.1021/ac010385j. [DOI] [PubMed] [Google Scholar]
- 12.Ge Y, Lawhorn BG, ElNaggar M, Strauss E, Park JH, Begley TP, McLafferty FW. J Am Chem Soc. 2002;124:672–678. doi: 10.1021/ja011335z. [DOI] [PubMed] [Google Scholar]
- 13.Kelleher RL, Zubarev RA, Bush K, Furie B, Furie BC, McLafferty FW, Walsh CT. Anal Chem. 1999;71:4250–4253. doi: 10.1021/ac990684x. [DOI] [PubMed] [Google Scholar]
- 14.Wyttenbach T, Vonhelden G, Bowers MT. J Am Chem Soc. 1996;118:8355–8364. [Google Scholar]
- 15.Valentine SJ, Anderson JG, Ellington AD, Clemmer DE. J Phys Chem B. 1997;101:3891–3900. [Google Scholar]
- 16.Kinnear BS, Hartings MR, Jarrold MF. J Am Chem Soc. 2002;124:4422–4431. doi: 10.1021/ja012150v. [DOI] [PubMed] [Google Scholar]
- 17.Clemmer DE, Jarrold MF. J Mass Spectrom. 1997;32:577–592. [Google Scholar]
- 18.Winger BE, Light-Wahl KJ, Rockwood AL, Smith RD. J Am Chem Soc. 1992;114:5897–5898. [Google Scholar]
- 19.McLafferty FW, Guan ZQ, Haupts U, Wood TD, Kelleher NL. J Am Chem Soc. 1998;120:4732–4740. [Google Scholar]
- 20.Stephenson JL, Schaaff TG, McLuckey SA. J Am Soc Mass Spectrom. 1999;10:552–556. doi: 10.1016/S1044-0305(99)00026-4. [DOI] [PubMed] [Google Scholar]
- 21.Williams ER. J Mass Spectrom. 1996;31:831–842. doi: 10.1002/(SICI)1096-9888(199608)31:8<831::AID-JMS392>3.0.CO;2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loo RRO, Smith RD. J Mass Spectrom. 1995;30:339–347. [Google Scholar]
- 23.Gross DS, Schnier PD, Rodriguez-Cruz SE, Fagerquist CK, Williams ER. Proc Natl Acad Sci USA. 1996;93:3143–3148. doi: 10.1073/pnas.93.7.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jarrold MF. Acc Chem Res. 1999;32:360–367. [Google Scholar]
- 25.Jockusch RA, Lemoff AS, Williams ER. J Am Chem Soc. 2001;123:12255–12265. doi: 10.1021/ja0106873. [DOI] [PubMed] [Google Scholar]
- 26.Schnier PD, Klassen JS, Strittmatter EE, Williams ER. J Am Chem Soc. 1998;120:9605–9613. doi: 10.1021/ja973534h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gabelica V, De Pauw E. J Am Soc Mass Spectrom. 2002;13:91–98. doi: 10.1016/s1044-0305(01)00335-x. [DOI] [PubMed] [Google Scholar]
- 28.Loo JA, He JX, Cody WL. J Am Chem Soc. 1998;120:4542–4543. [Google Scholar]
- 29.Hunter CL, Mauk AG, Douglas DJ. Biochemistry. 1997;36:1018–1025. doi: 10.1021/bi961993+. [DOI] [PubMed] [Google Scholar]
- 30.Kitova EN, Bundle DR, Klassen JS. J Am Chem Soc. 2002;124:5902–5913. doi: 10.1021/ja017213o. [DOI] [PubMed] [Google Scholar]
- 31.Gross DS, Zhao YX, Williams ER. J Am Soc Mass Spectrom. 1997;8:519–524. doi: 10.1016/S1044-0305(97)00010-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Felitsyn N, Kitova EN, Klassen JS. Anal Chem. 2001;73:4647–4661. doi: 10.1021/ac0103975. [DOI] [PubMed] [Google Scholar]
- 33.Fitzgerald MC, Chernushevich I, Standing KG, Whitman CP, Kent SBH. Proc Natl Acad Sci USA. 1996;93:6851–6856. doi: 10.1073/pnas.93.14.6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Light-Wahl KJ, Schwartz BL, Smith RD. J Am Chem Soc. 1994;116:5271–5278. [Google Scholar]
