

Abstract
In vertebrate meiosis, unfertilized eggs are arrested in metaphase II by cytostatic factor (CSF), which is required to maintain mitotic cyclin-dependent kinase activity. Fertilization triggers a transient increase in cytosolic free Ca2+, which leads to CSF inactivation and ubiquitin-dependent cyclin destruction through the anaphase promoting complex or cyclosome (APC/C). The Ca2+/calmodulin-dependent protein kinase II (CaMKII) and the Polo-like kinase Plx1 are essential factors for Ca2+-induced meiotic exit, but the critical targets of these kinases were unknown. The APC/C inhibitor Emi2 or XErp1 has recently been characterized as a pivotal CSF component, required to maintain metaphase II arrest and rapidly destroyed in response to Ca2+ signaling through phosphorylation by Plx1 and ubiquitination by the SCFβTrCP complex. An important question is how the increase in free Ca2+ targets Plx1 activity toward Emi2. Here, we demonstrate that CaMKII is required for Ca2+-induced Emi2 destruction, and that CaMKII functions as a “priming kinase,” directly phosphorylating Emi2 at a specific motif to induce a strong interaction with the Polo Box domain of Plx1. We show that the strict requirement for CaMKII to phosphorylate Emi2 is a specific feature of CSF arrest, and we also use phosphatase inhibitors to demonstrate an additional mode of Emi2 inactivation independent of its destruction. We firmly establish the CSF component Emi2 as the first-known critical and direct target of CaMKII in CSF release, providing a detailed molecular mechanism explaining how CaMKII and Plx1 coordinately direct APC/C activation and meiotic exit upon fertilization.
Keywords: fertilization, meiosis, ubiquitin ligases
During meiosis in vertebrate systems, mature oocytes arrest at metaphase II to await fertilization and prevent parthenogenesis. Sperm penetration initiates a calcium wave that spreads throughout the egg and triggers the metaphase II-anaphase II transition, segregation of chromosomes, extrusion of the second polar body, and the commencement of alternating cycles of DNA replication and cell division (reviewed in ref. 1). In 1971, Masui and Markert (2) published a historic paper describing a cytoplasmic activity responsible for mitotic arrest that was able to inhibit cleavage when injected into the dividing embryo. This activity has come to be known as “cytostatic factor,” or CSF. A later seminal paper by Sagata et al. (3) in 1989 identified Mos, an activator of the mitogen-activated protein kinase (MAPK) pathway, as a critical factor for CSF establishment. How this enzyme cascade arrests the egg during meiosis II is largely unknown.
Over the years, it has become clear that the downstream effect of the Mos/MAPK pathway is inhibition of the E3 ubiquitin ligase responsible for the destruction of mitotic cyclin, the anaphase-promoting complex or cyclosome (APC/C) (reviewed in ref. 4). Some have proposed that the MAPK effector p90Rsk is sufficient to establish CSF arrest (5–7), perhaps through activating known APC/C inhibitors such as the spindle checkpoint protein Bub1 (8, 9) or the interphase APC/C inhibitor Emi1 (10, 11). By using immunological techniques, spindle checkpoint components and Emi1 have each been shown to be required to maintain CSF arrest (12, 13), but recent studies have questioned the validity of each of these intermediaries between Mos/MAPK and the APC/C (14–16).
Similarly, the biochemical pathway that facilitates meiotic exit upon fertilization has known upstream initiators whose ultimate effect is APC/C activation and destruction of cyclin, but the precise mechanism by which CSF release occurs has remained enigmatic (reviewed in ref. 4). It has been known for some time that the critical effector of the cytosolic free Ca2+ increase that occurs in Xenopus oocytes upon fertilization is the Ca2+/calmodulin-dependent protein kinase II (CaMKII) (17), and the ability of CaMKII to trigger CSF release has recently been validated in mammalian oocytes as well (18). In Xenopus oocytes, the Polo-like kinase Plx1 is also required for Ca2+-induced meiotic exit (19–22), but exactly how CaMKII and Plx1 conspire to activate the APC/C has been unknown.
