

Abstract
The tumour suppressor p53 is a tetrameric protein that is phosphorylated in its BOX-I transactivation domain by checkpoint kinase 2 (CHK2) in response to DNA damage. CHK2 cannot phosphorylate small peptide fragments of p53 containing the BOX-I motif, indicating that undefined determinants in the p53 tetramer mediate CHK2 recognition. Two peptides derived from the DNA-binding domain of p53 bind to CHK2 and stimulate phosphorylation of full-length p53 at Thr 18 and Ser 20, thus identifying CHK2-docking sites. CHK2 can be fully activated in trans by the two p53 DNA-binding-domain peptides, and can phosphorylate BOX-I transactivation-domain fragments of p53 at Thr 18 and Ser 20. Although CHK2 has a basal Ser 20 kinase activity that is predominantly activated towards Thr 18, CHK1 has constitutive Thr 18 kinase activity that is predominantly activated in trans towards Ser 20. Cell division cycle 25C (CDC25C) phosphorylation by CHK2 is unaffected by the p53 DNA-binding-domain peptides. The CHK2-docking site in the BOX-V motif is the smallest of the two CHK2 binding sites, and mutating certain amino acids in the BOX-V peptide prevents CHK2 activation. A database search identified a p53 BOX-I-homology motif in p21WAF1 and although CHK2 is inactive towards this protein, the p53 DNA-binding-domain peptides induce phosphorylation of p21WAF1 at Ser 146. This provides evidence that CHK2 can be activated allosterically towards some substrates by a novel docking interaction, and identify a potential regulatory switch that may channel CHK2 into distinct signalling pathways in vivo.
Introduction
The tumour-suppressor protein p53 is activated as a transcription factor in response to a variety of genotoxic and metabolic stresses, resulting in either cell-cycle arrest or apoptosis (Vogelstein et al., 2000). Well-characterized gene products that mediate the tumour-suppressor function of p53 include the cyclin kinase inhibitor p21WAF1 and the pro-apoptotic BCL2 antagonist BAX. Several functional domains of p53 are involved in promoting transactivation, including an amino-terminal activation domain that binds p300, a core sequence-specific DNA-binding domain, a tetramerization domain and a carboxy-terminal regulatory domain, the phosphorylation and acetylation of which regulates p53-dependent transcription. DNA-damage-activated protein kinases phosphorylate up to three clustered sites in the BOX-I transactivation domain of p53. These protein kinases form part of the evolutionarily conserved ataxia-telangiectasia-mutated (ATM)–checkpoint kinase 2 (CHK2) DNA-damage signalling cascades that activate p53 (Wahl & Carr, 2001). CHK2 can phosphorylate p53 at Thr 18 or Ser 20 (Shieh et al., 2000). Phosphorylation at the Thr 18 site attenuates MDM2 (mouse double minute 2) binding to p53 (Craig et al., 1999; Schon et al., 2002) and relieves p53 from negative control by MDM2. In addition, phosphorylation at Thr 18 or Ser 20 by CHK2 stabilizes p300 binding to the LxxLL activation domain of p53, and promotes DNA-dependent acetylation of p53 by p300 (Dornan et al., 2003). Therefore, phosphorylation of the p53 activation domain seems to act as a switch to convert p53 from an MDM2-binding protein to a p300-binding protein, leading to enhanced DNA-dependent acetylation of p53 at promoters (Lane & Hupp, 2003).
The role of CHK2 in modifying the p53 pathway is controversial. Original studies indicated that CHK2 forms stable complexes with p53 in cells (Falck et al., 2001), deletion of CHK2 reduces the specific activity of p53 (Wu et al., 2002), and CHK2 and p53 cooperate in the DNA-damage response (Hirao et al., 2000; Takai et al., 2002). Sequestration of CHK2 to spatially restricted nuclear regions impairs its ability to stimulate p53 activity (Lukas et al., 2003). However, neither mutation of the murine p53 Chk2 site nor deletion of Chk2 alter p53 stabilization after damage (Takai et al., 2002; Wu et al., 2002). The CHK2 phosphorylation site at Ser 20 is one of the least conserved sites on p53, the transactivation domain of p53 does not have a consensus CHK2 phosphorylation site, and the p53 activation-domain fragment is not a CHK2 substrate (O'Neill et al., 2002), questioning whether CHK2 is a p53 kinase. In this report, we have reconstituted the minimal components required for CHK2 to function as a p53 kinase, and we resolve the issue of how CHK2 can target a protein that does not have a CHK2 consensus phosphorylation site. The phosphorylation of p53 by CHK2 requires kinase docking to a protein–protein interaction module in the DNA-binding domain of p53. These data have led to the identification of an 'activated' CHK2 consensus-site that expands the types of potential CHK2 substrates and highlights the growing realization that protein-kinase docking sites can be an important component of kinase specificity (Biondi & Nebreda, 2003).
