

Abstract
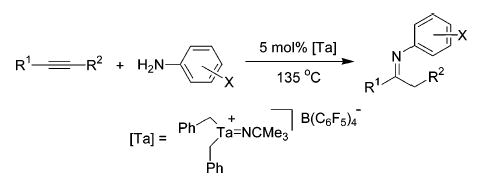
Several tantalum imido complexes have been synthesized and shown to efficiently catalyze the hydroamination of internal and terminal alkynes. An unusual hydroamination/hydroarylation reaction of norbornene catalyzed by a highly electrophilic cationic tantalum imido complex is also reported. Factors affecting catalyst activity and selectivity are discussed along with mechanistic insights gained from stoichiometric reactions.
Catalytic hydroamination is a potentially powerful synthetic method by which valuable nitrogen-containing products (e.g., amines, imines, and enamines) may be obtained in a single step from readily available unsaturated hydrocarbons. Several recent reviews have highlighted the considerable current interest in this transformation.1 Catalysts derived from both early and late transition metals as well as lanthanides have shown significant activity for the addition of N–H bonds across a variety of alkynyl and activated alkenyl moieties;2–7 however, a general and selective protocol for the hydroamination of unactivated alkenes remains unknown.
Hydroamination methods using complexes of the group 4 metals have been described by the Bergman group3 as well as by Doye,1e,4 Odom,5 Beller,6 and others.7 In each of these examples, metal-imido (M==NR) species have been proposed as key intermediates in the catalytic cycle. We noticed that, in contrast to the extensive application of group 4 metals to hydroamination catalysis, no examples existed of catalysis by group 5 analogues.8 Cationic group 5 imido complexes seemed particularly promising as potential hydroamination catalysts, since these compounds are isoelectronic to the group 4 catalysts and the enhanced polarity of the metal imide linkage of such compounds would likely result in increased catalytic activity.
We report herein the synthesis of new neutral and cationic imidotantalum complexes and their application to the catalytic hydroamination of alkynes. These tantalum imido species also catalyze an unusual hydroamination/hydroarylation reaction between norbornene and aniline. The hydroamination of norbornene represents one of the first reports of an intermolecular alkene hydroamination catalyzed by an early transition metal.9 The scope of this method, a comparison between neutral and cationic tantalum catalysts, and mechanistic insights gained from stoichiometric reactions are discussed below.
Neutral trialkyltantalum imido complexes 1a and 1b were synthesized by treatment of the trichlorotantalum precursor (py)2Cl3Ta==NCMe3 (py = pyridine) with 3 equiv of the corresponding Grignard reagent (Scheme 1). Benzyl anion abstraction from 1a with Ph3CB(C6F5)4 produced an insoluble orange solid that was tentatively assigned as an oligomer of the 10e− cationic complex 2. This formulation was supported by reaction of 2 with diphenylacetylene, which afforded the fully characterized azametallacyclobutene complex 3.
Scheme 1.
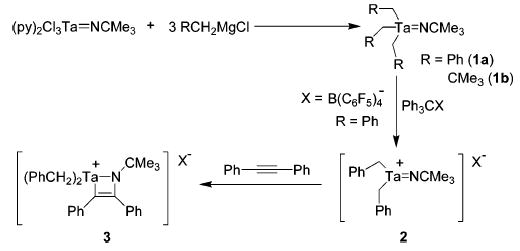
Synthesis of Neutral and Cationic Alkyl Tantalum Imido Complexes
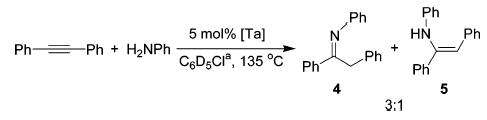
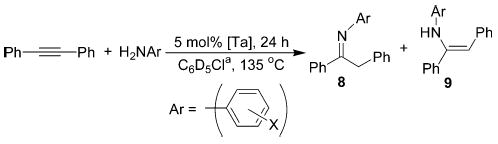
In a preliminary experiment, a mixture of diphenylacetylene and aniline in the presence of 5 mol % of 2 at 135 °C gave the products of hydroamination (imine/enamine = 3:1) in 26% yield (1H NMR). Under analogous conditions, complex 3 gave similar results. Improved reactivity and >95% yield were achieved through in situ generation of 2 (Table 1, entry 2).10 All subsequent experiments with the cationic complex were therefore performed by premixing 1a and Ph3CB(C6F5)4 with the substrate–amine mixture.
