

Abstract
Efficient targeting of proteins for degradation from the secretory pathway is essential to homeostasis. This occurs through endoplasmic reticulum (ER)-associated degradation (ERAD). In this study, we establish that a human ubiquitin ligase (E3), gp78, and a specific E2, Ube2g2, are both critically important for ERAD of multiple substrates. gp78 exhibits a complex domain structure that, in addition to the RING finger, includes a ubiquitin-binding Cue domain and a specific binding site for Ube2g2. Disruption of either of these domains abolishes gp78-mediated ubiquitylation and protein degradation, resulting in accumulation of substrates in their fully glycosylated forms in the ER. This suggests that gp78-mediated ubiquitylation is an early step in ERAD that precedes dislocation of substrates from the ER. The in vivo requirement for both an E2-binding site distinct from the RING finger and a ubiquitin-binding domain intrinsic to an E3 suggests a previously unappreciated level of complexity in ubiquitin ligase function. These results also provide proof of principle that interrupting a specific E2-E3 interaction can selectively inhibit ERAD.
Keywords: autocrine motility factor receptor, ubiquitin protein ligase, ubiquitin, unfolded protein response, MmUBC7
Misfolded, unassembled, and highly regulated proteins must all be eliminated from the endoplasmic reticulum (ER) by ER-associated degradation (ERAD). ERAD is a set of tightly coupled processes that include ubiquitylation, retrograde movement from ER to cytoplasm, deglycosylation, and interaction with cytosolic intermediaries, including p97/CDC48 complexes. The order of these events is obscure, although it is clear that ERAD substrates are ultimately degraded by proteasomes (reviewed in refs. 1-3).
Specific ubiquitin-conjugating enzymes (E2s) and ubiquitin protein ligases (E3s) have been implicated in ERAD. In Saccharomyces cerevisiae, a polytopic ER RING finger E3, known as Hrd1p/Der3p, which is implicated in sterol-regulated proteasomal degradation of hydroxymethylglutaryl-CoA reductase, functions with Ubc7p in ERAD (4, 5). Ubc7p is a cytosolic E2 that is recruited to the ER membrane by Cue1p, a 203-aa N-terminal-anchored ER protein (6-8). The molecular basis by which Cue1p recruits Ubc7p is not yet known. Cue1p is the founding member of a group of proteins that contain a conserved ≈40-aa region known as the Cue domain (9). Cue domains of several proteins bind ubiquitin, although it is not evident that Cue1p itself binds ubiquitin (10, 11). More recently, another ER membrane E3, Doa10p, has been shown to target specific substrates. Doa10p also utilizes Ubc7p that is recruited by Cue1p (12, 13).
In mammals, murine MmUBC7, encoded by the UBE2G2 gene and known in humans as Ube2g2, is a Ubc7p ortholog, and the E2 most clearly implicated in ERAD. Catalytically inactive MmUBC7 inhibits ERAD of transmembrane substrates, including the T cell antigen receptor (TCR) subunits TCR-α and CD3-δ (14, 15) and others (16, 17). A human ortholog of Hrd1p/Der3p, HsHrd1, has now been identified (18). Overexpression studies suggest HsHrd1 may play a role in ERAD. However, unlike yeast, there is no evidence that HsHrd1 mediates sterol-regulated proteasomal degradation of hydroxymethylglutaryl-CoA reductase. Analogies to yeast Hrd1p/Der3p further break down, because there is no identifiable human Cue1p ortholog. More recently, a mammalian Doa10p ortholog, TEB4, has been identified that may play a role in MHC regulation (19, 20).
gp78, which has similarity to Hrd1p in its transmembrane domain and RING finger, is the first-described and most well documented human ERAD E3 intrinsic to the ER. This protein, also known as the tumor autocrine motility factor receptor (21), includes a ≈350-aa cytoplasmic tail that contains a RING finger, which is essential for E3 activity. When overexpressed, gp78 targets itself and heterologous substrates, such as CD3-δ and apolipoprotein B, for degradation (22, 23). Most recently, gp78 has been implicated in the regulated degradation of hydroxymethylglutaryl-CoA reductase (24). gp78 can use MmUBC7/Ube2g2 as its E2 (22). Strikingly, gp78 binds Ube2g2 in a RING finger-independent manner. There is 20% identity to yeast Cue1p in 198 amino acids distal to the gp78 RING finger, which includes a Cue domain consensus sequence (22). This overall homology raises the possibility that the gp78 cytoplasmic tail could double as the mammalian Cue1p equivalent in recruiting Ube2g2 to heterologous ERAD E3s. However, the molecular determinants responsible for the gp78-Ube2g2 interaction are unknown. It is also not known whether recruitment of Ube2g2 via this RING finger-independent region is important for ERAD or gp78 function in vivo.