The APC/C inhibitor Emi2 or XErp1, an immunologically cross-reactive homolog of Emi1, has newly emerged as the most likely candidate to inhibit the APC/C during CSF arrest (23, 24). Emi2 is the first known CSF component that is both required for maintaining CSF arrest once established and is rapidly destroyed in response to Ca2+ signaling. We have previously shown that, after Ca2+ addition to extracts from CSF-arrested eggs, Emi2 is destroyed in a process similar to the destruction pathway for Emi1 (25, 26): Plx1 phosphorylates the DSGYSDS motif of Emi2, which is then recognized for ubiquitination by the SCFβTrCP complex (24). This Ca2+-induced destruction of Emi2 is essential for APC/C activation and meiotic exit, making Emi2 a pivotal component of CSF activity. In this article, we further detail the mechanism of Ca2+-induced Emi2 destruction and report that CaMKII specifically directs Plx1 activity toward Emi2 by functioning as a “priming kinase,” creating a phosphoepitope on Emi2 that recruits the Polo Box domain (PBD) of Plx1, allowing subsequent phosphorylation of Emi2's degron and its recognition by SCFβTrCP for ubiquitination and destruction. We also uncover another mode of mitotic Emi2 destruction that is CaMKII-independent, as well as an additional mode of Emi2 inactivation that is independent of its proteolytic destruction.
Results
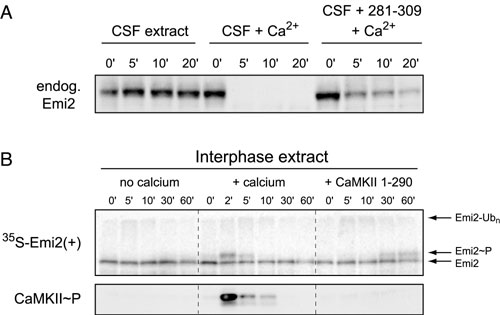
Activation of CaMKII in CSF Extract Is both Necessary and Sufficient to Trigger Emi2 Destruction. Because CaMKII activation is necessary for Ca2+-induced cyclin degradation in CSF extracts and addition of a constitutively active CaMKII mutant is sufficient to drive cyclin degradation in the absence of Ca2+ (17), we first sought to demonstrate that CaMKII activity regulates Emi2 stability to explain how CaMKII causes activation of the APC/C. By using a peptide from the autoinhibitory domain of CaMKII (residues 281–309) to inhibit CaMKII (17, 27, 28), we tested whether CaMKII activity is a necessary component of Ca2+-induced Emi2 destruction. As expected, addition of the 281–309 peptide blocked the appearance of active, phosphorylated CaMKII and blocked Ca2+-induced destruction of Emi2 and cyclin B (Fig. 1A and Fig. 5A, which is published as supporting information on the PNAS web site). Addition of in vitro-translated, constitutively active CaMKII (residues 1–290 of rat CaMKIIα) to CSF extract bypassed the requirement for Ca2+ addition, resulting in the spontaneous destruction of Emi2 and cyclin B (Fig. 1B). Of note, addition of Ca2+ or active CaMKII to interphase egg extract did not cause destruction of Emi2 because Plx1, which phosphorylates the Emi2 degron recognized by βTrCP, is not active during interphase. However, activation of CaMKII in interphase extracts did cause a corresponding reduction in Emi2 gel mobility (Fig. 5B), supporting the possibility that CaMKII directly phosphorylates Emi2.
Fig. 1.

Destruction of Emi2 in CSF extract requires CaMKII. (A) Ca2+-induced destruction of Emi2 during CSF release requires CaMKII activation. Radiolabeled IVT Emi2 was incubated in CSF extract with or without Ca2+ addition and with or without 281–309 peptide. Activation of CaMKII and destruction of cyclin B and Emi2 were monitored by immunoblot or autoradiography. (B) Addition of active CaMKII to CSF extract is sufficient to drive destruction of Emi2 and cyclin B2 without Ca2+ addition. A constitutively active fragment of CaMKII or mock IVT was added to CSF extract. Destruction of Emi2 and cyclin B2 was monitored by immunoblot.
CaMKII Directly Phosphorylates Emi2 to Induce Its Association with Plx1. Because Polo-like kinases are targeted to substrates through the phosphoprotein-binding Polo Box domain (29), we hypothesized that phosphorylation of Emi2 by CaMKII might stimulate interaction between Emi2 and Plx1. We tested whether binding of Emi2 and Plx1 in CSF extract requires Ca2+/CaMKII activity. MBP-Emi2N was incubated in CSF extract for 5 min with or without Ca2+ addition, after which the protein was purified on amylose resin and analyzed by immunoblot for bound Plx1. Emi2 only associated with Plx1 after Ca2+ addition, and use of the 281–309 peptide to block CaMKII activation prevented this interaction (Fig. 2A). The MBP-Emi2N in this experiment was not destroyed in the 5-min window because the amount added, 600 nM, saturates the capacity of the Emi2 destruction pathway (see Fig. 6, which is published as supporting information on the PNAS web site). Finally, using purified components, we reproduced this reaction in vitro by showing that phosphorylation of Emi2 by CaMKII induced a strong interaction of MBP-Emi2N with a GST fusion of the Plk1 PBD (Fig. 2B) but not with GST alone (data not shown). Cdc2 also efficiently phosphorylated MBP-Emi2N in vitro, as judged by the clear shift in Emi2 mobility, but this phosphorylation did not stimulate PBD interaction. Taken together, these results indicate that CaMKII specifically targets Plx1 activity toward Emi2 by acting as a priming kinase, directly phosphorylating Emi2 and stimulating its interaction with the Polo Box domain of Plx1.