Table 1.
Hydroamination of Diphenylacetylene with Aniline Using Tantalum Imido Complexes
| entry | [Ta] | time (h) | % yieldb |
|---|---|---|---|
| 1 | (PhCH2)3Ta==NCMe3, 1a | 30 | >95 |
| 2 | [(PhCH2)Ta==NCMe3]+, 2 | 8 | >95 |
| 3 | Np3Ta==NCMe3, 1b | 12 | >95 |
| 4 | (Et2N)3Ta==NCMe3 | 30 | >95 |
| 5 | Ta(NMe2)5 | 30 | >95 |
| 6 | Cl3Ta==NCMe3 | 30 | NR |
Identical results were obtained when the reaction was run in C6D6 or C7D8.
Yields are given as NMR yields.
Several neutral Ta complexes were also evaluated as catalysts for the hydroamination of diphenylacetylene with aniline. As shown in Table 1, compounds 1a, 1b, (Et2N)3-Ta==NCMe3, and Ta(NMe2)511 were all competent catalysts, affording the desired products as thermodynamic imine/enamine mixtures. While all the catalysts screened provided high yields, the enhanced reactivity associated with neutral tris(neopentyl) complex 1b and cationic 2 made these compounds attractive for further study.
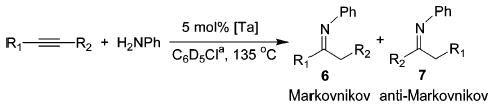
Several alkynes were treated with aniline in the presence of 1b and 2 in order to determine the scope of this method with respect to the alkyne component (Table 2). Both complexes catalyze the hydroamination of all substrates investigated; dialkylacetylenes react more slowly than diphenylacetylene (entries 1 and 2), while terminal alkynes are converted significantly faster (entries 3 and 4). The hydroamination of 2-hexyne (entry 2) proceeded with no regioselectivity. High levels of Markovnikov selectivity are observed with other substrates (entries 3 and 4), but may be caused by selective decomposition of the anti-Markovnikov product.12 Surprisingly, 1-phenylpropyne was a difficult substrate for these catalytic systems (entry 5). However, the hydroamination of 1-phenylpropyne with 2, while 1b fails to react, suggests that 2 is the more potent catalyst. Although 2 exhibits greater activity toward 1-phenylpropyne, neutral catalyst 1b appears to provide reaction products for this survey of alkynes in consistently higher yields.
Table 2.
Hydroamination of Various Alkynes with Aniline
| % yieldb |
|||||
|---|---|---|---|---|---|
| entry | R1, R2 | 6/7 | time (h) | 1b | 2 |
| 1 | Et, Et | NA | 24 | >95 (83c) | >95 |
| 2 | n-Pr, Me | 1:1 | 24 | >95 | 71 |
| 3 | Ph, H | only 6 | 2 | 77 (65c) | 66 |
| 4 | n-Pr, H | only 6 | 2 | 70 | 62 |
| 5 | Ph, Me | only 7 | 24 | NR | 19 |
Identical results were obtained when the reaction was run in C6D6 or C7H8.
Yields are given as NMR yields. Hydrolysis to the corresponding ketone was used to confirm these assignments.
Isolated yield of imine reduction product.
Several substituted anilines were examined in the reaction with diphenylacetylene to determine the tolerance of the two catalysts for the amine component (Table 3). Both 1b and 2 were efficient catalysts for the reaction with para-substituted anilines. Ortho-substituted anilines proved to be more challenging substrates, showing little or no reaction with 1b and giving moderate yields with 2. The rate of catalyst decomposition in these cases appears to be competitive with the rate of hydroamination.13 The reaction in entry 5 can be driven to completion if additional 2 is introduced after 24 h. Very low yields of hydroaminated products along with products of catalyst decomposition were obtained in reactions with benzylamine and n-butylamine. Sterically demanding tert-butylamine was unreactive in the presence of both catalysts.
Table 3.