Results
Peptide consensus sites for CHK2 that have homology to CDC25, but not to the BOX-I activation domain of p53, have been developed (Fig. 1A). Small peptides from the p53 activation domain were not phosphorylated by CHK2, but CHK2 was an effective kinase for tetrameric p53, and phosphorylated Thr 18 and Ser 20 (Fig. 1B). These data suggest that CHK2 is activated towards p53 by other determinants in the tetramer. Overlapping peptides from p53 (Fig. 1C) were assayed for CHK2 protein binding to identify such CHK2-docking sites. Two high-affinity CHK2 binding regions were identified in the BOX-II (peptide 5) and BOX-V (peptide 18) domains of p53, with a more localized binding site present in the BOX-V domain (Fig. 1C). If CHK2 requires docking to these two sites in the DNA-binding domain of p53 to promote BOX-I-domain phosphorylation, then the peptides might competitively inhibit CHK2 phosphorylation. Surprisingly, however, both of the BOX-II-domain and BOX-V-domain peptides stimulated, rather than inhibited, phosphorylation of p53 at Ser 20 (Fig. 1D, compare lane 2 with lanes 5 and 8). The BOX-I peptide that contained the phosphorylation site for CHK2 did not inhibit p53 phosphorylation (Fig. 1D, compare lanes 2 and 3), consistent with the fact that this small peptide is not phosphorylated by CHK2 (O'Neill et al., 2002; see below). The Thr 18 kinase activity of CHK2 was also stimulated by the BOX-II-domain and BOX-V-domain peptides (Fig. 1E, compare lane 1 with lanes 2 and 3). Using [32Pγ]ATP as a substrate, the BOX-II-domain and BOX-V-domain peptides stimulated phosphorylation of p53 (Fig. 1F, compare lane 1 with lanes 2 and 3, lower band) under conditions in which CHK2 autophosphorylation was unaffected (Fig. 1F, lanes 1–3, upper band). These data indicate that the kinase activity of CHK2 is stimulated by the BOX-II-domain and BOX-V-domain peptides. The human CHK2 used in these assays was purified from insect cells and the enzyme was highly phosphorylated at the ATM-activating site of Thr 68, indicating that CHK2 was in an activated conformation (see supplementary information online).
Figure 1.
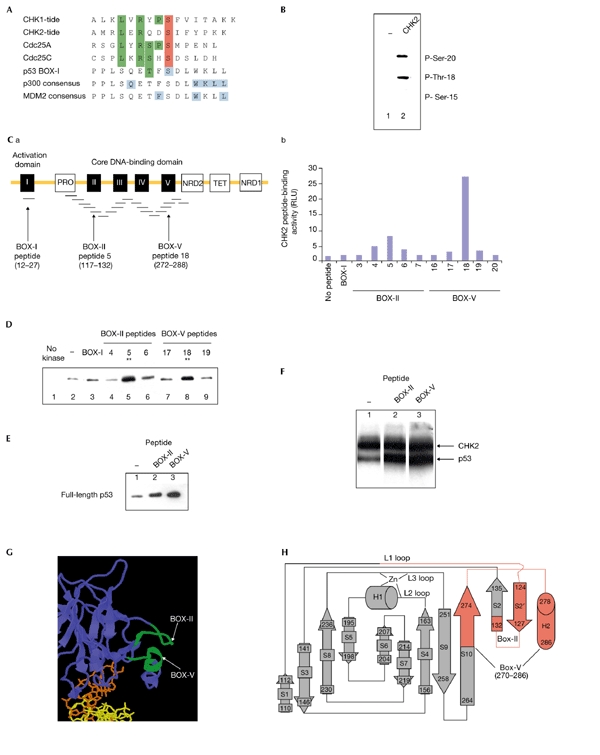