Fig. 2.

CaMKII directly phosphorylates Emi2 as a priming kinase to induce association of Plx1 with Emi2. (A) Ca2+ addition to CSF extract induces Plx1/Emi2 association. An MBP fusion of the Emi2 N terminus (or MBP alone) was incubated in CSF extract for five min with or without Ca2+ addition and with or without 281–309 peptide. Recombinant protein was purified on amylose resin, and associated Plx1 was detected by immunoblotting. (B) Emi2 binds the PBD of Plk1 after phosphorylation by CaMKII. Indicated MBP proteins were phosphorylated in vitro with CaMKII, Cdc2, or mock treatment, incubated with a GST fusion of the Plk1 PBD, and purified on amylose resin. Captured proteins were detected by Coomassie stain.
A Specific Motif in Emi2 Is Phosphorylated by CaMKII, Binds Plx1, and Is Required for Emi2 Destruction During CSF Release. Given the direct interaction between Emi2 and Plx1 induced by CaMKII, we scanned the primary sequence of Emi2 for motifs that match the consensus sequences for both phosphorylation by CaMKII (R/K-X-X-S/T) and strong interaction with the Polo Box domain (S-pS/pT) (29), namely R/K-X-S-S/T. We found two such motifs in the Emi2 N terminus well conserved among vertebrate Emi2 orthologs: 192RSST195 and 333RLST336 (Fig. 3A). We mutated these motifs and tested whether these mutant proteins, which we call 192-5* and 333-6*, functioned as CaMKII substrates, Plx1 binding sites, and determinants of CSF activity. We first established that each of these motifs can be phosphorylated by CaMKII in vitro, because each mutation reduced the amount of 32P incorporation from CaMKII phosphorylation (Fig. 7, which is published as supporting information on the PNAS web site). We then tested whether mutation of these motifs caused failure to bind the Plk1 PBD after phosphorylation by CaMKII using purified components. In this in vitro assay, only simultaneous mutation of both motifs abrogated PBD binding, whereas mutation of either motif alone still allowed strong PBD interaction, indicating that each motif can independently bind the PBD after CaMKII phosphorylation (Fig. 3B).
Fig. 3.

Emi2's 192RSST motif is critical for CaMKII-induced Plx1 association and is required for Emi2 destruction and CSF release. (A) A schematic of the primary structure of Xenopus Emi2 with candidate CaMKII target/Polo Box-binding sites and equivalent sequences among vertebrate Emi2 orthologs depicted. (B) Both the 192RSST and 333RLST motifs of Emi2 can bind the PBD in vitro after phosphorylation by CaMKII. Indicated variants of the Emi2 N terminus were phosphorylated by CaMKII and processed as in Fig. 2B to assay Polo Box-binding. (C) The 192RSST motif of Emi2 is chiefly responsible for Ca2+-induced Plx1 association in CSF extract. Indicated variants of the Emi2 N terminus were incubated in CSF extract for 5 min with or without Ca2+ addition. Recombinant protein was purified on amylose resin, electrophoresed, and Coomassie stained. Associated Plx1 was detected by immunoblotting. (D) CaMKII-induced Emi2 destruction requires the 192RSST motif of Emi2. Radiolabeled IVT Emi2 was preincubated in CSF extract for 5 min before addition of IVT-active CaMKII fragment or mock IVT. Emi2 stability was monitored by autoradiography. (E) The 192RSST motif in Emi2 is required for Ca2+-induced Emi2 destruction in CSF extract. Radiolabeled IVT Emi2 variants were incubated in CSF extract with Ca2+ addition, and Emi2 stability and gel mobility were monitored by autoradiography. (F) Expression of the Emi2 192–5* mutant in intact oocytes prohibits Ca2+-induced CSF release and meiotic exit. Oocytes were injected with in vitro-transcribed myc-Emi2 mRNA, matured with progesterone, and treated with ionophore A23187 at 3 h post-germinal vesicle breakdown to induce Ca2+ release and meiotic exit. Anti-Emi2 or anti-myc antibodies were used to detect endogenous or expressed Emi2, respectively, and cyclin B levels and H1 kinase activity were monitored at 5, 15, and 30 min after ionophore treatment.