E2-binding sites and sites of potential ubiquitin interaction have been observed in several other E3s (25-28). None of these have yet been shown to be functionally important. In this study, we establish essential roles for gp78 and Ube2g2 in ERAD and demonstrate that both gp78 and Ube2g2 are required before retrotranslocation of substrate from the ER into the cytosol. A discrete and highly specific Ube2g2-binding region (G2BR) in gp78, distinct from the Cue domain, which mediates the interaction of this E2-E3 pair, is identified. This E2-binding site and the gp78 Cue domain, which we show to bind ubiquitin, are both essential for gp78-mediated ERAD.
Results
Endogenous gp78 and Ube2g2 Are Critical for ERAD. Short-hairpin RNAs (shRNAs) specific for human gp78 (gp78-sh1 and gp78-sh2) or Ube2g2 (Ube2g2-sh1) were evaluated in the fibrosarcoma HT1080 (Fig. 1A) and in human embryonic kidney 293T (Fig. 1B). These shRNAs specifically knocked down their targets but not their closest E3 and E2 relatives, HsHrd1 and Ube2g1. Each shRNA markedly increased hemagglutinin (HA)-tagged CD3-δ (HA-CD3-δ) (Fig. 1 A Top) despite the presence of other putative ERAD machinery. In comparison, a control shRNA was without effect. This increase was shown to be due to stabilization of CD3-δ by cycloheximide chase and by 35S pulse-chase metabolic labeling (Fig. 6, which is published as supporting information on the PNAS web site). Similarly, these shRNAs stabilized TCR-α (Fig. 1B), providing evidence that TCR-α degradation depends on gp78 and Ube2g2. These results establish important roles for both endogenous gp78 and Ube2g2 in the degradation of two distinct ERAD substrates.
Fig. 1.

Essential roles for gp78/autocrine motility factor receptor (AMFR) and Ube2g2 in ERAD. (A) HT1080 cells were cotransfected with the indicated shRNA plasmids and HA-tagged CD3-δ. After 48 h, cell lysates were analyzed by immunoblotting (IB) after resolution of equal amounts of lysates on duplicate gels. (B) Human embryonic kidney 293T cells were cotransfected with shRNA plasmids and TCR-α. gp78 and TCR-α were first immunoprecipitated before SDS/PAGE. Transfection efficiency was monitored by GFP.
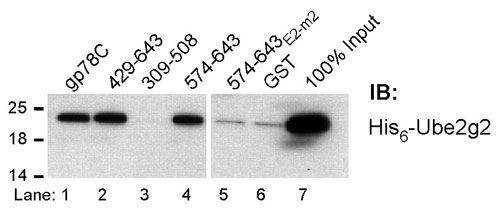
gp78 Encodes a Discrete Ube2g2-Binding Site. Both RING finger and HECT domain E3s are generally believed to interact with cognate E2s through their ubiquitin ligase domains. In the few instances where E2-binding sites on E3s distinct from ligase domains have been demonstrated, no functional role for this binding has been determined (25-27). It was previously shown that gp78 (schematized in Fig. 2A) could bind Ube2g2/MmUBC7 through undetermined regions in its C-terminal cytoplasmic tail (22). To determine the molecular determinants for this interaction, GST fusions of gp78 were evaluated for Ube2g2 binding. A summary of results of binding of in vitro translated Ube2g2 to these fusions is shown in Fig. 2B. Amino acids 574-611 bound Ube2g2 comparably to the complete C-terminal tail (gp78C) (Fig. 2C, lanes 3 and 4). Amino acids distal to 600 were dispensable for binding, whereas amino acids from 595 to 600 were required (Fig. 2C, lanes 6 and 7). This narrowed the E2-binding site to 574-600. Because yeast Cue1p binds Ubc7p (8), site-specific mutagenesis within this region was guided by alignment of gp78 with Cue1p (Fig. 2B Lower). Mutations in regions of homology (E2m-1, E2m-3, E2-m4, and E2-m5; Fig. 2B Lower and Fig. 2C, lane 8) did not disrupt binding. Surprisingly, the only mutation that resulted in loss of binding was the six amino acids, 579-584, which correspond to the gap in the alignment between gp78 and Cue1p (E2-m2; Fig. 2B Lower and Fig. 2C, lane 9). These findings narrow the minimal G2BR to 22 amino acids from 579 to 600. The binding of Ube2g2 to gp78 was further determined to be direct (Fig. 7, which is published as supporting information on the PNAS web site). Because active E2s are linked to ubiquitin by thiolester bonds, we addressed whether the G2BR binds Ube2g2∼ubiquitin. Bacterially expressed Ube2g2 bound to a fragment of gp78 containing the G2BR (574-643) both in a free form or when loaded with ubiquitin (Fig. 2D), showing that G2BR can recruit active E2 to the ER.
Fig. 2.

