

Abstract
The screening of new antibiotics against several bacterial strains often reveals unexpected occurrences of natural drug resistance. Two examples of this involve specific inhibitors of Staphylococcus aureus isoleucyl-transfer-RNA synthetase 1 (IleRS1) and, more recently, Streptococcus pneumoniae methionyl-tRNA synthetase 1 (MetRS1). In both cases, resistance is due to the presence of a second gene that encodes another synthetase (IleRS2 or MetRS2). Here, we show that both S. pneumoniae MetRS2 and S. aureus IleRS2 have closely related homologues in the Gram-positive bacterium Bacillus anthracis, the causative agent of anthrax. Furthermore, similar to drug-resistant pathogens, strains of B. anthracis and its closest relative, B. cereus, also have wild-type ileS1 and metS1 genes. Clostridium perfringens, the causative agent of gangrene, also has two metS genes, whereas Oceanobacillus iheyensis isolated from deep-sea sediments has a single ileS2-type gene. This study shows the importance of understanding complex evolutionary networks of ancient horizontal gene transfer for the development of novel antibiotics.
Introduction
Genomic studies of bacterial pathogens have led to the discovery of many proteins that, in theory, should fulfil the criteria for being novel, broad-spectrum bactericidal targets that are not made invulnerable by known mechanisms of antibiotic resistance (Akerley et al., 2002; Freiberg et al., 2001). Aminoacyl-transfer-RNA synthetases, which function in protein synthesis by attaching an amino acid to its cognate tRNA molecule, are examples of excellent targets for novel classes of antibiotics, as they are essential for bacterial viability, omnipresent, highly divergent between mammals and bacteria, and have well-characterized biochemistries and structures (Woese et al., 2000; Brown, 1998). Pseudomonic acid A (also know as mupirocin), a naturally occurring compound, is a potent inhibitor of the class-I isoleucyl-tRNA synthetase (IleRS) and is used as a topical antibiotic for Staphylococcus aureus infections (Sutherland et al., 1985). More recently, synthetic small-molecule inhibitors of methionyl-tRNA synthetase (MetRS), another class-I synthetase, have been developed, and have potent Gram-positive antibacterial activities (Jarvest et al., 2002).
Horizontal gene transfer (HGT) has had important role in the evolution of aminoacyl-tRNA synthetases (Woese et al., 2000; Brown, 1998) and is also a factor in the occurrence of antibiotic resistance in clinical isolates. Mupirocin-resistant S. aureus strains (minimal inhibitory concentration (MIC) > 512 μg ml−1) usually have a second, divergent copy of IleRS (IleRS2), which can be either plasmid-borne or located in the genome (Dyke et al., 1991; Gilbart et al., 1993; Hodgson et al., 1994). Previous phylogenetic analyses have suggested that S. aureus IleRS2 is closely related to eukaryotic homologues, whereas 'wild-type' IleRS1 clusters with proteins from other low-GC-content Gram-positive bacteria, such as those of the genera Streptococcus, Enterococcus and Bacillus (Brown et al., 1998). However, recent HGT between S. aureus and a eukaryote is unlikely, as S. aureus IleRS2 was an outgroup to all known eukaryotes, including simple protists (Brown et al., 1998). Whereas other bacterial species, such as Mycobacterium tuberculosis (Sassanfar et al., 1996), also have ileS2-like genes, none belong to the low-GC-content Gram-positive group, and other than mupirocin- resistant S. aureus strains, no bacterial species have both ileS1 and ileS2 simultaneously. The source organism of S. aureus ileS2 has not been confirmed because the branch-points in that portion of the tree are not well-resolved (Brown et al., 1998).
Profiling of MetRS inhibitors against collections of Streptococcus pneumoniae clinical isolates has identified a few highly resistant strains (Gentry et al., 2003). The chromosomes of these strains all have a second gene, metS2, which encodes a highly diverged MetRS2, which is more closely related to arch-aeal synthetases than to the wild-type locus from S. pneumoniae (Gentry et al., 2003). Here, we determine the evolutionary origins of IleRS2 and MetRS2 and reveal previously undocumented occurrences of these putative antibiotic resistance genes in a diverse array of soil-dwelling and deepsea bacteria. This study is a practical example of the importance of understanding the evolution and diversity of bacterial genomes in the selection and development of novel antimicrobial targets.