Addition of Substituted Anilines to Diphenylacetylene
| % yieldb |
||||
|---|---|---|---|---|
| entry | X | 8/9 | 1b | 2 |
| 1 | H | 3:1 | 98 | 96 (75c) |
| 2 | 4-Me | 4:1 | 79 | 74 |
| 3 | 4-OMe | 7:1 | 31 | 72 (83c) |
| 4 | 4-Cl | 4:1 | >95 | >95 (66c) |
| 5 | 2,6-Me2 | only 8 | 7 | 69 |
Identical results were obtained when the reaction was run in C6D6 or C7D8.
Yields are given as NMR yields. Hydrolysis to the corresponding ketone was used to confirm these assignments.
Isolated yield of the ketone hydrolysis product.
In an attempt to extend the scope of Ta-catalyzed hydroaminations beyond alkynes, both 1b and 2 were examined as catalysts for the addition of anilines to allenes and olefins. Catalyst 1b failed to exhibit any activity toward allenes; however, 2 catalyzed the hydroamination of both propadiene and cyclonona-1,2-diene (Scheme 2).
Scheme 2.
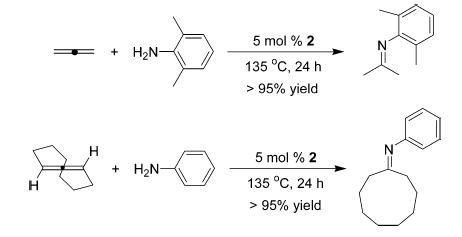
Hydroamination of Allene and Cyclononadiene
Treatment of a mixture of norbornene and aniline with a catalytic amount of 2 afforded products both of olefin hydroamination (10) and hydroarylation (11) in a ratio of 1:2 (Scheme 3).9,14,15 This is one of the first examples of intermolecular alkene hydroamination with an early transition metal catalyst. Unoptimized, this reaction only provides 32% yield of the desired amines. A significant amount of polymer and other higher molecular weight byproducts were observed in this reaction and are thought to be responsible for the overall low yields of the desired products. An interesting aspect of this transformation is the possibility of selectively activating N–H bonds versus C–H bonds. Variation of the ratio of aniline to norbornene failed to alter the 1:2 ratio of products; however, preliminary studies with substituted anilines have shown promising results.16
Scheme 3.
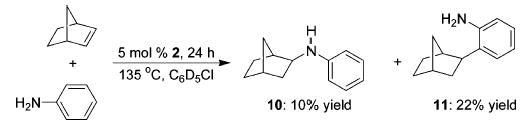
Addition of Aniline to Norbornene
Two key steps in the catalytic cycles proposed for hydroamination with group 4 complexes are the formation of an intermediate azametallacycle and its subsequent protonation by amine.3a,c,d,4b To assess whether similar steps could be involved in cationic Ta-catalyzed hydroaminations, a study was carried out using stoichiometric amounts of isolable azametallacyclobutene complex 3 and 2,6-dimethylaniline (Scheme 4). Upon mixing at room temperature, the amine immediately coordinates to the electrophilic tantalum center of 3, as indicated by the appearance of two distinctive diastereotopic N–H resonances (δ = −1.05, −1.23) in the 1H NMR spectrum of the reaction mixture (12).17 Interestingly, this complex is stable at room temperature for approximately 24 h before the Ta–C bond of the metallacycle is protonated and the diastereotopic N–H resonances disappear. Furthermore, metallacycle 3 catalyzes the addition of aniline to diphenylacetylene with efficiency identical to that of 2. Overall, these results suggest that cationic tantalum-catalyzed hydroaminations proceed through a catalytic cycle similar to that proposed for group 4 analogues.
Scheme 4.
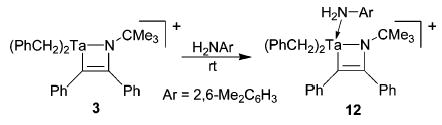
Treatment of 3 with 2,6-Dimethylaniline
In summary, several neutral and cationic tantalum imido complexes have been identified as effective catalysts for the hydroamination of alkynes, allenes, and norbornene. Cationic tantalum complex 2 has shown enhanced reactivity toward more challenging substrates such as ortho-substituted anilines and allenes, in agreement with our original hypothesis. Interestingly, the cationic complex is also one of the first two early metal complexes shown to catalyze the intermolecular hydroamination of norbornene. Stoichiometric reactions have indicated that the cationic tantalum catalyzed processes are occurring through a mechanism similar to that known for group 4 catalysts. Work is currently in progress to increase the lifetimes, activities, and substrate scope of these and related catalysts.