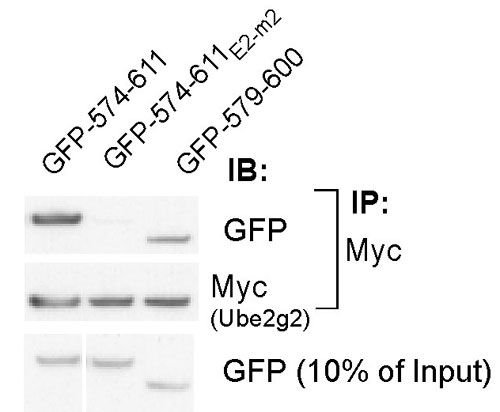
Localization and specificity of Ube2g2 binding to gp78. (A) Schematic representation of full-length gp78 showing RING finger (Rf) and Cue domain (Cue). (B) (Upper) Schematic of gp78 cytoplasmic domain with RING finger, Cue domain and minimal G2BR indicated. GST fusions tested for Ube2g2 binding are shown below with summary of results at right. (Lower) Alignment of gp78 G2BR with the analogous region of Cue1p. Mutations and their effects on E2 binding are shown below the gp78 sequence. (C) Ube2g2 was translated in reticulocyte lysate with 35S-Met and incubated with the indicated GST fusions bound to beads and assessed for binding. (D) Bacterially expressed Ube2g2 was reacted with E1 with or without ubiquitin and bound to the indicated GST fusions. Washed samples were eluted from beads in the absence (NR) or presence (R) of reducing agent (2-mercaptoethanol) before SDS/PAGE and IB with anti-Ube2g2. (E) Human embryonic kidney (HEK) 293T cells were cotransfected with Myc-Ube2g2 and gp78 or the indicated mutants. Rf-m is an inactivating RING finger mutation. Subscripts 595 and 611 are truncations after the indicated amino acids. E2-m2 corresponds to the mutation in the G2BR described in B. Samples were treated with MG132 for 8 h before lysis. Immunoprecipitation (IP) (Upper) with anti-Myc was followed by IB, as indicated. Lysate corresponding to 10% of the material used for IP was directly immunoblotted for gp78 (Lower). (F) HEK 293T were transfected with plasmid encoding the indicated amino acids of gp78 fused to the C terminus of GFP. Anti-GFP IPs (Left) or whole-cell lysates corresponding to ≈10% of the amount used for IP (Right) were immunoblotted with antibodies raised against the indicated E2s.
To confirm Ube2g2 binding to G2BR in vivo, mutations and C-terminal truncations of full length gp78 were tested for binding to coexpressed Myc epitope-tagged Ube2g2 (Myc-Ube2g2) in cells (Fig. 2E). Mutations of critical RING finger residues (gp78Rf-m) did not affect gp78's association with Ube2g2 (Fig. 2E, compare lanes 4 and 5). In contrast, both a mutation and a truncation that disrupts the G2BR (gp78E2-m2 and gp78595) lost E2 binding. A truncation after 611 (gp78611), which leaves the G2BR intact, retained binding.
To determine the specificity of E2 binding in cells, GFP fusions encompassing the E2-binding region were expressed (Fig. 2F). Endogenous Ube2g2 was coimmunoprecipitated, whereas no binding to its closest relative, Ube2g1, was observed. Further, the minimal G2B2, amino acids 579-600, was sufficient to bind Ube2g2 in cells (Fig. 8, which is published as supporting information on the PNAS web site). These results define a discrete 22-aa Ube2g2-binding site in gp78.
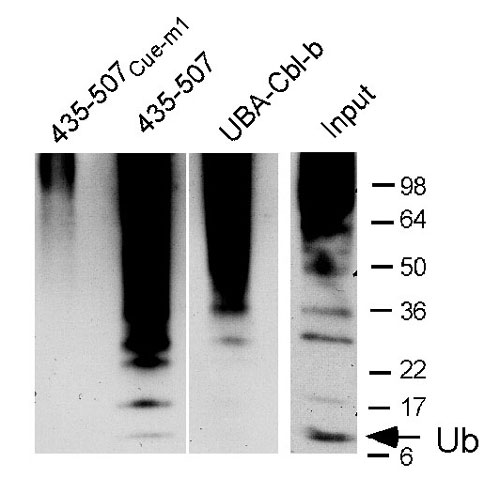
The gp78 Cue Domain Binds Ubiquitin. An increasing number of ubiquitin-binding proteins are being identified. Postulated roles for these include functions in ubiquitylation-deubiquitylation and chaperone-like functions in endocytosis and proteasomal targeting (reviewed in refs. 28-30). To date, the UBA of Cbl-b is the only known example of a ubiquitin-binding domain in an E3, but its deletion has no effect on the well described function of this E3 in receptor tyrosine kinase ubiquitylation (28). To assess whether the gp78 Cue domain binds ubiquitin, the cytoplasmic tail of gp78 was evaluated for binding of lysine 48 (K48)-linked tetraubiquitin (31). The GST-gp78C fusion bound polyubiquitin chains, as did an N-terminal truncated version that lacks the RING finger but retains the Cue domain (429-643) (Fig. 3A). The UBA of Cbl-b (UBA-Cbl-b) also bound tetraubiquitin. In contrast, no binding to the cytoplasmic domain of yeast Cue1p (Cue1pC), which includes its Cue domain, was observed. Based on sequences predicted to be important for ubiquitin binding to other Cue domains (32, 33), two sets of mutations were generated in the gp78 Cue domain. Either of these, Cue-m1 and Cue-m2, resulted in loss of ubiquitin binding, as did the double mutant, Cue-m1,2 (Fig. 3A). A GST fusion of the isolated gp78 Cue domain (435-507) was sufficient to bind tetraubiquitin (Fig. 3B); binding was eliminated by the Cue-m1 mutation (435-507Cue-m1). Thus, unlike Cue1p, the gp78 Cue domain shows a substantial capacity to bind polyubiquitin. The GST fusion of the gp78 Cue domain also bound ubiquitylated species from cell lysate and has the capacity to bind monoubiquitin, albeit to a lesser extent than ubiquitin oligomers (Fig. 9, which is published as supporting information on the PNAS web site, and data not shown).