In contrast, Ca2+-induced Emi2/Plx1 association in CSF extract relied mostly on the 192RSST motif, whereas the 333RLST motif contributed only slightly to Plx1 binding (Fig. 3C). Accordingly, we found that the Emi2 192-5* mutant was completely stable after addition of either Ca2+ or active CaMKII in CSF extract, whereas Ca2+-induced destruction of the 333-6* mutant proceeded with normal kinetics (Fig. 3 D and E). (The trace amount of stable IVT Emi2 used here is insufficient to prevent cyclin B destruction.) Similar mutations in the human Emi2 protein (product of the FBXO43 gene) behaved essentially identically in that residues 231–234 (equivalent to 192–195 in xEmi2) are required for Ca2+-induced destruction, whereas residues 383–386 (equivalent to 333–336 in xEmi2) are dispensable (Fig. 8, which is published as supporting information on the PNAS web site). Interestingly, the 192–5* mutant did not undergo the dramatic reduction in gel mobility after Ca2+ addition seen with the stable DS32AA degron mutant (Fig. 3E). This mobility supershift is Plx1-dependent (22), so the failure of the 192–5* mutant to experience this supershift is consistent with its failure to bind and be phosphorylated by Plx1. The correlation between Plx1-dependent phosphorylation and supershift in Emi2 gel mobility is also observed in Fig. 3C.
Finally, to test the requirement for Emi2's 192RSST motif in vivo, we injected mRNAs encoding Emi2 variants into oocytes to observe their effects on meiotic exit. Although wild-type Emi2 did not prevent ionophore-induced meiotic exit, expression of the nondestructible 192RSST mutant blocked CSF release (Fig. 3F). These results clearly establish the 192RSST motif of Emi2 as the first-known critical molecular target of CaMKII during fertilization and provide a detailed mechanistic understanding of how CaMKII and Plx1 conspire to activate the APC/C to direct cyclin degradation and facilitate meiotic exit.
Emi2 Destruction in Δ90 Extracts Is CaMKII-Independent. An interesting question is whether the requirement for Ca2+/CaMKII to direct Plx1 activity toward Emi2 is a general feature of Emi2 during each mitosis or whether it is a specialized requirement during meiosis II. To begin to address this question, we prepared extracts from interphase eggs, induced a permanent mitotic state using Δ90 (nondestructible) cyclin B, and assayed the stability of Emi2 in these extracts. We found that Emi2 is actually destroyed in these mitotic extracts, although at a slow rate with a half-life of 30–60 min (Fig. 4A). The DS32AA degron mutant was not ubiquitinated, implying that the destruction occurred through the known Plx1- and βTrCP-dependent pathway. However, Emi2 destruction in Δ90 extract appeared to be independent of CaMKII activity based on three observations. First, autophosphorylated, active CaMKII was not detected by immunoblot. Second, addition of the 281–309 inhibitory peptide did not prevent Emi2 ubiquitination (whether added before or after Δ90 addition). Third, the Emi2 192–5* mutant was also ubiquitinated, indicating that phosphorylation and Plx1-binding at this site is unnecessary for Emi2 destruction in Δ90 extract. For the nondestructible DS32AA mutant, we also observed the Plx1-dependent supershift in Emi2 gel mobility, although only to a limited extent. Thus, we can infer that Emi2 is able to associate with Plx1 in these mitotically arrested extracts, probably through being “primed” at suboptimal Polo-binding sites by Cdc2 or some other mitotic kinase because the Plx1 interaction seems weak or attenuated. Interestingly, using Ca2+ to stimulate CaMKII activity in Δ90 extract reduced the half-life of wild-type Emi2 from ≈30 min to ≈10 min, indicative of more efficient targeting of Plx1 activity toward Emi2. Similarly, Ca2+ addition to Δ90 extract caused a complete shift of the nondestructible DS32AA mutant to the hyperphosphorylated form, another Plx1-dependent event. The fact that Plx1 and Emi2 moderately interact in Δ90 extract without CaMKII activity suggests that the stability of Emi2 in CSF extract is a unique property of CSF arrest, where other activities are required to maintain Emi2 in a fully stable configuration.
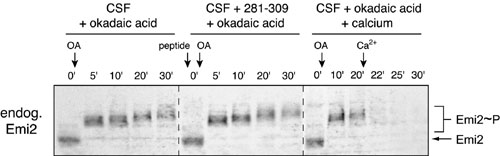
Fig. 4.