Checkpoint kinase 2 docking to the DNA-binding domain of p53. (A) Low homology between p53 and checkpoint kinase 2 (CHK2) substrates. Consensus phosphorylation sites have been defined for both CHK1 and CHK2 (CHK1-tide and CHK2-tide; O'Neill et al., 2002). The homology of these consensus-site peptides to cell division cycle 25 (CDC25) and p53 are as indicated. The key residues required for p300 and MDM2 (mouse double minute 2) binding to the p53 BOX-I domain (Dornan et al., 2003) are shown in blue. Green shading indicates homolgous sites, red shading indicates phosphoacceptor sites. (B) CHK2 modifies two sites on p53. An immunochemical assay using phospho-specific monoclonal antibodies to the BOX-I phosphorylation sites (Craig et al., 1999) was used to assay CHK2. The addition of CHK2 to reactions with p53 tetramers (compare lane 1 with lane 2) results in the specific phosphorylation of p53 at Ser 20 and Thr 18, but not at Ser 15. (C) Two peptides derived from the core domain of p53 bind to CHK2. (Ca) Overlapping peptides derived from p53 were tested for binding to CHK2. The five conserved BOX domains of p53 are shown in black. The CHK2 phosphorylation sites are in the BOX-I domain, whereas the CHK2-docking peptides are in the BOX-II and BOX-V domains. (Cb) Direct binding of CHK2 was tested for all p53 peptides, and relative binding of CHK2 to the BOX-II and BOX-V peptides in an ELISA (enzyme-linked immunosorbent assay) format (ELISA method as in Dornan et al., 2003) is shown as relative light units (RLUs). (D,E) The BOX-II-domain and BOX-V-domain peptides stimulate CHK2 activity. Kinase reactions containing p53 were assembled without CHK2 (lane 1) or with CHK2 (lanes 2–9). The indicated overlapping peptides from the BOX-I, BOX-II and BOX-V domains were added, and reaction products were analysed for p53 phosphorylation at Ser 20 (D) and Thr 18 (E) using an immunochemical blotting assay. (F) The BOX-II-domain and BOX-V-domain peptides stimulate CHK2 activity. Kinase reactions containing [32Pγ]ATP, full-length p53 and CHK2 were assembled without peptide (lane 1) or with the indicated peptides from BOX-II and BOX-V (lanes 2 and 3). Reaction products were analysed for p53 phosphorylation after electrophoresis by autoradiography. The upper band shows CHK2 autophosphorylation and the lower band shows p53 phosphorylation. (G) The minimal adjacent locations of the BOX-II-domain and BOX-V-domain peptides (in the S2′ and H2 motifs) within the core domain of p53 are shown in green. (H) The entire BOX-II and BOX-V peptide sequences that bind CHK2 (C) extend further into the core domain and reside within the same plane (shown in red, including part of the S10 β-sheet, the S2 and S2' β-sheets and the helix H2).