Results and Discussion
To determine the evolutionary history of MetRS2 and IleRS2, we searched public databases of both complete and partial genome sequences. Surprisingly, both synthetases had two highly similar homologues in the genomes of Bacillus anthracis (Read et al., 2003) and B. cereus (Ivanova et al., 2003) a near clonal relative. In addition, the completed genome sequence of Clostridium perfringens contains homologues of MetRS1 and MetRS2, but only a typical IleRS1 (Shimizu et al., 2002). Notably, the genome sequence of Oceanobacillus iheyensis contains an IleRS2 homologue similar to that of S. aureus, no IleRS1, and a typical MetRS1 (Takami et al., 2002). All of these species are low-GC-content Gram-positive bacteria. B. anthracis and C. perfringens live in soil and are the causative agents of anthrax and gangrene, respectively. B. cereus is also soil-dwelling, and is an occasional food contaminant (Helgason et al., 2000). On the other hand, O. iheyensis is an alkaliphilic, highly halotolerant bacillus, which was recently isolated from ocean sediments at a depth of 1,050 m (Lu et al., 2001).
As B. anthracis is the only species that has both the antibiotic resistance genes metS2 and ileS2, and because it is a potential bio-weapon agent, comparisons with the respective loci counterparts in S. pneumoniae and S. aureus merit further discussion. Both genes, metS2 and ileS2, are located on the main chromosome, as the sequenced strain has been cured of its two virulence plasmids, pOX1 and pOX2 (Baillie & Read, 2000). Furthermore, homology searches of the complete sequences of both plasmids (Okinaka et al., 1999) with MetRS2, IleRS2 and the amino-acid sequences of all aminoacyl-tRNA synthetases from B. subtilis did not show any significant hits. The amino-acid sequences of the predicted open reading frames (ORFs) of the ileS2 genes of S. aureus and B. anthracis are 61% similar (Fig. 1A). B. anthracis IleRS2 is only 9 amino acids longer than the S. aureus ORF (1,024 amino acids), and this is mainly due to a few extra leading and trailing amino acids and a small insertion in the carboxy-terminal half of the protein. Comparisons of encoding nucleotide sequences show a high level of sequence identity (59%), with the B. anthracis ileS2 gene having a higher GC content (38%) than S. aureus (29%). However, there were no discernable similarities in the DNA sequences of regions flanking the ileS2 gene. Similarly, MetRS2 from S. pneumoniae and B. anthracis have highly conserved amino-acid sequences, as they are 65% similar and differ in their total lengths by only one amino acid (Fig. 1B). Comparisons of encoding nucleotide sequences show a high level of sequence identity (61%), with GC composition being similar between B. anthracis (37%) and S. pneumoniae (32%). Homologous alignments of nucleotides flanking the metS2 locus could only be extended 15 nucleotides upstream and could not be extended downstream. The lack of conserved flanking sequences suggests that metS2 and ileS2 genes were either transferred independently of any known extrachromosomal vector (that is, of any plasmid, prophage or insertional sequence element), or that detectable flanking-region DNA homology has been erased by genetic drift.
Figure 1.

Alignments of horizontally transferred aminoacyl-transfer-RNA synthetases from anthrax and Gram-positive cocci. (A) Isoleucyl-transefer-RNA synthetases (IleRS2s) from Bacillus anthracis (Baan_ileS2) and Staphylococcus aureus (Stau_ileS2), which is resistant to the antibiotic mupirocin. Identical residues are shaded. (B) Methionyl-tRNA synthetases (MetRS2s) from B. anthracis (Baan_metS2) and Streptococcus pneumoniae (Stpn_metS2), which is resistant to MetRS inhibitory compounds. Letters above the alignments indicate the positions of the HIGH and KMSKS signature motifs of class I tRNA synthetases.
Phylogenetic trees constructed using various methods all support a monophyletic group of IleRS2 proteins from B. anthracis, B. cereus, O. iheyensis and S. aureus (Fig. 2). The overall position of the IleRS2 clade, which is intermediate between the Archaea and eukaryotes, is consistent with previous analyses (Brown et al., 1998), although more ileS2-type genes have now been discovered in other species. The intermixing of archaeal and bacterial lineages suggests that HGT has been extensive for this gene. However, other than B. anthracis and mupirocin-resistant S. aureus, no species show the simultaneous occurrence of IleRS1 and IleRS2. B. anthracis IleRS1 clusters strongly with homologues from three other Bacillus species, as well as those of other low-GC-content Gram-positive bacteria, including cocci.