Multiple activities contribute to Emi2 stability and function in CSF extract. (A) Emi2 is destroyed in Δ90 mitotic extract in a CaMKII-independent manner, and Ca2+ addition stimulates its rate of destruction. Radiolabeled IVT Emi2 variants were incubated in Δ90 extract with or without Ca2+ addition and with or without 281–309 peptide. Emi2 stability and gel mobility were monitored by autoradiography, and CaMKII activation was detected by immunoblotting. The phospho-CaMKII blot is representative of all ±Ca2+ pairs except those involving the 281–309 peptide, for which no signal for phospho-CaMKII was detected. (B) Phosphatase inhibition in CSF extract causes cyclin B destruction independent of CaMKII activation and Emi2 destruction. CSF extracts were stimulated with either Ca2+ or okadaic acid with or without 281–309 peptide. The stability and gel mobility of radiolabeled IVT Emi2 were monitored by autoradiography. Destruction of cyclin B and activation of CaMKII were monitored by immunoblotting. (C) Emi2 destruction can be induced in okadaic acid-treated CSF extracts, which requires CaMKII and the 192RSST and 32DSGYSDS motifs of Emi2. CSF extracts previously stimulated to destroy cyclin by phosphatase inhibition with or without 281–309 peptide were further stimulated by Ca2+ addition. Stability and gel mobility of radiolabeled IVT Emi2 variants were monitored by autoradiography.
A Phosphatase Activity Maintains Emi2 Activity During CSF Arrest. One way to prevent unwanted association of Emi2 with Plx1 during CSF arrest would be for a phosphatase to remove unwanted priming phosphorylation of Emi2 by mitotic kinases. The phosphatase inhibitors okadaic acid and microcystin cause spontaneous cyclin destruction in CSF extract without Ca2+ addition (21, 30). This destruction is APC/C-dependent because cyclin B lacking the RXXL sequence recognized by the APC/C remains stable (30) and because addition of the APC/C inhibitor Emi1 blocks the destruction (13). Thus, we hypothesized that phosphatase inhibition might allow CaMKII-independent priming of Emi2, phosphorylation by Plx1, and SCFβTrCP-dependent destruction. To our surprise, we found that addition of okadaic acid to CSF extract caused hyperphosphorylation but not destruction of Emi2, whereas cyclin B was destroyed as expected (Fig. 4B and Fig. 9, which is published as supporting information on the PNAS web site). Similar results were obtained by using 1 μM microcystin as phosphatase inhibitor (data not shown). This result implies that a second mode of Emi2 inactivation, independent of destruction, can occur under some circumstances. This inactivation did not require CaMKII activity because okadaic acid did not cause appreciable activation of CaMKII and because the 281–309 peptide did not block the effects of okadaic acid. Interestingly, addition of Ca2+ to okadaic acid-treated extracts still induced ubiquitination of Emi2, and this ubiquitination still required CaMKII and the 192RSST and 32DSGYSDS motifs of Emi2 (Figs. 4C and 9). These results suggest that phosphatase activity helps maintain Emi2 in an active configuration during CSF arrest, whereas an additional activity keeps Emi2 in a stable configuration.
Discussion
How the increase in cytosolic-free Ca2+ induced by fertilization triggers resumption of the meiotic process and mitotic cyclin destruction has been a long-standing puzzle for biologists. Lorca et al. (17) demonstrated that the central effector of this Ca2+ increase is CaMKII, but how CaMKII activates the APC/C was unknown. In this article, we clearly delineate the fundamental mechanism by which the Ca2+ spike after fertilization provokes destruction of the APC/C inhibitor Emi2, thus allowing APC/C-dependent meiotic exit. We show here that, during CSF arrest, activation of CaMKII is both necessary for Ca2+-induced Emi2 destruction and sufficient to induce spontaneous Emi2 destruction. We have demonstrated that Emi2 interacts with Plx1 in a Ca2+/CaMKII-dependent manner, and we have identified a specific motif in Emi2 that recruits Plx1 after phosphorylation by CaMKII and is required for Emi2 destruction and CSF release. Our results are in close agreement with newly published findings of Rauh et al. (31).
Although these results implicate the 192RSST motif of Emi2 as a critical molecular target of CaMKII during CSF release, we do not exclude that CaMKII may have other targets in meiotic exit, either through Emi2-dependent or Emi2-independent pathways. Similarly, it is possible that Plx1 has other important targets during CSF release besides Emi2. Nevertheless, we believe that the target of primary importance for both kinases is Emi2.