The BOX-II-domain and BOX-V-domain peptides are not contiguous in the primary sequence of p53 (Fig. 1C), but reside on the same face in the crystal structure of p53, which may form the basis for a three-dimensional docking site for CHK2 (Fig. 1G,H). Protein kinases, including phosphoinositide-dependent protein kinase 1 (PDK1), mitogen-activated protein kinase (MAPK), and glycogen synthase kinase 3 (GSK3) also have high-affinity docking sites that are distinct from the substrate phosphorylation site (Biondi & Nebreda, 2003). Two mechanisms may account for CHK2 stimulation after binding the BOX-II-domain and BOX-V-domain peptides (Fig. 2A): first, CHK2 docking to a high-affinity site might tether the kinase to p53, allowing it to phosphorylate a low-affinity, non-consensus phosphorylation site. Second, CHK2 might be activated as a p53 kinase after docking by an allosteric or induced-fit mechanism. The effect of these CHK2-docking peptides in trans on the phosphorylation of a small glutathione-S-transferase (GST)-tagged N-terminal fragment of p53 (amino acids 1–66; p53N1–66) that lacks the CHK2-docking sites was examined (Fig. 2B). A [32Pγ]ATP kinase assay showed some basal CHK2-dependent 32P incorporation into p53N1–66 (Fig. 2B, lane 1). However, the inclusion of BOX-II-domain or BOX-V-domain peptides stimulated the phosphorylation of p53N1–66 (Fig. 2B, compare lane 1 with lanes 2 and 3). Under these conditions, the basal autophosphorylation of CHK2 was unaffected (Fig. 2B, compare lane 1 with lanes 2 and 3; CHK2 band). The inclusion of both CHK2-docking peptides together induced stoichiometric phosphorylation of p53N1–66 by CHK2 (Fig. 2C, compare lanes 3 and 4 with lane 5), without altering the basal autophosphorylation of CHK2 (Fig. 2C, compare lane 1 with lane 5, CHK2 band). This multi-peptide addition effectively reconstituted CHK2 activity towards a p53 fragment that lacks the CHK2-docking sites, and indicates that allosteric effects mediate CHK2 phosphorylation of p53 (Fig. 2A, model 2). The specific activity of CHK2 and 'peptide-activated' CHK2 was tested towards its classic substrate, CDC25C. A [32Pγ]ATP kinase assay showed equivalent phosphorylation of CDC25C by either CHK2 (Fig. 2D, lane 2) or 'peptide-activated' CHK2 (Fig. 2D, compare lane 2 with lanes 3–5).
Figure 2.
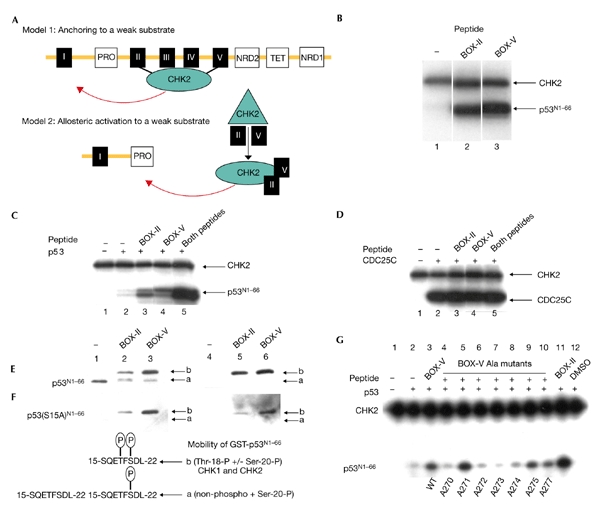
Checkpoint kinase 2 is activated in trans by p53 DNA-binding-domain peptides. (A) Two models account for the stimulation of checkpoint kinase 2 (CHK2) by the BOX-II-domain and BOX-V-domain peptides. According to the anchoring model (model 1), the BOX-II-domain and BOX-V-domain peptides only anchor CHK2 and should have no effect in trans on CHK2 activity towards the p53 fragments. Alternatively, according to the allosteric model (model 2), the BOX-II-domain and BOX-V-domain peptides can activate CHK2 towards the p53 activation-domain fragment that lacks the CHK2-core domain sites. (B) The BOX-II-domain and BOX-V-domain peptides activate CHK2. Kinase reactions containing [32Pγ]ATP, p53N1–66 and CHK2 were assembled without peptide (lane 1) or with the indicated peptides from BOX-II and BOX-V (lanes 2 and 3). Reaction products were analysed after electrophoresis by autoradiography for p53N1–66 phosphorylation and CHK2 autophosphorylation. The upper band shows CHK2 autophosphorylation and the lower band shows p53N1–66 phosphorylation. (C) The BOX-II-domain and BOX-V-domain peptides synergistically activate CHK2. Kinase reactions contained [32Pγ]ATP and CHK2 only (lane 1), or contained [32Pγ]ATP, CHK2 and p53N1–66 either without peptide (lane 2), with the indicated peptides from BOX-II and BOX-V (lanes 3 and 4), or with both peptides from BOX-II and BOX-V (lane 5). Reaction products were analysed after electrophoresis by autoradiography for p53N1–66 phosphorylation (lower band) and for CHK2 autophosphorylation (upper band). (D) The BOX-II-doman and BOX-V-domain peptides do not affect CHK2 phosphorylation of CDC25. Kinase reactions contained [32Pγ]ATP and CHK2 only (lane 1), or contained [32Pγ]ATP, CHK2 and CDC25C either without peptide (lane 2), with the indicated peptides from BOX-II and BOX-V (lanes 3 and 4), or with both peptides from BOX-II and BOX-V (lane 5). Reaction products were analysed for CDC25 phosphorylation (lower band) and CHK2 autophosphorylation (upper band) after electrophoresis by autoradiography. (E) The BOX-II-domain and BOX-V-domain peptides activate CHK2 phosphorylation of p53 at Thr 18. Kinase reactions containing p53N1–66 and CHK2 were assembled without peptide (lanes 1 and 4) or with peptides (lanes 2, 3, 5 and 6). Reaction products were analysed for p53N1–66 phosphorylation at Ser 20 (lanes 1–3) or Thr 18 (lanes 4–6) using an immunochemical blotting assay. Protein band 'a' is unphosphorylated p53N1–66 or p53N1–66 phosphorylated at Ser 20, whereas protein band 'b' is a kinase supershift that indicates Thr 18 phosphorylation. (F) The BOX-II-domain and BOX-V-domain peptides activate CHK2 phosphorylation of p53(S15A)N1–66 at Ser 20 and Thr 18. Kinase reactions containing p53(S15A)N1–66 and CHK2 were assembled without peptide (lanes 1 and 4) or with peptides (lanes 2, 3, 5 and 6). Reaction products were analysed for p53 phosphorylation at Ser 20 (lanes 1–3) or Thr 18 (lanes 4–6) phosphorylation using an immunochemical blotting assay. (G) Mutation of the BOX-V peptide attenuates its activity as a CHK2 activator. Reactions were assembled with p53N1–66, [32Pγ]ATP, CHK2 and the BOX-V or BOX-II peptides, as indicated in the figure. BOX-V peptides containing alanine mutations at the indicated codons (270–277) were added, and the relative phosphorylation of p53 and CHK2 is indicated.
The activation of CHK2 by BOX-II-domain and BOX-V-domain peptides towards p53N1–66 was examined using the immunochemical Ser 20 and Thr 18 phosphorylation assay. CHK2 showed basal Ser 20 kinase activity (Fig. 2E, lane 1), whereas no residual activity was detected towards Thr 18 (Fig. 2E, lane 4). The BOX-II and BOX-V peptides stimulated Ser 20 phosphorylation (Fig. 2E, lanes 2 and 3) and activated Thr 18 kinase activity (Fig. 2E, lanes 5 and 6). A conformational change induced by activated CHK2 on p53N1–66 phosphorylation at Thr 18 is inferred by the mobility shift from band 'a' to band 'b' in Fig. 2E. Together with the results from the 32P kinase assay above, these data indicate that the predominant site of phosphorylation by activated CHK2 is at Thr 18. Although there is no contaminating Ser 15 kinase in CHK2 (Fig. 1B), p53(S15A)N1–66 was also used as a substrate, as this eliminates any contribution of residual Ser 15 phosphorylation in driving Thr 18 phosphorylation. The BOX-II and BOX-V peptides activated CHK2 towards both the Ser 20 and Thr 18 sites of p53 (protein band 'b' in Fig. 2F; compare lanes 2 and 3, and 5 and 6, respectively, with lanes 1 and 4). However, the Ser 15 residue seems to be important for CHK2 phosphorylation at Ser 20, as CHK2 has no basal Ser 20 kinase activity towards p53(S15A)N1–66. CHK2 immunoprecipitated from irradiated human cells is modified on Thr 68 (see supplementary information online) and can also be activated towards the Thr 18 site of p53N1–66 by the BOX-V peptide (see supplementary information online), indicating that wild-type CHK2 has biochemical characteristics similar to recombinant CHK2.
The related enzyme CHK1 was also tested for the presence of a p53-docking activity to determine whether the allosteric effector site could be expanded to a related CHK2 family member (see supplementary information online). CHK1 shows constitutive Thr 18 kinase activity towards p53N1–66 or p53(S15A)N1–66, whereas CHK1 is not active as a Ser 20 kinase on p53 fragments unless bound by the BOX-II or BOX-V peptides. These data indicate that the allosteric effect of the p53 DNA-binding domain peptides can be extended to two calcium–calmodulin-kinase superfamily members: CHK2 has basal Ser 20 kinase activity and is predominantly activated towards Thr 18, whereas CHK1 has constitutive Thr 18 kinase activity and is predominantly activated towards Ser 20.