Figure 2.
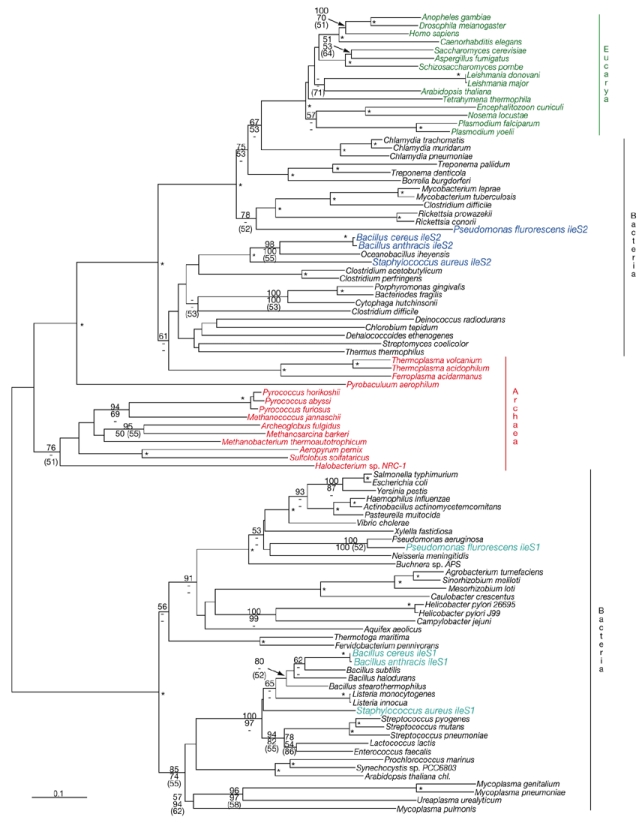
Phylogeny of isoleucyl-transfer-RNA synthetase protein sequences. Those species with genes for both isoleucyl-transfer-RNA synthetase 1 (IleRS1) and IleRS2 (Staphylococcus aureus, Pseudomonas flurorescens, Bacillus anthracis and B. cereus) are indicated in light and dark blue, respectively. Eukaryotic (green), archaeal (red) and bacterial (black) species are also indicated. Trees were constructed using the neighbour-joining (NJ) method (http://evolution.genetics.washington.edu/phylip.html). Numbers along the branches show the percentage occurrence of nodes in 1,000 bootstrap replicates of NJ (numbers in plain text) and maximum parsimony (MP; italicized numbers) analyses (Swofford, 1999) or greater than 50% of 1,000 maximum likelihood (ML; Strimmer and von Hasseler, 1996) quartet puzzling steps (numbers in parentheses). Asterisks represent support for nodes in more than 70% of NJ and MP bootstrap replications and 60% of ML puzzling steps. Scale bar, 0.1 estimated amino-acid substitutions per site.
Phylogenetic trees of MetRS support the monophyly of C. perfringens, B. anthracis, B. cereus and S. pneumoniae MetRS2 (Fig. 3). The MetRS tree topology is generally similar to the IleRS tree in having various bacterial species with eukaryotic or archaeal metS2-like genes. Similar to the occurrence of IleRS, no species other than these four have simultaneously occurring metS1 and metS2 genes. As well as amino-acid substitutions, all MetRS2-type proteins, including those of C. perfringens, B. anthracis, B. cereus and S. pneumoniae, differ from MetRS1-type proteins in having an inserted region of ∼27 amino acids between residues 151 and 178 of Escherichia coli. In the MetRS1 portion of the tree, all four species cluster with other low-GC-content Gram-positive bacteria.
Figure 3.