The ability of okadaic acid to induce APC/C activation independent of Emi2 destruction suggests that there is a distinct mechanism to inactivate Emi2. Although Emi2 experiences a Plx1-dependent supershift in gel mobility, it is unlikely that this phosphorylation accounts for Emi2 inactivation for two reasons. First, the nondestructible Emi2 DS32AA mutant experiences this hyperphosphorylation in response to Ca2+ but retains APC/C inhibitory activity. Second, phosphatase inhibition bypasses the requirement for Plx1 in cyclin destruction (20). Phosphatase inhibition may lower Emi2's intrinsic APC/C-inhibitory activity, or the APC/C may simply become refractory to inhibition by Emi2.
Taken at face value, the requirement for CaMKII to induce Emi2/Plx1 association seems sufficient to explain the stability of Emi2 in CSF extract. However, the fact that Emi2 is destroyed in Δ90 extract independent of CaMKII indicates that Plx1 may interact with Emi2 without prior CaMKII phosphorylation, perhaps through priming phosphorylation by other kinases, such as Cdc2, at suboptimal Polo Box-binding sites on Emi2. The extreme stability of Emi2 in CSF extract in the presence of active Plx1 leads us to speculate that other factors, besides the absence of CaMKII activity, actively cooperate to stabilize Emi2 during CSF arrest. It appears that Emi2's degron in CSF extract is somehow protected from phosphorylation, even under conditions of phosphatase inhibition that allow Plx1-dependent hyperphosphorylation of Emi2 at other sites. This finding is in stark contrast to the situation in Δ90 cyclin B-arrested mitotic extracts, in which Plx1 phosphorylates Emi2's degron irrespective of CaMKII and the 192RSST motif. We speculate that, during CSF arrest, the Emi2 degron is buried due to Emi2's conformation or interaction with another protein. In this situation, phosphorylation of Emi2's degron by Plx1 is strictly dependent on prior phosphorylation of its 192RSST motif by CaMKII, which may alter Emi2's conformation or protein–protein interactions in addition to generating a high-affinity Plx1-binding site. We suspect that Emi2 stability and function during CSF arrest result from a dynamic balance of several biochemical activities.
Materials and Methods
Reagents. Constructs for wild-type and degron mutant Xenopus Emi2 in the pCS2+myc vector have been described (24), as well as the construct for active CaMKII fragment (13). Emi2 motifs were mutated by using QuikChange site-directed mutagenesis protocol. Mutations introduced were 192RSST → ASAA and 333RLST → RLAA. The Emi2 N terminus (residues 1–350) was PCR subcloned into pMAL-c2X. Emi2 mRNA was in vitro-transcribed by using the mMessage Machine kit (Ambion, Austin, TX). Radiolabeled proteins were in vitro-translated (IVT) by using the TNT system (Promega). The CaMKII 281–309 peptide was purchased from Calbiochem (208711). MBP-Emi2N was expressed in bacteria and purified by standard procedures. GST-hPlk1 residues 326–603 (GST-PBD) was a gift from M. Yaffe (Massachusetts Institute of Technology, Cambridge, MA). Purified CaMKII and calmodulin were gifts from A. Hudmon (Yale University, New Haven, CT). Cyclin B/Cdc2 with reaction buffer was purchased from New England Biolabs. Antibodies used for immunoblots were mouse anti-Plk1 (17-3300; Zymed Laboratories), mouse anti-Xenopus cyclin B2 (gift from T. Hunt, Imperial Cancer Research Fund, London, U.K.), rabbit anti-phospho-T286 CaMKII (3361; Cell Signaling Technology, Beverly, MA), rabbit anti-myc (sc-789; Santa Cruz Biotechnology), and our own rabbit anti-xEmi2. For anti-xEmi2, sera from rabbits immunized with MBP-Emi2N were affinity-purified on columns of GST-Emi2N immobilized on CNBr-Sepharose resin, with acid elution. Anti-xEmi2 antibodies were used at 0.6 μg/ml for immunoblotting.
Destruction Assays. CSF, interphase, and Δ90-cyclin B-arrested egg extracts were prepared, and assays for protein destruction were performed as described (10, 13, 32). CaCl2 was added to 800 μM to induce CaMKII activation. IVT Emi2 protein was added at 1:20, and IVT CaMKII 1-290 was added at 1:10. The CaMKII 281–309 peptide was used at 0.4 mM final concentration and preincubated with extract for 5′ before addition of CaCl2. Okadaic acid was used at 2 μM. The polyubiquitinated species (denoted Emi2-Ubn in figures) is practically immobile and collects as a band at the top of the resolving gel. The radiolabeled Emi2-Ubn band can be visualized when the gel is dried onto Whatman paper, whereas endogenous Emi2-Ubn cannot be seen in immunoblots because it transfers out of the gel poorly.