Fig. 3.

gp78 binds ubiquitin through its Cue domain. (A and B) Equal amounts of GST fusions were incubated with K48-Ub4 and binding determined by IB with antiubiquitin. gp78CCue-m1 includes the complete C-terminal region of gp78 with an MFP to GGR mutation of 467 to 469. gp78Cue-m2 contains a VLQDL to RLQVD mutation of 476 to 480. gp78CCue-m1,2 contains both sets of mutations. Cue1pC and UBA-Cbl-b correspond to GST fusions of the cytoplasmic domain of Cue1p and the UBA of Cbl-b, respectively. 435-507 and 435-507Cue-m1 are GST fusions of amino acids 435-507 of gp78 either without or with the Cue-m1 mutation.
The G2BR and Cue Domain Are Essential for ERAD Function of gp78. Mutations of the RING finger (gp78Rf-m), Cue domain (gp78Cue-m1,2) and G2BR (gp78E2-m2) were evaluated for their functional effects within full length gp78. Each of these gp78 mutants exhibited increased levels compared with WT gp78 (Fig. 4A Top). As expected, WT gp78 decreased CD3-δ levels compared with vector control (Fig. 4A Middle), whereas the RING finger mutation resulted in increased CD3-δ, suggesting a critical role for this domain in ERAD. Mutation of the Cue domain also markedly increased CD3-δ. Expression of gp78 mutated in the G2BR (gp78E2-m2) neither increased nor decreased CD3-δ levels compared with vector control (compare lanes 1, 2, and 5). gp78 and CD3-δ turnover was further evaluated by cycloheximide chase. Consistent with Fig. 4A, mutations of the RING finger, Cue domain, and G2BR each stabilized gp78 (Fig. 4 B and C Top). Similarly, a truncation in which the G2BR is interrupted (595) stabilized gp78. A truncation that retained E2 binding (611) but lost the reported binding of gp78 to p97 (34) (Fig. 10, which is published as supporting information on the PNAS web site) was indistinguishable from WT gp78 in its own degradation (Fig. 4B). Double mutants of the G2BR and either the RING finger (Rf-m/E2-m2) or Cue domain (Cue-m1,2/E2-m2) also stabilized gp78 (Fig. 4 B and C).
Fig. 4.

Multiple domains of gp78 are required to target itself and a heterologous ERAD substrate for degradation. (A-C) Cells were cotransfected with the indicated forms of gp78 together with CD3-δ and lysates analyzed by IB. (B and C) Cells were treated with cycloheximide (CHX), as indicated. (D) Graphic representation of CD3-δ levels from B and C (full length gp78 transfections only). Cue-m1,2 is the average of data shown in B and C. (E) Cells were cotransfected with TCR-α and GFP fusions of gp78 and treated with MG132 where indicated. (F) Cells transfected with CD3-δ and Mdm2 were cotransfected with Flag-tagged amino acids 574-643 of gp78 where indicated. In lanes 3 and 6, 2 μg of Ube2g2 plasmid was cotransfected; in lanes 4 and 7, 4 μg was used. MG132 treatment was for 8 h.
As expected, expression of WT gp78 accelerated CD3-δ degradation (Fig. 4B Middle). Truncation at amino acid 611, which loses p97 binding but retains the Ube2g2 interaction, was indistinguishable from WT gp78 in accelerating CD3-δ degradation. In contrast, mutations in either the RING finger or the Cue domain markedly inhibited CD3-δ degradation. However, when the G2BR was truncated (595) or mutated either by itself or together with mutations in either the RING mutation or the Cue domain, there was minimal effect on CD3-δ degradation compared with vector control (Fig. 4 B and C, quantified in Fig. 4D). These results demonstrate that the RING finger, Cue domain, and G2BR are all critical to the ERAD function of gp78. The binding of p97 by gp78 has been proposed to be important for substrate degradation based on the effects of a truncation at amino acid 595 (34). Our findings with the 611 truncation, which retains Ube2g2 binding but loses the capacity to bind p97, and with the G2BR mutation (E2-m2), which loses E2 binding but retains the ability to bind p97 (Fig. 10; summarized in Table 1, which is published as supporting information on the PNAS web site), suggest that the importance attributed to p97 binding may instead reflect the failure of the 595 truncation mutant to recruit Ube2g2. Moreover, the lack of any effect on degradation of heterologous substrate seen with disruption of the gp78 G2BR, compared with cells not expressing any form of exogenous gp78, suggests that the dominant negative effect we observe with RING finger and Cue domain mutants is due to sequestration of endogenous Ube2g2 by gp78 inactivated in these other critical domains.