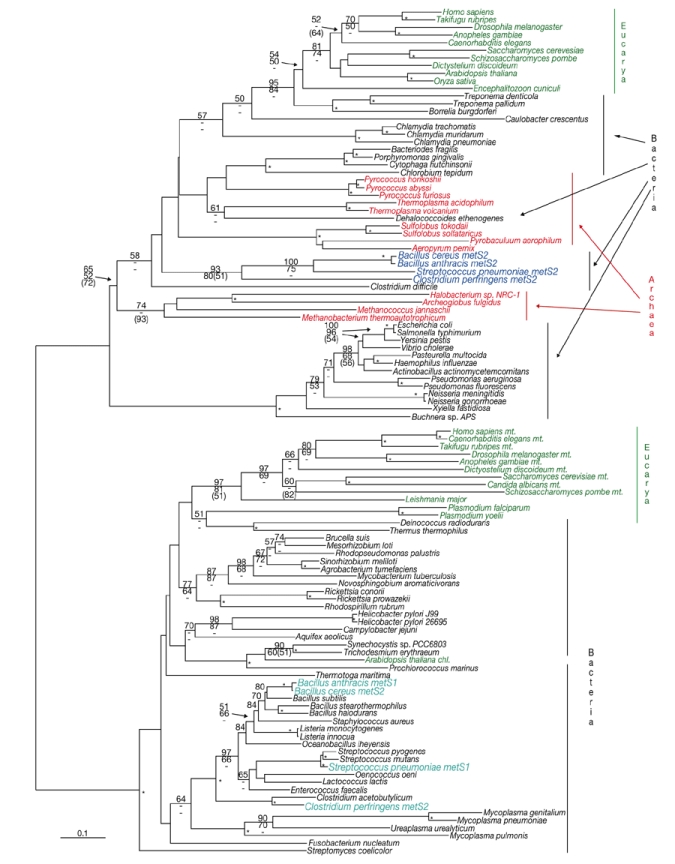
Phylogeny of methionyl-transfer-RNA synthetase protein sequences. Species with genes for both methionyl-transfer-RNA synthetase 1 (MetRS1) and MetRS2 (Streptococcus pneumoniae, Bacillus anthracis, B. cereus and Clostridium perfringens) are indicated (in light and dark blue, respectively). Phylogenetic-tree reconstruction methods and nomenclature are given in the legend to Fig. 2. For some eukaryotic species, mitochondria-targeted (mt.) or chloroplast-targeted (chl.) MetRS isoforms have been identified. Scale bar, 0.1 estimated amino-acid substitutions per site.
To determine whether the existence of two ileS and metS genes occur widely in B. anthracis strains, we carried out a PCR-based survey of eight strains of B. cereus, a close relative of B. anthracis (Helgason et al., 2000), for the genes ileS1, ileS1, metS1 and metS2, using locispecific primers. For all strains, DNA fragments longer than 900 bp corresponding to each of the four genes were amplified. The identities of most fragments were confirmed by DNA sequencing. Gene orthologues from B. anthracis and B. cereus loci had 92–98% DNA sequence identity. Furthermore, BLAST searches (Altschul et al., 1997) of all four sequenced B. anthracis strains (A2012, Ames, KrugerB and WesternNA; Read et al., 2002, 2003) consistently showed ileS1/ileS2 and metS1/metS2 gene pairs. Given the overall intraspecific genetic diversity of B. cereus and B. anthracis (Keim et al., 1999), the occurrence of two copies of IleRS and MetRS-encoding genes are probably universal in these species.
By contrast, occurrences of the ileS2 gene in S. aureus and of the metS2 gene in S. pneumoniae are highly strain-specific. Published complete genome sequences of two strains each of S. aureus (Kuroda et al., 2001) and S. pneumoniae (Hoskins et al., 2001; Tettelin et al., 2001) do not have either ileS2 or metS2. Furthermore, a PCR survey showed that the metS2 locus occurs in 144 of 315 clinical S. pneumoniae isolates tested, and a copy of the metS1 gene is present in all isolates (Gentry et al., 2003). Species of the B. anthracis/B. cereus group may have transferred ileS2 and metS2 genes into different strains of S. aureus or S. pneumoniae, respectively, or possibly into an ancestral lineage of both of these species, followed by a series of gene losses during the subsequent diversification of strains within each taxon. Alternatively, ileS2 and metS2 genes might have been acquired by the ancestral species of B. anthracis and B. cereus from the respective Gram-positive coccal species, only to be lost several times from different strains of the host species.