Acknowledgments
We would like to thank Dr. Lutz Ackermann for sharing information about his titanium hydroamination system and for publishing concurrently. This work was supported by the National Institutes of Health (Grant No. GM-25459 to R.G.B.) and the National Science Foundation (Grant No. CHE-0072819 to J.A.).
Footnotes
Supporting Information Available: Experimental procedures, relevant NMR spectra, and characterization data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a Muller TE, Beller M. Chem Rev. 1998;98:675–703. doi: 10.1021/cr960433d. [DOI] [PubMed] [Google Scholar]; b Beller M, Breindl C, Eichberger M, Hartung CG, Seayad J, Thiel OR, Tillack A, Trauthwein H. Synlett. 2002;10:1579–1594. [Google Scholar]; c Pohlki F, Doye S. Chem Soc Rev. 2003;32:104–114. doi: 10.1039/b200386b. [DOI] [PubMed] [Google Scholar]; d Bytschkov I, Doye S. Eur J Org Chem. 2003:935–946. [Google Scholar]; e Yamamoto Y, Radhakrishnan U. Chem Soc Rev. 1999;28:199–207. [Google Scholar]; f Seayad J, Tillack A, Hartung CG, Beller M. Adv Synth Catal. 2002;344:795–813. [Google Scholar]; (e) For an extensive list of references also see: Heutling A, Doye S.J Org Chem 2002671961–1964. [DOI] [PubMed] [Google Scholar]
- 2.a Ryu JS, Marks TJ, McDonald FE. J Org Chem. 2004;69:1038–1052. doi: 10.1021/jo035417c. [DOI] [PubMed] [Google Scholar]; b Hong S, Kawaoka AM, Marks TJ. J Am Chem Soc. 2003;125:15878–15892. doi: 10.1021/ja036266y. [DOI] [PubMed] [Google Scholar]; c Kim YK, Livinghouse T. Angew Chem, Int Ed. 2002;41:3645–3647. doi: 10.1002/1521-3773(20021004)41:19<3645::AID-ANIE3645>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]; d Utsunomiya M, Hartwig JF. J Am Chem Soc. 2004;126:2702–2703. doi: 10.1021/ja031542u. [DOI] [PubMed] [Google Scholar]; e Yamashita M, Vicario JVC, Hartwig JF. J Am Chem Soc. 2003;125:16347–16360. doi: 10.1021/ja037425g. [DOI] [PubMed] [Google Scholar]; f Kanzelberger M, Zhang X, Emge TJ, Goldman AS, Zhao J, Incarvito C, Hartwig JF. J Am Chem Soc. 2003;125:13644–13645. doi: 10.1021/ja037363u. [DOI] [PubMed] [Google Scholar]; g Fadini L, Togni A. A Chem Commun. 2003:30–31. doi: 10.1039/b210680a. [DOI] [PubMed] [Google Scholar]; h Dorta R, Egli P, Zurcher F, Togni A. J Am Chem Soc. 1997;119:10857–10858. [Google Scholar]; i Brunet JJ, Cadena M, Chu NC, Diallo O, Jacob K, Mothes E. Organometallics. 2004;23:1264–1268. [Google Scholar]; j Lauterwasser F, Hayes PG, Brase S, Piers WE, Schafer LL. Organometallics. 2004;23:2234–2237. [Google Scholar]; k Hultzsch KC, Hampel F, Wagner T. Organometallics. 2004;23:2601–2612. [Google Scholar]
- 3.a Baranger AM, Walsh PJ, Bergman RG. J Am Chem Soc. 1993;115:2753–2763. [Google Scholar]; b Walsh PJ, Baranger AM, Bergman RG. J Am Chem Soc. 1992;114:1708–1719. [Google Scholar]; c Johnson JS, Bergman RG. J Am Chem Soc. 2001;123:2923–2924. doi: 10.1021/ja005685h. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Straub BF, Bergman RG. Angew Chem, Int Ed. 2001;40:4632–4635. doi: 10.1002/1521-3773(20011217)40:24<4632::aid-anie4632>3.0.co;2-v. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Ackermann L, Bergman RG. Org Lett. 2002;4:1475–1478. doi: 10.1021/ol0256724. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Ackermann L, Bergman RG, Loy RN. J Am Chem Soc. 2003;125:11956–11963. doi: 10.1021/ja0361547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a Haak E, Siebeneicher H, Doye S. Org Lett. 2000;2:1935–1937. doi: 10.1021/ol006011e. [DOI] [PubMed] [Google Scholar]; b Haak E, Bytschkov I, Doye S. Angew Chem, Int Ed. 1999 38;8:3389–3391. [PubMed] [Google Scholar]; c Pohlki F, Doye S. Angew Chem, Int Ed. 2001;40:2305–2308. [PubMed] [Google Scholar]