Consistent with the sequestration hypothesis, expression of the Ube2g2-binding region in isolation inhibited degradation of TCR-α and CD3-δ (Fig. 4 E and F Top, compare lanes 1 and 2). Furthermore, accumulation of CD3-δ can be reversed by coexpressing increasing amounts of Ube2g2 (Fig. 4F, lanes 3 and 4). This effect is specific for ERAD substrates, because the p53 E3, Mdm2, which targets itself for degradation and is not known to use Ube2g2, was unaffected (Fig. 4F Middle).
The Cue Domain, G2BR, and RING Finger Are All Essential for gp78-Mediated Ubiquitylation. There are a number of ways in which gp78 might be contributing to ERAD. One possibility is that gp78, or its various domains, is required for ubiquitylation subsequent to retrotranslocation. To examine this possibility, the location of accumulated CD3-δ in cells in which either gp78 or Ube2g2 was knocked down was evaluated. Loss of either gp78 or Ube2g2 resulted in CD3-δ accumulation only in the membrane fraction (Fig. 5A Top). Similarly, when Cue mutant or RING finger mutant gp78 was expressed, CD3-δ accumulated in the membrane fraction (Fig. 11, which is published as supporting information on the PNAS web site). Additionally, the CD3-δ that accumulates migrates at a molecular weight consistent with its three N-linked oligosaccharides being intact (≈27 kDa) and treatment with N-glycanase results in the removal of these oligosaccharides (not shown). Thus, gp78 is required for early steps in ERAD before retrotranslocation out of the ER membrane.
Fig. 5.

The G2BR and Cue domain are required for in vivo E3 activity. (A) Membrane (M) and cytoplasmic (C) fractions from HT1080 cells transfected with HA-CD3-δ and the indicated shRNAs were prepared by hypotonic lysis and equal cell equivalents of each resolved by SDS/PAGE. Membrane fractions were subject to a high salt wash to further remove nonintegral membrane proteins. (B) Cells transfected as indicated were treated with MG132 for 5 h before IP with anti-gp78 followed by IB as indicated for gp78 or HA-CD3-δ. (Lower) IB of whole cell lysate representing 10% of the material used for IP. Similar co-IP was obtained when IPs were washed in either Triton X-100 buffer (shown) or RIPA buffer. (C) Cells were transfected with WT or mutant gp78 plasmids as indicated. Samples were treated with MG132 for 8 h and lysates immunoprecipitated with anti-gp78 followed by IB with either antiubiquitin (Upper) or anti-gp78 (Lower). Arrow indicates migration of unmodified gp78. (D) Cells were transfected with CD3-δ and gp78 as indicated. In lane 8, plasmid encoding Ube2g2 was coexpressed. Cells were treated with MG132 as in C before lysis and IP with anti-HA. Arrow indicates migration of CD3-δ.
The accepted essential feature of RING finger E3s is a capacity to interact with substrate and to mediate transfer of ubiquitin from E2 to substrate. E2 interactions are generally believed to occur primarily through the RING finger itself. It is possible that the Cue domain might function in substrate recruitment by binding already ubiquitylated substrates. In this model, gp78 would function primarily as an E4, enhancing the processivity of ubiquitin chain elongation after initiation of ubiquitylation by heterologous E3s. Contrary to predictions of this model, the interaction between gp78 and substrate was unaffected by mutations in the Cue domain or by disruption of the G2BR or the RING finger (Fig. 5B). Moreover, CD3-δ that coimmunoprecipitated with gp78 did not exhibit evidence of ubiquitylation (Fig. 5B).
These observations lead to the possibility that the primary function of the Cue domain and the G2BR is to function in a coordinated manner with the RING finger in mediating ubiquitylation. Consistent with this hypothesis, mutations in each of these domains resulted in a significant reduction in gp78 self-ubiquitylation in vivo (Fig. 5C). Mutations in either the Cue domain or the RING finger also resulted in a marked decrease in CD3-δ ubiquitylation (Fig. 5D). The G2BR mutation neither inhibited nor stimulated CD3-δ ubiquitylation. This is consistent with a lack of sequestration of endogenous Ube2g2 by the G2BR mutant, thus allowing for CD3-δ ubiquitylation by endogenous gp78 or other ERAD E3s. The importance of an intact G2BR in decreasing ubiquitylation by sequestering Ube2G2 is underscored by the restoration of CD3-δ ubiquitylation to a level seen with vector control when exogenous Ube2g2 was coexpressed with Cue mutant gp78 (Fig. 5D, compare lanes 3, 7, and 8). These findings demonstrate that the RING finger, Cue domain, and the G2BR are all integral to the process whereby gp78 mediates both its own ubiquitylation and that of heterologous substrate.