Speculation
Circumstantial evidence suggests that resistance to naturally produced antibiotics was the selection agent for the HGT of these genes. Mupirocin is a naturally produced antibiotic, which is also known as pseudomonic acid A (Fuller et al., 1971). Plasmids that confer high-level resistance to mupirocin were isolated nearly twenty years before the clinical use of this drug (Rahman et al., 1990). However, the MetS class of inhibitors are synthetic experimental compounds that have never been clinically used (Jarvest et al., 2002). Therefore, recent acquisition due to clinical use of antibiotics is unlikely. Our results suggest that some strains of Gram-positive coccal pathogens have recently shared genes with B. cereus and B. anthracis species. Furthermore, metS1 and metS2 occur in C. perfringens, another soil-dwelling Gram- positive bacterium. Past HGT events between different groups of species, perhaps facilitated by co-habitation of hosts and/or soil environments, are now becoming apparent due to genomic sequencing and intensive studies of antibiotic-resistance mechanisms. The environmental connection to O. iheyensis, which harbours an ileS2 gene, is less obvious, as this species was isolated from deepsea sediments at a depth of over 1,000 m. However, our knowledge of bacterial diversity is limited, and newly discovered species might reveal further evolutionary linkages. We suggest that comparative genomic studies of all bacterial species, not only pathogens, potentially have great importance in the prediction of future drug-resistance mechanisms, as well as in the development of new therapies against community pathogens and bacterial agents used for biowarfare.
Methods
Sequence analysis.
The distributions of ileS1, ileS2, metS1 and metS2 were determined from eight B. cereus strains: National Collection of Industrial and Marine Bacteria (NCIMB) strain 11934, American Type Culture Collection (ATCC) strain 2 (AMC 800, NRS 342), and GlaxoSmithKline strains BRL1243, BRL1244, Harlow, I, LDH23125 and 1239. Primers for the amplification of the metS1 and metS2 loci were designed to anneal to DNA sequences conserved between S. pneumoniae and B. anthracis, and ileS1 and ileS2 primers were designed to anneal to nucleotide sequences conserved between S. aureus and B. anthracis. Primer sequences and PCR amplification conditions can be found in the supplementary information online.
Evolutionary analysis.
We found other methionyl-tRNA synthetase and isoleucyl-tRNA synthetase sequences in public databases, including partial genome sequences (NCBI), using S. pneumoniae MetRS2 and S. aureus IleRS2 (GenBank accession numbers AY198311 and P41368, respectively) as query sequences. Translated ORFs and complete DNA sequences were searched using the programs BLASTP and TBLASTN, respectively (Altschul et al., 1997). Preliminary sequence data for B. cereus and B. anthracis strains were obtained from Integrated Genomics (IG; http://www.integratedgenomics.com) and The Institute for Genomic Research (TIGR; http://www.tigr.org), respectively.
We aligned individual protein datasets using the program CLUSTALW v.1.7 with default settings (Thompson et al., 1994). Subsequently, we manually refined multiple sequence alignments using the program SEQLAB from the GCG v10.0 software package (Genetics Computer Group). Regions with residues that could not be unambiguously aligned or that contained insertions or deletions were removed before phylogenetic analyses. The GCG package (OLDDISTANCES program) was used to calculate pairwise amino-acid and nucleotide similarities between B. anthracis proteins and genes and their respective orthologues in S. aureus and S. pneumoniae, based on the length of the shortest sequence, without gaps, and using the BLOSUM62 matrix and DNADISTANCE. The lengths of edited IleRS2 and MetRS2 alignments were 423 and 274 amino acids, respectively.
Phylogenetic trees were constructed using neighbour-joining (NJ), maximum parsimony (MP) and maximum likelihood quartet puzzling (QP) methods. NJ trees were based on pairwise distances between amino-acid sequences that were calculated using the Dayhoff matrix implemented in the PHYLIP 3.6 package (http://evolution.genetics.washington.edu/phylip.html). Confidence limits of phylogenetic branching points in NJ and MP analyses were assessed from 1,000 bootstrap replications. MP was carried out using the software package PAUP 4.0b5 (Swofford, 1999). The numbers and lengths of minimal MP trees were estimated from 100 random sequence additions. ML tree topologies were constructed using the software PUZZLE 4.0 (Strimmer & von Hasseler, 1996), using 1,000 puzzling steps, the JTT substitution matrix, estimation of rate heterogeneity using the gamma distribution model with eight rate categories, and the α-parameter estimation from the dataset.