- 35.Rostom AA, Sunde M, Richardson SJ, Schreiber G, Jarvis S, Bateman R, Dobson CM, Robinson CV. Proteins: Struct, Funct, Genet. 1998:3–11. doi: 10.1002/(sici)1097-0134(1998)33:2+<3::aid-prot2>3.3.co;2-8. [DOI] [PubMed] [Google Scholar]
- 36.Schwartz BL, Bruce JE, Anderson GA, Hofstadler SA, Rockwood AL, Smith RD, Chilkoti A, Stayton PS. J Am Soc Mass Spectrom. 1995;6:459–465. doi: 10.1016/1044-0305(95)00191-F. [DOI] [PubMed] [Google Scholar]
- 37.Smith RD, Light-Wahl KJ, Winger BE, Loo JA. Org Mass Spectrom. 1992;27:811–821. [Google Scholar]
- 38.Versluis C, van der Staaij A, Stokvis E, Heck AJR, de Craene B. J Am Soc Mass Spectrom. 2001;12:329–336. doi: 10.1016/S1044-0305(00)00227-0. [DOI] [PubMed] [Google Scholar]
- 39.Zhang ZG, Krutchinsky A, Endicott S, Realini C, Rechsteiner M, Standing KG. Biochemistry. 1999;38:5651–5658. doi: 10.1021/bi990056+. [DOI] [PubMed] [Google Scholar]
- 40.Pearson KM, Pannell LK, Fales HM. Rapid Commun Mass Spectrom. 2002;16:149–159. doi: 10.1002/rcm.554. [DOI] [PubMed] [Google Scholar]
- 41.Knapp JA, Pace CN. Biochemistry. 1974;13:1289–1294. doi: 10.1021/bi00703a036. [DOI] [PubMed] [Google Scholar]
- 42.Konermann L, Douglas DJ. Biochemistry. 1997;36:12296–12302. doi: 10.1021/bi971266u. [DOI] [PubMed] [Google Scholar]
- 43.Gauthier JW, Trautman TR, Jacobson DB. Anal Chim Acta. 1991;246:211–225. [Google Scholar]
- 44.Schnier PD, Jurchen JC, Williams ER. J Phys Chem B. 1999;103:737–745. doi: 10.1021/jp9833193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clemmer DE, Hudgins RR, Jarrold MF. J Am Chem Soc. 1995;117:10141–10142. [Google Scholar]
- 46.Wood TD, Chorush RA, Wampler FM, Little DP, O’Connor PB, McLafferty FW. Proc Natl Acad Sci U S A. 1995;92:2451–2454. doi: 10.1073/pnas.92.7.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mao Y, Woenckhaus J, Kolafa J, Ratner MA, Jarrold MF. J Am Chem Soc. 1999;121:2712–2721. [Google Scholar]
- 48.Schnier PD, Gross DS, Williams ER. J Am Soc Mass Spectrom. 1995;6:1086–1097. doi: 10.1016/1044-0305(95)00532-3. [DOI] [PubMed] [Google Scholar]
- 49.Iavarone AT, Jurchen JC, Williams ER. Anal Chem. 2001;73:1455–1460. doi: 10.1021/ac001251t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suckau D, Shi Y, Beu SC, Senko MW, Quinn JP, Wampler FM, McLafferty FW. Proc Natl Acad Sci U S A. 1993;90:790–793. doi: 10.1073/pnas.90.3.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Campbell S, Rodgers MT, Marzluff EM, Beauchamp JL. J Am Chem Soc. 1995;117:12840–12854. [Google Scholar]
- 52.Wyttenbach T, Bowers MT. J Am Soc Mass Spectrom. 1999;10:9–14. [Google Scholar]
- 53.Wells, J. M. C., P. A.; McLuckey, S. A. Proceedings of the 50th ASMS Conference on Mass Spectrometry and Allied Topics, Orlando, FL, June 2–6, 2002.