Discussion
ERAD involves ubiquitylation, retrograde movement from ER to cytoplasm, deglycosylation, and interaction with cytosolic intermediaries (reviewed in refs. 1-3). Interestingly, gp78 has been identified as a PNGase-binding partner (35), and more recently a complex containing a polytopic protein, Derlin-1, has been proposed to function as an ERAD retrotranslocation channel (36, 37). Despite these advances, it is as yet unclear how a complex involving E2, E3, substrate, and retrotranslocation channels is assembled, and how the multiple events in ERAD are functionally and temporarily related.
We now show that gp78 associates with its substrate, CD3-δ, independent of its E3 activity. We also demonstrate that gp78 and Ube2g2 can stably and directly interact in a RING finger-independent manner, and that loss of either this E3 or E2 results in CD3-δ accumulation in its fully glycosylated form and only in the membrane fraction; this occurs despite the fact that these cells have other putative ERAD machinery such as HsHrd1. Similarly, overexpression of inactive gp78 results in CD3-δ accumulation only in the membrane fraction. This finding suggests that ubiquitylation mediated by gp78 is an early step in ERAD before extraction of substrate from the ER.
Among characterized E3s, gp78 has the previously unreported in vivo requirements for both a non-RING finger E2-binding site (G2BR) and a ubiquitin-binding Cue domain. Our findings show that G2BR functions to recruit Ube2g2. This would position Ube2g2, potentially loaded with ubiquitin, in proximity to the gp78 RING finger, which then facilitates transfer of ubiquitin from E2 to substrate. The essential role for the Cue domain in ubiquitylation of both gp78 and heterologous substrate indicates that this domain must be involved in substrate ubiquitylation. Whether it functions primarily by stabilizing activated ubiquitin during transfer to substrate, protecting nascent polyubiquitin chains from deubiquitylating enzymes, or serving as a scaffold to enhance chain processivity remains to be determined.
The inhibition of ERAD that occurs when Ube2g2 is recruited to the ER by mutant gp78 raises two mechanistic points. First, the capacity to bind E2 loaded with ubiquitin is insufficient to confer E3 activity when the RING finger is inactivated. This supports the view that the RING finger is critical as an allosteric activator in the transfer of ubiquitin from E2 to substrate and not simply an E2 docking site. The second point pertains to analogies to yeast Cue1p, for which there is no apparent mammalian ortholog. The major established function of Cue1p is to recruit Ubc7p to the ER membrane to function with ERAD E3s (8, 13). CD3-δ stabilization by sequestration of endogenous Ube2g2 by gp78 mutants suggests that Ube2g2 bound by gp78 needs to function in cis with active gp78 and is not available to other ERAD E3s including endogenous gp78 and HsHrd1, which has been reported to affect the rate of CD3-δ degradation in overexpression studies (18). Therefore, the Ube2g2-binding site on gp78 does not apparently serve a general Cue1p-like function for ERAD E3s. It remains to be determined whether other ERAD E3s, such as HsHrd1, directly associate with Ube2g2 or use E2 recruited by yet-to-be-identified ER membrane proteins.
The finding that both ubiquitin- and E2-binding sites, independent of a ubiquitin ligase domain, are required for gp78 activity in cells suggests a previously unappreciated degree of complexity in E3 function. This raises the possibility that similar, as yet unknown, interactions are likely critical to the in vivo function of other E3s. Assessment of binding of E2s to ubiquitin ligase domains in general, and RING fingers in particular, is generally problematic, indicating these interactions are of low affinity (reviewed in ref. 38). Therefore, additional higher-affinity binding sites provided by nonligase domain E2-binding sites could increase E3 activity in vivo by enhancing E2 availability. Such sites could exist in as-yet-uncharacterized domains of E3s or be provided in trans by other associated proteins. The latter is the case for Hrd1p/Der3p and Doa10p, where Cue1p recruits Ubc7p through currently unidentified interactions sites. The HECT domain E3, Nedd4, and yeast RING finger E3, Ubr1p, are examples of E3s where E2-binding sites have been found intrinsic to E3s but distinct from ligase domains (25-27). For Nedd4, the exact site of interaction and its significance are unknown. For Ubr1p, mutations that lose detectable binding to the full-length form of its E2 have no substantial effect on the N-end rule pathway (27). In light of our observations, a reevaluation of the role of the non-RING finger Ubr1p E2-binding site on other Ubr1p substrates might be warranted.
The Cue domain of gp78, unlike that of Cue1p, binds ubiquitin (10). Structural studies suggest that the difference between the Cue1p Cue domain and those that bind ubiquitin derives in part from the lack of the tripeptide sequence Met-Phe-Pro in Cue1p (32, 33). gp78 contains this sequence, mutation of which abolishes ubiquitin binding (gp78Cue-m1). The only other E3 known to bind ubiquitin is Cbl-b, which contains a UBA. However, unlike gp78, where the Cue domain is essential for function, no demonstrable effect of deletion of the Cbl-b UBA on Cbl-b-mediated EGF receptor ubiquitylation has been observed (28). Regardless, it seems likely that roles for ubiquitin-binding domains in E3 activity will be a more general finding than currently appreciated. Such binding sites might be intrinsic to the E3 or could be provided in trans through other interacting proteins. A recent example of the latter is the E2-like molecule, Mms2, where ubiquitin binding correlates with the ability of the interacting RING finger E3, Rad5p, to generate polyubiquitin chains (39).