As the BOX-V-domain peptide is the most potent CHK2 and CHK1 activator under limiting conditions (Fig. 2F; and see supplementary information online) and represents a relatively small CHK2-docking interface (Fig. 1C), the effects of individual amino-acid mutations on the BOX-V-peptide activation of CHK2 were examined. The mutation of amino acids to alanine at positions 270, 272, 273, 274 and 277 in the S10 β-sheet (Fig. 2G, compare lane 4 and lanes 6–8 with lane 1) attenuated CHK2 activation by the BOX-V peptide. This region is adjacent to the binding site of the RNA-MDM2 isoform (Shimizu et al., 2002) and suggests that this domain may be a multiprotein-binding site. Although mutation of the BOX-V docking sites of CHK2 prevents p53 phosphorylation (Fig. 2G), mutating single amino acids on full-length p53 did not block phosphorylation by CHK2 (data not shown), presumably because of the relatively large docking interface of CHK2 on p53 (Fig. 1H).
The ability of CHK2 to alter its phosphorylation activity depending on enzyme docking opens the door to identifying novel BOX-I-domain-like CHK2 substrates. CHK2 cannot phosphorylate the minimal BOX-I-domain peptide (amino acids 12–27; Fig. 3B, left bar), but the specific activity of CHK2 was increased by more than 16-fold by the inclusion of BOX-II-domain and/or BOX-V-domain peptides (Fig. 3B, right bars). These data indicate that CHK2 can be stimulated towards a minimal BOX-I-peptide motif. A database search for proteins with homology to the p53 BOX-I domain has identified the cyclin-dependent kinase (CDK) inhibitor p21WAF1 (Fig. 3A). This region of p21WAF1 is a regulatory domain, the phosphorylation of which at Ser 146 by atypical protein kinase Cs (PKCs) can regulate the half-life of the protein in insulin-stimulated cells (Scott et al., 2002). The Ser 146 residue of p21WAF1 and the Thr 18 position of p53 are equivalent in the homology line-up. We examined whether CHK2 was inactive towards p21WAF1 and whether BOX-II-domain and BOX-V-domain peptides activated CHK2 towards the Ser 146 site of p21WAF1. Although CHK2 activity was latent towards p21WAF1 protein (Fig. 3C, lane 2), the BOX-II and BOX-V peptides activated CHK2 towards p21WAF1 (Fig. 3C, compare lane 2 with lanes 3 and 4). The combined addition of both BOX-II-domain and BOX-V-domain peptides synergistically activated CHK2 towards p21WAF1 (Fig. 3C, compare lanes 3 and 4 with lane 5). Thus, proteins with BOX-I-homology-domain motifs can be CHK2 substrates after docking-mediated kinase activation.
Figure 3.
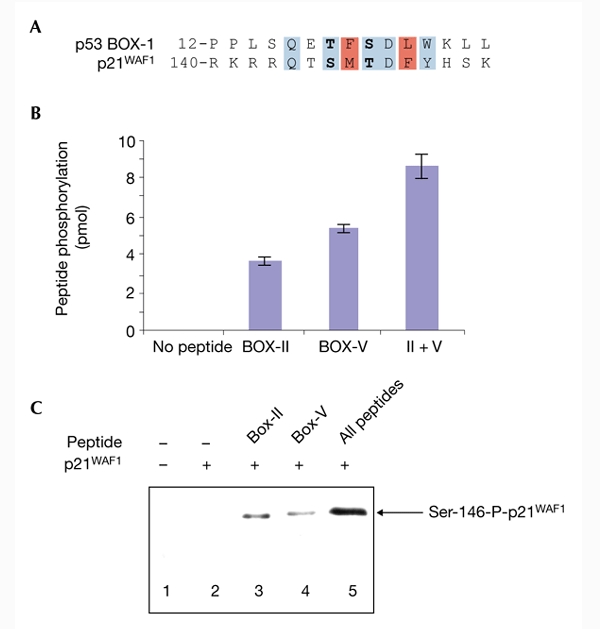
p53-core-domain peptides induce checkpoint-kinase-2-dependent phosphorylation of p21WAF1. (A) Homology of the p53 BOX-I-homology domain to p21WAF1. A database search of proteins with homology to the BOX-I domain of p53 identified p21WAF1. The Thr 18 and Ser 20 phosphoacceptor sites of p53 (bold) correspond to the Ser 146 and Thr 148 phosphoacceptor sites of p21WAF1. (B) The BOX-II-domain and BOX-V-domain peptides activate checkpoint kinase 2 (CHK2) phosphorylation of a BOX-I domain peptide. Kinase reactions contained the p53 BOX-I peptide, [32Pγ]ATP and CHK2 (all bars), either without CHK2-docking peptides (left bar) or with the BOX-II-domain and BOX-V-domain peptides, individually or together (II + V; right bars). Reaction products were spotted onto filter paper; kinase activity is represented as picomoles of 32P incorporation (Scott et al., 2002). (C) CHK2 activation towards p21WAF1. The BOX-II-domain and BOX-V-domain peptides synergistically activate CHK2 phosphorylation of p21WAF1. Kinase reactions contained [32Pγ]ATP and CHK2 only (lane 1), or [32Pγ]ATP, CHK2 and p21WAF1 with or without the indicated peptides from BOX-II and BOX-V (lanes 2–5). Reaction products were analysed after electrophoresis by autoradiography for p21WAF1 phosphorylation (Scott et al., 2002).