For IleRS, there were six minimal-length MP trees, 7,619 steps in length with a consistency index (CI) of 0.3336 and a retention index (RI) of 0.6498, and all trees had a clade consisting of B. anthracis, B. cereus, O. iheyensis and S. aureus IleRS2. For MetRS, 14 minimal-length MP trees were recovered, all 6,562 steps in length with a CI of 0.3153 and a RI of 0.6307, and all trees had a clade consisting of C. perfringens, B. anthracis, B. cereus and S. pneumoniae MetRS2. See supplementary information online for multiple-sequence alignments.
GenBank accession numbers.
B. cereus sequences were deposited in GenBank under the accession numbers AY280849–AY280855 and AY282419–AY282436.
Supplementary information is available at EMBO reports online (http://www.nature.com/embor/journal/vaop/ncurrent/extref/4-embor881-s1.pdf http://www.nature.com/embor/journal/vaop/ncurrent/extref/4-embor881-s2.pdf http://www.nature.com/embor/journal/vaop/ncurrent/extref/4-embor881-s3.pdf).
Supplementary Material
supplementary information
supplementary information
Supplementary information
Acknowledgments
We thank W. Marshall, R. Nelson, M. Italia and J. Peterson for software support, and R. Jarvest, for his comments on the manuscript. The support of GlaxoSmithKline R&D, Genetics Research, Bioinformatics Division is gratefully acknowledged.
References
- Akerley B.J., Rubin E.J., Novick V.L., Amaya K., Judson N. & Mekalanos J.J. (2002) A genomescale analysis for identification of genes required for growth or survival of Haemophilus influenzae. Proc. Natl Acad. Sci. USA, 99, 966–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W. & Lipman D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillie L. & Read T.D. (2000) Bacillus anthracis, a bug with attitude! Curr. Opin. Microbiol., 4, 78–91. [DOI] [PubMed] [Google Scholar]
- Brown J.R. (1998) in Thermophiles—the Keys to Molecular Evolution and the Origin of Life? (eds Wiegel, J. & Adams, M.), 217–230. Taylor & Francis Group Ltd., London, UK. [Google Scholar]
- Brown J.R., Zhang J. & Hodgson J.E. (1998) A bacterial antibiotic resistance gene with eukaryotic origins. Curr. Biol., 8, R365–R367. [DOI] [PubMed] [Google Scholar]
- Dyke K.G.H., Curnock S.P., Golding M. & Noble W.C. (1991) Cloning of the gene conferring resistance to mupirocin in Staphylococcus aureus. FEMS Microbiol. Lett., 77, 195–198. [DOI] [PubMed] [Google Scholar]
- Freiberg C., Wieland B., Spaltmann F., Ehlert K., Brötz H. & Labischinski H. (2001) Identification of novel essential Escherichia coli genes conserved among pathogenic bacteria. J. Mol. Microbiol. Biotechnol., 3, 483–489. [PubMed] [Google Scholar]
- Fuller A.T., Mellows G., Woolford M., Banks G.T., Barrow K.D. & Chain E.B. (1971) Pseudomonic acid: an antibiotic produced by Pseudomonas fluorescens. Nature, 234, 416–417. [DOI] [PubMed] [Google Scholar]
- Gentry D.R., Ingraham K.A., Stanhope M.J., Rittenhouse S., Jarvest R.L., O'Hanlon P.J., Brown J.R. & Holmes D.J. (2003) Variable sensitivity to bacterial methionyl-tRNA synthetase inhibitors reveals sub populations of Streptococcus pneumoniae with two distinct methionyl tRNA synthetase genes. Antimicrob. Agents Chemother., 47, 1784–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbart J., Perry C.R. & Slocombe B. (1993) High-level mupirocin resistance in Staphylococcus aureus: evidence for two distinct isoleucyl-tRNA synthetases. Antimicrob. Agents Chemother., 37, 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helgason E.O., Økstad A., Caugant D.A., Johansen H.A., Fouet A., Mock M., Hegna I. & Kolstø A.B. (2000) Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis—one species on the basis of genetic evidence. Appl. Environ. Microbiol., 66, 2627–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson J.E., Curnock S.P., Dyke K.G., Morris R., Sylvestor D.R. & Gross M.S. (1994) Molecular characterization of the gene encoding high-level mupirocin resistance in Staphylococcus aureus J2870. Antimicrob. Agents Chemother., 38, 1205–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins J.A. et al. (2001) The genome of the bacterium Streptococcus pneumoniae strain R6. J. Bacteriol., 183, 5709–5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova N. et al. (2003) Genome sequence of Bacillus cereus and comparative analysis with Bacillus anthracis. Nature, 423, 87–91. [DOI] [PubMed] [Google Scholar]