- 5.a Shi Y, Ciszewski JT, Odom AL. Organometallics. 2001;20:3967–3969. [Google Scholar]; b Cao C, Ciszewski JT, Odom AL. Organometallics. 2001;20:5011–5013. [Google Scholar]; c Li Y, Shi Y, Odom AL. J Am Chem Soc. 2004;126:1794–1803. doi: 10.1021/ja038320g. [DOI] [PubMed] [Google Scholar]
- 6.a Tillack A, Castro IG, Hartung CG, Beller M. Angew Chem, Int Ed. 2002;41:2541–2543. doi: 10.1002/1521-3773(20020715)41:14<2541::AID-ANIE2541>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]; b Khedkar V, Tillack A, Beller M. Org Lett. 2003;5:4767–4770. doi: 10.1021/ol035653+. [DOI] [PubMed] [Google Scholar]
- 7.a Ong TG, Yap GPA, Richeson DS. Organometallics. 2002;21:2839–2841. [Google Scholar]; b Li C, Thomson RK, Gillon B, Patrick BO, Schafer LL. Chem Commun. 2003:2462–2463. doi: 10.1039/b304176j. [DOI] [PubMed] [Google Scholar]; c Zhang Z, Schafer LL. Org Lett. 2003;5:4733–4736. doi: 10.1021/ol0359214. [DOI] [PubMed] [Google Scholar]; d Knight PD, Munslow I, O’Shaughnessy PN, Scott P. Chem Commun. 2004:894–895. doi: 10.1039/b401493f. [DOI] [PubMed] [Google Scholar]
- 8.During the course of this work, a V(IV) hydroaminationcatalyst was reported: Lorber C, Choukroun R, Vendier L.Organometallics 2004231845–1850. [Google Scholar]
- 9.See the accompanying paper in this issue for independent work on a Ti-catalyzed system for the hydroamination of norbornene: Ackermann L, Kaspar LT, Gschrei CJ.Org Lett 200462515–2518. [DOI] [PubMed] [Google Scholar]
- 10.The cause of higher activity in the in situ generated complex is presently unknown; unfortunately, neither the cationic complex nor any subsequent intermediates can be observed using NMR spectroscopy due to constraints of instantaneous reactivity and insolubility, respectively.
- 11.This compound has already been shown to catalyze the hydroamination of 1-hexyne. See ref 8.
- 12.When this reaction is performed at 75 °C, production of the anti-Markovnikov enamine can be observed by 1H NMR but decomposes before the reaction goes to completion. Low yields of product caused by oligomerization of phenylacetylene have also been observed by Odom and co-workers (see ref 5a). Low yields and Markovnikov selectivity were also observed by Vendier and co-workers (see ref 8).
- 13.Preliminary results indicate that the catalyst decomposes to a tantalum imido cluster within 24 h.
- 14.Compound 1b showed no reactivity when treated with a mixture of norbornene and aniline.
- 15.This reaction has previously been observed with a Rh catalyst. Brunet JJ, Commenges G, Neibecker D, Philippot K.J Organomet Chem 1994469221–228. [Google Scholar]
- 16.Preliminary GC–MS results indicate that electron-withdrawing substituents on aniline favor formation of 10 while electron-donating substituents favor formation of 11.
- 17.An analogous experiment with 2,6-dimethylaniline-N-d2 confirmed the assignment of the protons in question.