An observation of potential therapeutic significance is that sequestration of endogenous Ube2g2 blocks ERAD. This raises the possibility that disrupting the function of Ube2g2, or specifically interfering with interactions with its cognate E3(s), may be a way to specifically block ERAD. This could potentially either enhance expression of proteins, such as the Δ508 form of cystic fibrosis transmembrane conductance regulator, or selectively stimulate ER stress and apoptosis in susceptible tumors, such as multiple myeloma and adenocarcinomas with active secretory systems. In this regard, our findings provide an important proof of principle, demonstrating that interfering with E2 and E3 enzymes involved in ERAD, whether through G2BR-like molecules or other means, represents a way to specifically target ERAD.
Materials and Methods
Details regarding cell lines, expression plasmids, shRNAs in pSuper, site-specific mutagenesis, antibodies, and binding assays are found in Supporting Text, which is published as supporting information on the PNAS web site. Transfections were carried out ≈24 h after plating, and cells were then harvested after ≈48 h. Further details are found in Supporting Text. MG132 was used at 50 μM; cycloheximide was used at 50 μg/ml. For subcellular fractionation, cells were lysed by passage through a 27-gauge needle followed by removal of unbroken cells and nuclei by a 1,000 × g spin. To separate membrane and cytosolic fractions, supernatants were pelleted by ultracentrifugation. Further details are found in Supporting Text.
Supplementary Material
Acknowledgments
We thank Gaye Wilson, Barry O'Keefe, and Shengyun Fang for assistance in generating reagents and Michael Glickman, Michael Kuehn, Zlatka Kostova, Stan Lipkowitz, Kevin Lorick, and Larry Samelson for invaluable discussions and comments. This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Author contributions: A.M.W. designed research; B.C., J.M., Y.C.T., A.H.C., and M.C. performed research; B.C., J.M., and Y.C.T. contributed new reagents/analytic tools; B.C., J.M., Y.C.T., M.C., and A.M.W. analyzed data; and A.M.W. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ER, endoplasmic reticulum; ERAD, ER-associated degradation; G2BR, Ube2g2-binding region; TCR, T cell antigen receptor; shRNA, short-hairpin RNA; IB, immunoblotting; IP, immunoprecipitation; HA, hemagglutinin.
References
- 1.Hampton, R. Y. (2002) Curr. Opin. Cell Biol. 14, 476-482. [DOI] [PubMed] [Google Scholar]
- 2.Kostova, Z. & Wolf, D. H. (2003) EMBO J. 22, 2309-2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jarosch, E., Lenk, U. & Sommer, T. (2003) Int. Rev. Cytol. 223, 39-81. [DOI] [PubMed] [Google Scholar]
- 4.Bays, N. W., Gardner, R. G., Seelig, L. P., Joazeiro, C. A. & Hampton, R. Y. (2001) Nat. Cell Biol. 3, 24-29. [DOI] [PubMed] [Google Scholar]
- 5.Deak, P. M. & Wolf, D. H. (2001) J. Biol. Chem. 276, 10663-11069. [DOI] [PubMed] [Google Scholar]
- 6.Biederer, T., Volkwein, C. & Sommer, T. (1996) EMBO J. 15, 2069-2076. [PMC free article] [PubMed] [Google Scholar]
- 7.Hiller, M. M., Finger, A., Schweiger, M. & Wolf, D. H. (1996) Science 273, 1725-1728. [DOI] [PubMed] [Google Scholar]
- 8.Biederer, T., Volkwein, C. & Sommer, T. (1997) Science 278, 1806-1809. [DOI] [PubMed] [Google Scholar]
- 9.Ponting, C. P. (2000) Biochem. J. 351, 527-535. [PMC free article] [PubMed] [Google Scholar]
- 10.Shih, S. C., Prag, G., Francis, S. A., Sutanto, M. A., Hurley, J. H. & Hicke, L. (2003) EMBO J. 22, 1273-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Donaldson, K. M., Yin, H., Gekakis, N., Supek, F. & Joazeiro, C. A. (2003) Curr. Biol. 13, 258-262. [DOI] [PubMed] [Google Scholar]
- 12.Swanson, R., Locher, M. & Hochstrasser, M. (2001) Genes Dev. 15, 2660-2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huyer, G., Piluek, W. F., Fansler, Z., Kreft, S. G., Hochstrasser, M., Brodsky, J. L. & Michaelis, S. (2004) J. Biol. Chem. 279, 38369-38378. [DOI] [PubMed] [Google Scholar]