Conclusion
The substrates of protein kinases have classically been defined by the ability of a small fragment derived from the full-length protein to serve as an effective substrate in a phosphotransferase reaction. However, growing evidence indicates that protein kinases have high-affinity docking sites that catalyse substrate phosphorylation. Such docking sites have been observed previously for CDK2 (Luciani et al., 2000), PDK1, MAPK and GSK3 (Biondi & Nebreda, 2003). The docking site for a protein kinase consists of a small, linear sequence of amino acids that forms the basis for an anchoring motif that can control substrate phosphorylation. A docking site usually has no sequence homology to the substrate of the kinase, can be attached to the substrate itself or on an adaptor protein, and can itself be a site of phosphorylation. CHK2 action after DNA damage is related, at least in part, to the direct phosphorylation of p53. However, p53 does not have a canonical CHK2 phosphorylation site (O'Neill et al., 2002). The identification of a CHK2-docking site in p53 identifies an allosteric mechanism for CHK2 that may have widespread significance for understanding CHK2 kinase function, substrate specificity determination and cell-cycle checkpoint regulation.
Methods
Reagents, proteins, antibodies and enzyme assays.
All chemicals were from Sigma unless otherwise indicated. Tetrameric forms of p53 were expressed as previously reported (Luciani et al., 2000). The CHK2 and CHK1 complementary DNAs were a gift from S. Elledge. The p53N1–66 fusion protein (GST-tagged) was generated by amplifying the cDNA encoding the N-terminal 66 amino acids of p53 and subcloning this into the isopropyl-β-D-thiogalactoside (IPTG)-inducible expression vector pGEX-2TK (Pharmacia). CHK2 and CHK1 were cloned into pFASTBAC-HTb (which contains a His6 tag) and His-tagged CHK2 and CHK1 were expressed in and purified from Sf9 cells using Ni2+ agarose and ion-exchange chromatography. Peptides derived from the p53 coding region were obtained from Chiron Mimitopes. p53 tetramer or peptide kinase assays were carried out as described previously (Luciani et al., 2000), adding empirically determined amounts of CHK2 and p53 (400 ng), p53N1–66 and p53(S15A)N1–66 (1 μg) or CDC25 (1 μg). Immunoblotting for Thr 18 and Ser 20 phosphorylation was performed using the monoclonal antibodies FPS18 and FPS20 (Craig et al., 1999). Antibodies to CHK2 and the Thr-68-modified CHK2 were obtained from Santa Cruz Biotechnology. p21 protein was purified as indicated and the Ser-146-phospho-specific antibody was used as a probe for p21 phosphorylation (Scott et al., 2002).
Supplementary information is available at EMBO reports online (http://www.nature.com/embor/journal/vaop/ncurrent/extref/4-embor901-s1.pdf).
Supplementary Material
Supplementary information online
Acknowledgments
K.L.B. is supported by a Cancer Research UK Senior Fellowship and by the Association for International Cancer Research. T.R.H. is supported by a Programme Grant from Cancer Research UK, a UK Medical Research Council Career Establishment Grant, and the Association for International Cancer Research.