In Vitro Phosphorylation and PBD-Binding Assays. CaMKII reaction mixture was 50 mM Pipes pH 7.4, 150 mM NaCl, 10 mM MgCl2, 2 mM CaCl2, 1 mM DTT, 1 mM ATP, 2.5 μM calmodulin, and 2.5 ng/μL CaMKII. Mock reactions lacked only CaMKII. Twenty-microliter reactions containing 2 μg of substrate were incubated at 30°C for 1 h. For GST-PBD binding, salt concentration was raised to 300 mM, and Igepal was added to 0.5%. GST-PBD (1.5 μg) was added, and reactions were incubated on ice for 45 min. Reactions were diluted with 100 μl of cold wash buffer (50 mM Tris, pH 7.5/300 mM NaCl/20 mM β-glycerophosphate/5 mM EDTA/0.5% Igepal) and rotated with 10 μl amylose resin at 4°C for 45 min. Beads were washed four times with 150 μl of cold wash buffer and analyzed by SDS/PAGE and Coomassie staining for bound proteins.
Plx1 Capture Assay in Extract. One microgram of MBP-Emi2N was combined with 20 μl of CSF extract with or without CaCl2 addition, with or without CaMKII 281–309 peptide, and incubated at room temperature for 5 min. Reactions were diluted with 200 μl of cold wash buffer and microcentrifuged for 10 min, and the supernatants were rotated with 10 μl of amylose resin for 45 min at 4°C. Beads were processed and analyzed for bound proteins as described above and by immunoblotting.
Handling of Xenopus Oocytes. Oocytes were obtained and processed for H1 kinase activity and immunoblotting as described (33). Oocytes were injected with 10 ng of myc-Emi2 mRNA in a volume <50 nl. Maturation was induced by treating oocytes with 10 μg/ml progesterone. Eggs were activated with A23187 ionophore (Sigma).
Supplementary Material
Acknowledgments
We thank Anthony Hudmon and Howard Schulman (Surromed, Menlo Park, CA) for CaMKII reagents and discussion, Tim Hunt for cyclin B2 antibody, and Michael Yaffe for GST-PBD protein. This work was supported by a Stanford Presidential Fellowship (to D.V.H.) and by Public Health Service Grants 5T32 CA09302-27 (to J.J.T.) and RO1 GM60439 and GM54811 (to P.K.J.).
Conflict of interest statement: No conflicts declared.
Abbreviations: APC/C, anaphase-promoting complex/cyclosome; CaMKII, Ca2+/calmodulin-dependent protein kinase II; CSF, cytostatic factor; IVT, in vitro-translated; PBD, Polo Box domain; Plx1, Xenopus Polo-like kinase 1; MAPK, mitogen-activated protein kinase.
References
- 1.Runft, L. L., Jaffe, L. A. & Mehlmann, L. M. (2002) Dev. Biol. 245, 237–254. [DOI] [PubMed] [Google Scholar]
- 2.Masui, Y. & Markert, C. L. (1971) J. Exp. Zool. 177, 129–145. [DOI] [PubMed] [Google Scholar]
- 3.Sagata, N., Watanabe, N., Vande Woude, G. F. & Ikawa, Y. (1989) Nature 342, 512–518. [DOI] [PubMed] [Google Scholar]
- 4.Tunquist, B. J. & Maller, J. L. (2003) Genes Dev. 17, 683–710. [DOI] [PubMed] [Google Scholar]
- 5.Bhatt, R. R. & Ferrell, J. E., Jr. (1999) Science 286, 1362–1365. [DOI] [PubMed] [Google Scholar]
- 6.Gross, S. D., Schwab, M. S., Lewellyn, A. L. & Maller, J. L. (1999) Science 286, 1365–1367. [DOI] [PubMed] [Google Scholar]
- 7.Gross, S. D., Schwab, M. S., Taieb, F. E., Lewellyn, A. L., Qian, Y. W. & Maller, J. L. (2000) Curr. Biol. 10, 430–438. [DOI] [PubMed] [Google Scholar]
- 8.Schwab, M. S., Roberts, B. T., Gross, S. D., Tunquist, B. J., Taieb, F. E., Lewellyn, A. L. & Maller, J. L. (2001) Curr. Biol. 11, 141–150. [DOI] [PubMed] [Google Scholar]
- 9.Tunquist, B. J., Schwab, M. S., Chen, L. G. & Maller, J. L. (2002) Curr. Biol. 12, 1027–1033. [DOI] [PubMed] [Google Scholar]