- 14.Yang, M., Omura, S., Bonifacino, J. S. & Weissman, A. M. (1998) J. Exp. Med. 187, 835-846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tiwari, S. & Weissman, A. M. (2001) J. Biol. Chem. 276, 16193-16200. [DOI] [PubMed] [Google Scholar]
- 16.Webster, J. M., Tiwari, S., Weissman, A. M. & Wojcikiewicz, R. J. (2003) J. Biol. Chem. 278, 38238-38246. [DOI] [PubMed] [Google Scholar]
- 17.Kim, B. W., Zavacki, A. M., Curcio-Morelli, C., Dentice, M., Harney, J. W., Larsen, P. R. & Bianco, A. C. (2003) Mol. Endocrinol. 17, 2603-2612. [DOI] [PubMed] [Google Scholar]
- 18.Kikkert, M., Doolman, R., Dai, M., Avner, R., Hassink, G., van Voorden, S., Thanedar, S., Roitelman, J., Chau, V. & Wiertz, E. (2004) J. Biol. Chem. 279, 3525-3534. [DOI] [PubMed] [Google Scholar]
- 19.Hassink, G. C., Kikkert, M., van Voorden, S., Lee, S. J., Spaapen, R., van Laar, T., Coleman, C. S., Bartee, E., Fruh, K., Chau, V., et al. (2005) Biochem. J. 388, 647-655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bartee, E., Mansouri, M., Hovey Nerenberg, B. T., Gouveia, K. & Fruh, K. (2004) J. Virol. 78, 1109-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nabi, I. R., Watanabe, H., Silletti, S. & Raz, A. (1991) EXS 59, 163-177. [DOI] [PubMed] [Google Scholar]
- 22.Fang, S., Ferrone, M., Yang, C., Jensen, J. P., Tiwari, S. & Weissman, A. M. (2001) Proc. Natl. Acad. Sci. USA 98, 14422-14427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang, J. S., Kim, T., Fang, S., Yamaguchi, J., Weissman, A. M., Fisher, E. A. & Ginsberg, H. N. (2003) J. Biol. Chem. 278, 23984-23988. [DOI] [PubMed] [Google Scholar]
- 24.Song, B. L., Sever, N. & Debose-Boyd, R. A. (2005) Mol. Cell 19, 829-840. [DOI] [PubMed] [Google Scholar]
- 25.Madura, K., Dohmen, R. J. & Varshavsky, A. (1993) J. Biol. Chem. 268, 12046-12054. [PubMed] [Google Scholar]
- 26.Hatakeyama, S., Jensen, J. P. & Weissman, A. M. (1997) J. Biol. Chem. 272, 15085-15092. [DOI] [PubMed] [Google Scholar]
- 27.Xie, Y. & Varshavsky, A. (1999) EMBO J. 18, 6832-6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davies, G. C., Ettenberg, S. A., Coats, A. O., Mussante, M., Ravichandran, S., Collins, J., Nau, M. M. & Lipkowitz, S. (2004) Oncogene 23, 7104-7115. [DOI] [PubMed] [Google Scholar]
- 29.Madura, K. (2002) Cell Cycle 1, 235-244. [PubMed] [Google Scholar]
- 30.Hicke, L. & Dunn, R. (2003) Annu. Rev. Cell Dev. Biol. 19, 141-172. [DOI] [PubMed] [Google Scholar]
- 31.Thrower, J. S., Hoffman, L., Rechsteiner, M. & Pickart, C. M. (2000) EMBO J. 19, 94-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kang, R. S., Daniels, C. M., Francis, S. A., Shih, S. C., Salerno, W. J., Hicke, L. & Radhakrishnan, I. (2003) Cell 113, 621-630. [DOI] [PubMed] [Google Scholar]
- 33.Prag, G., Misra, S., Jones, E. A., Ghirlando, R., Davies, B. A., Horazdovsky, B. F. & Hurley, J. H. (2003) Cell 113, 609-620. [DOI] [PubMed] [Google Scholar]
- 34.Zhong, X., Shen, Y., Ballar, P., Apostolou, A., Agami, R. & Fang, S. (2004) J. Biol. Chem. 279, 45676-45684. [DOI] [PubMed] [Google Scholar]
- 35.Park, H., Suzuki, T. & Lennarz, W. J. (2001) Proc. Natl. Acad. Sci. USA 98, 11163-11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ye, Y., Shibata, Y., Yun, C., Ron, D. & Rapoport, T. A. (2004) Nature 429, 841-847. [DOI] [PubMed] [Google Scholar]
- 37.Lilley, B. N. & Ploegh, H. L. (2004) Nature 429, 834-840. [DOI] [PubMed] [Google Scholar]
- 38.Lorick, K. L., Tsai, Y. C., Yang, Y. & Weissman, A. M. (2005) in Protein Degradation, eds. Mayer, R. J., Ciechanover, A. & Rechsteiner, M. (Wiley-VCH, Weinhein, Germany), Vol. 1, pp. 44-104. [Google Scholar]
- 39.Tsui, C., Raguraj, A. & Pickart, C. M. (2005) J. Biol. Chem. 280, 19829-19835. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}