- Jarvest R.L. et al. (2002) Nanomolar inhibitors of Staphylococcus aureus methionyl tRNA synthetase with potent antibacterial activity against Gram positive pathogens. J. Med. Chem., 45, 1959–1962. [DOI] [PubMed] [Google Scholar]
- Keim P., Klevytska A.M., Price L.B., Schupp J.M., Zinser G., Smith K.L., Hugh-Jones M.E., Okinaka R., Hill K.K. & Jackson P.J. (1999) Molecular diversity in Bacillus anthracis. J. Appl. Microbiol., 87, 215–217. [DOI] [PubMed] [Google Scholar]
- Kuroda M. et al. (2001) Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet, 357, 1225–1240. [DOI] [PubMed] [Google Scholar]
- Lu J., Nogi Y. & Takami H. (2001) Oceanobacillus iheyensis gen. nov., sp. nov., a deepsea extremely halotolerant and alkaliphilic species isolated from a depth of 1050 m on the Iheya Ridge. FEMS Microbiol. Lett., 205, 291–297. [DOI] [PubMed] [Google Scholar]
- Okinaka R.T. et al. (1999) Sequence and organization of pOX1, the large Bacillus anthracis plasmid harboring the anthrax toxin genes. J. Bacteriol., 181, 6509–6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman M., Connolly S., Noble W.C., Cookson B. & Phillips I. (1990) Diversity of staphylococci exhibiting high-level resistance to mupirocin. J. Med. Microbiol., 33, 97–100. [DOI] [PubMed] [Google Scholar]
- Read T.D. et al. (2002) Comparative genome sequencing for discovery of novel polymorphisms in Bacillus anthracis. Science, 296, 2028–2033. [DOI] [PubMed] [Google Scholar]
- Read T.D. et al. (2003) The genome sequence of Bacillus anthracis Ames and comparison to closely related bacteria. Nature, 423, 81–86. [DOI] [PubMed] [Google Scholar]
- Sassanfar M., Kranz J.E., Gallant P., Schimmel P. & Shiba K. (1996) A eubacterial Mycobacterium tuberculosis tRNA synthetase is eukaryote-like and resistant to a eubacterialspecific antisynthetase drug. Biochemistry, 35, 9995–10003. [DOI] [PubMed] [Google Scholar]
- Shimizu T., Ohtani K., Hirakawa H., Ohshima K., Yamashita A., Shiba T., Ogasawara N., Hattori M., Kuhara S. & Hayashi H. (2002) Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. Proc. Natl Acad. Sci. USA, 99, 996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strimmer K. & von Hasseler A. (1996) Quartet puzzling: a quartet maxiumum likelihood method for reconstructing tree topologies. Mol. Biol. Evol., 13, 964–969. [Google Scholar]
- Sutherland R., Boon R.J., Griffin K.E., Masters P.J., Slocombe B. & White A.R. (1985) Antibacterial activity of mupirocin (pseudomonic acid), a new antibiotic for topical use. Antimicrob. Agents Chemother., 27, 495–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford D.L. (1999) PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. Sinauer Associates, Sunderland, Massachusetts, USA. [Google Scholar]
- Takami H., Takaki Y. & Uchiyama I. (2002) Genome sequence of Oceanobacillus iheyensis isolated from the Iheya Ridge and its unexpected adaptive capabilities to extreme environments. Nucleic Acids Res., 30, 3927–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tettelin H. et al. (2001) Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science, 293, 498–506. [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Higgins D.G. & Gibson T.J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positionspecific gap penalties and weight matrix choice. Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese C.R., Olsen G.J., Ibba M. & Söll D. (2000) Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol. Mol. Biol. Rev., 64, 202–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary information
supplementary information
Supplementary information