- 10.Reimann, J. D., Freed, E., Hsu, J. Y., Kramer, E. R., Peters, J. M. & Jackson, P. K. (2001) Cell 105, 645–655. [DOI] [PubMed] [Google Scholar]
- 11.Paronetto, M. P., Giorda, E., Carsetti, R., Rossi, P., Geremia, R. & Sette, C. (2004) EMBO J. 23, 4649–4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tunquist, B. J., Eyers, P. A., Chen, L. G., Lewellyn, A. L. & Maller, J. L. (2003) J. Cell Biol. 163, 1231–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reimann, J. D. & Jackson, P. K. (2002) Nature 416, 850–854. [DOI] [PubMed] [Google Scholar]
- 14.Dumont, J., Umbhauer, M., Rassinier, P., Hanauer, A. & Verlhac, M. H. (2005) J. Cell Biol. 169, 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohsumi, K., Koyanagi, A., Yamamoto, T. M., Gotoh, T. & Kishimoto, T. (2004) Proc. Natl. Acad. Sci. USA 101, 12531–12536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsurumi, C., Hoffmann, S., Geley, S., Graeser, R. & Polanski, Z. (2004) J. Cell Biol. 167, 1037–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lorca, T., Cruzalegui, F. H., Fesquet, D., Cavadore, J. C., Mery, J., Means, A. & Doree, M. (1993) Nature 366, 270–273. [DOI] [PubMed] [Google Scholar]
- 18.Madgwick, S., Levasseur, M. & Jones, K. T. (2005) J. Cell Sci. 118, 3849–3859. [DOI] [PubMed] [Google Scholar]
- 19.Liu, J. & Maller, J. L. (2005) Curr. Biol. 15, 1458–1468. [DOI] [PubMed] [Google Scholar]
- 20.Descombes, P. & Nigg, E. A. (1998) EMBO J. 17, 1328–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brassac, T., Castro, A., Lorca, T., Le Peuch, C., Doree, M., Labbe, J. C. & Galas, S. (2000) Oncogene 19, 3782–3790. [DOI] [PubMed] [Google Scholar]
- 22.Liu, J., Lewellyn, A. L., Chen, L. G. & Maller, J. L. (2004) J. Biol. Chem. 279, 21367–21373. [DOI] [PubMed] [Google Scholar]
- 23.Schmidt, A., Duncan, P. I., Rauh, N. R., Sauer, G., Fry, A. M., Nigg, E. A. & Mayer, T. U. (2005) Genes Dev. 19, 502–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tung, J. J., Hansen, D. V., Ban, K. H., Loktev, A. V., Summers, M. K., Adler, J. R., III, & Jackson, P. K. (2005) Proc. Natl. Acad. Sci. USA 102, 4318–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hansen, D. V., Loktev, A. V., Ban, K. H. & Jackson, P. K. (2004) Mol. Biol. Cell 15, 5623–5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Margottin-Goguet, F., Hsu, J. Y., Loktev, A., Hsieh, H. M., Reimann, J. D. & Jackson, P. K. (2003) Dev. Cell 4, 813–826. [DOI] [PubMed] [Google Scholar]
- 27.Colbran, R. J., Smith, M. K., Schworer, C. M., Fong, Y. L. & Soderling, T.R. (1989) J. Biol. Chem. 264, 4800–4804. [PubMed] [Google Scholar]
- 28.Matsumoto, Y. & Maller, J. L. (2002) Science 295, 499–502. [DOI] [PubMed] [Google Scholar]
- 29.Elia, A. E., Cantley, L. C. & Yaffe, M. B. (2003) Science 299, 1228–1231. [DOI] [PubMed] [Google Scholar]
- 30.Lorca, T., Fesquet, D., Zindy, F., Le Bouffant, F., Cerruti, M., Brechot, C., Devauchelle, G. & Doree, M. (1991) Mol. Cell. Biol. 11, 1171–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rauh, N. R., Schmidt, A., Bormann, J., Nigg, E. A. & Mayer, T. U. (2005) Nature 437, 1048–1052. [DOI] [PubMed] [Google Scholar]
- 32.Murray, A. W., Solomon, M. J. & Kirschner, M. W. (1989) Nature 339, 280–286. [DOI] [PubMed] [Google Scholar]
- 33.Furuno, N., Nishizawa, M., Okazaki, K., Tanaka, H., Iwashita, J., Nakajo, N., Ogawa, Y. & Sagata, N. (1994) EMBO J. 13, 2399–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}