References
- Biondi R. & Nebreda N. ( 2003) Signaling specificity of Ser/Thr protein kinases through docking site mediated interactions. Biochem. J., 371, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig A.L., Burch L., Vojtesek B., Mikutowska J., Thompson A. & Hupp T.R. ( 1999) Novel phosphorylation sites of human tumour suppressor protein p53 at Ser20 and Thr18 that disrupt the binding of mdm2 protein are modified in human cancers. Biochem. J., 342, 133–141. [PMC free article] [PubMed] [Google Scholar]
- Dornan D., Shimizu H., Perkins N.D. & Hupp T.R. ( 2003) DNA-dependent acetylation of p53 by the transcription coactivator p300. J. Biol. Chem., 278, 13431–13441. [DOI] [PubMed] [Google Scholar]
- Falck J., Lukas C., Protopopova M., Lukas J., Selivanova G. & Bartek J. ( 2001) Functional impact of concomitant versus alternative defects in the Chk2–p53 tumour suppressor pathway. Oncogene, 20, 5503–5510. [DOI] [PubMed] [Google Scholar]
- Hirao A., Kong Y.Y., Matsuoka S., Wakeham A., Ruland J., Yoshida H., Liu D., Elledge S.J. & Mak T.W. ( 2000) DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science, 287, 1824–1827. [DOI] [PubMed] [Google Scholar]
- Lane D.P. & Hupp T.R. ( 2003) Drug discovery and p53. Drug Discov. Today, 8, 347–355. [DOI] [PubMed] [Google Scholar]
- Lukas C., Falck J., Bartkova J., Bartek J. & Lukas J. ( 2003) Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nature Cell Biol., 5, 255–260. [DOI] [PubMed] [Google Scholar]
- Luciani M.G., Hutchins J.R., Zheleva D. & Hupp T.R. ( 2000) The C-terminal regulatory domain of p53 contains a functional docking site for cyclin A. J. Mol. Biol., 300, 503–518. [DOI] [PubMed] [Google Scholar]
- O'Neill T., Giarratani L., Chen P., Iyer L., Lee C.H., Bobiak M., Kanai F., Zhou B.B., Chung J.H. & Rathbun G.A. ( 2002) Determination of substrate motifs for human Chk1 and hCds1/Chk2 by the oriented peptide library approach. J. Biol. Chem., 277, 16102–16115. [DOI] [PubMed] [Google Scholar]
- Schon O., Friedler A., Bycroft M., Freund S.M. & Fersht A.R. ( 2002) Molecular mechanism of the interaction between MDM2 and p53. J. Mol. Biol., 323, 491–501. [DOI] [PubMed] [Google Scholar]
- Scott M.T., Ingram A. & Ball K.L. ( 2002) PDK1-dependent activation of atypical PKC leads to degradation of the p21 tumour modifier protein. EMBO J., 21, 6771–6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh S.Y., Ahn J., Tamai K., Taya Y. & Prives C. ( 2000) The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev., 14, 289–300. [PMC free article] [PubMed] [Google Scholar]
- Shimizu H., Burch L., Smith A., Dornan D., Ball K.L. & Hupp T.R. ( 2002) The conformationally flexible S9-S10 linker region in the core domain of p53 contains a novel MDM2 binding site whose mutation increases ubiquitination of p53 in vivo. J. Biol. Chem., 277, 28446–28458. [DOI] [PubMed] [Google Scholar]
- Takai H. et al. ( 2002) Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription. EMBO J., 21, 5195–5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B., Lane D. & Levine A.J. ( 2000) Surfing the p53 network. Nature, 408, 307–310. [DOI] [PubMed] [Google Scholar]
- Wahl G.M. & Carr A.M. ( 2001) The evolution of diverse biological responses to DNA damage: insights from yeast and p53. Nature Cell Biol., 3, E277–E286. [DOI] [PubMed] [Google Scholar]
- Wu Z., Earle J., Saito S., Anderson C.W., Appella E. & Xu Y. ( 2002) Mutation of mouse p53 Ser23 and the response to DNA damage. Mol. Cell Biol., 22, 2441–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information online