

Abstract
Cyclic AMP is a major trigger of the differentiation process of Trypanosoma brucei, a bloodstream parasite causing sleeping sickness. Its generation in trypanosomes is accomplished by a unique battery of membrane-bound adenylate cyclases (ACs). We have determined the high-resolution X-ray structures of the catalytic domains of two trypanosomal ACs (tACs), GRESAG4.1 and GRESAG4.3. The tAC domains are structurally highly related to the AC domains of higher eukaryotes, but also comprise a highly conserved structural element near the active site, the Δ-subdomain. A cavity below the Δ-subdomain might correspond to an allosteric regulator site as indicated by the stereospecific binding of a single (2S,3S)-1,4- dimercapto-2,3-butanediol molecule. In three different crystal forms, the tAC domains are exclusively observed in a monomeric, catalytically inactive state. Biochemical analysis and the mutagenesis profile of GRESAG4.1 confirmed a common catalytic mechanism of tACs that involves transient dimerization of the AC domain. A low dimerization tendency might play a regulatory role in T.brucei if the activation of tACs is similarly driven by ligand-induced dimerization as in membrane-bound guanylate cyclases.
Keywords: adenylyl cyclases/catalysis/crystal structure/monomer–dimer equilibrium/trypanosomes
Introduction
Adenylate cyclases (ACs) play a crucial role in the signal transduction pathways of many eukaryotic and eubacterial organisms (Barzu and Danchin, 1994; Sunahara et al., 1996) by converting ATP to the second messenger adenosine 3′,5′-monophosphate (cAMP). cAMP mediates cellular responses to numerous extracellular signals ranging from hormone or neurotransmitter stimuli to changes in nutritional conditions, and even constitutes itself an extracellular signal for the life cycle control of some eubacteria and protozoa. Based on their topologies, ACs can be subdivided into four classes (Tang and Gilman, 1992). Class I cyclases are predominantly found in metazoans and comprise a 12-transmembrane helix motif with two cytosolic regions that contain the catalytically active portion. In mammals, these rather complex cyclases occur in nine different isozymes (AC1–AC9) and represent the key effectors of many hormones that bind to G-protein-coupled receptors. Class II cyclases occur exclusively in protozoa and consist of an extracellular domain with putative receptor function, a single transmembrane span and a cytosolic catalytic region. Finally, class III cyclases are large, solely membrane-associated proteins in fungi while the class IV cyclases of eubacteria and mammalian testis are soluble proteins.
The enzymatic activities of ACs and of the related guanylate cyclases (GCs) originate from homologous catalytic domains that are 220–250 amino acids in length, respectively. Until now, most biochemical and biophysical studies focused on mammalian class I enzymes (Tang and Gilman, 1992; Sunahara et al., 1996; Hurley, 1998), which greatly benefitted from the construction of soluble fragments of these cyclases retaining most of their catalytic and regulatory properties (Tang and Gilman, 1995). In class I enzymes, the intramolecular association of two homologous AC/GC domains, C1A and C2A, to a pseudo-heterodimer is required for the formation of a catalytic site (Dessauer and Gilman, 1996; Whisnant et al., 1996). The binding of the diterpene forskolin or regulatory G-protein subunits modulates the activity of class I enzymes, presumably by affecting their C1/C2 association. Low- to medium-resolution X-ray crystallographic studies on a forskolin-linked, but mostly inactive C2A homodimer (Zhang et al., 1997b), and a catalytically competent, quarternary complex made of GSα, C1A, C2A and forskolin (Tesmer et al., 1997), demonstrated that the active site of class I ACs resides along the C1–C2 interface. Subsequently, it was concluded from biochemical data, sequence comparison and modelling studies that other nucleotidyl cyclases might function as hetero- or homodimeric entities as well (Liu et al., 1997).
Class II ACs occur as multiple isozymes in Trypanosoma brucei, a bloodstream parasite and the causative agent of sleeping sickness in humans and Nagana in cattle (Alexandre et al., 1990). Trypanosomal ACs (tACs) are either transiently expressed in the mammalian bloodstream like the ESAG4 isozymes (expression site associated gene 4) or constitutively throughout the life cycle like the isozymes GRESAG4.1, GRESAG4.2 and GRESAG4.3 (genes related to ESAG). In the bloodstream, T.brucei undergoes a transition from a proliferating long slender form to the non-dividing short stumpy form that is triggered by the activation of tACs and subsequent increases of cytosolic cAMP levels (Vassella et al., 1997). This bloodstream transition, a prerequisite for the transfer of the parasite from the mammalian host to the insect vector, is accompanied by further metabolic changes: long slender forms metabolize glucose only to pyruvate, which is then secreted. In contrast, the mitochondria of short stumpy forms acquire the capability to degrade pyruvate in the Krebs cycle, which becomes fully operational in the procyclic insect stage (Tielens and Van Hellemond, 1998; Matthews, 1999). Currently, it is not known whether cAMP signaling and the metabolic changes are directly linked to each other in vivo (Tielens and Van Hellemond, 1998), and what kind of activating ligands play a role therein. Apart from a 2- to 8-fold calcium-mediated stimulation of the tAC activity of bloodstream forms, tACs are known to be insensitive to the stimulation by G-proteins or other agents that commonly affect the catalytic activity of mammalian class I ACs (Rolin et al., 1996).
Here, we report the structures of the catalytically active AC domains of the trypanosomal isozymes GRESAG4.1 and GRESAG4.3 at 1.46 and 1.9 Å resolution, respectively. Unlike previous structures from class I ACs the catalytic domains of tACs crystallize only in their monomeric state. Furthermore, an internal cavity, occupied by (2S,3S)-1,4-dimercapto-2,3-butanediol (d-DTT) was located in the protein structure. The presence of this binding site and of a unique trypanosomal insertion, the Δ-subdomain, in a regulatory important region of the AC domain suggests that tACs might be allosterically controlled by small metabolites like some eubacterial ACs.
Results and discussion
Biochemical characterization of tACs
The gene fragments coding for the cytosolic region of GRESAG4.3 (residues A867–V1229) and ESAG4 (residues P897–R1269) were amplified from genomic DNA of the T.brucei strain 927, cloned and expressed in Escherichia coli, as previously described for GRESAG4.1 (Bieger and Essen, 2000). Unlike the recombinant C2A domains from mammalian ACs, which exhibit only marginal catalytic activities when assayed without accompanying C1A domains (Zhang et al., 1997a), the cytosolic regions of these tACs (mol. wt 40.2–41.9 kDa) are catalytically competent with specific activities of 4–5 nmol/min/mgI. The tAC fragments show classical Michaelis–Menten kinetics, with KMATP values of 17–25 µM and Vmax values of 3–10 nmol/min/mg (Table I), which are similar to non-stimulated, mammalian class I enzymes (Tang and Gilman, 1995; Dessauer and Gilman, 1996). Like the latter, the tAC fragments also exhibit catalytic activity only in the presence of divalent cations, with manganese being more effective as a cofactor than magnesium. For example, the maximal catalytic activity of GRESAG4.1-(A884–Y1241) is 9-fold higher at saturating manganese concentrations (≈6 mM) than in the presence of 40 mM magnesium (data not shown).
Table I. Kinetic characterization of the cytosolic regions of the tACs GRESAG4.1, GRESAG4.3 and ESAG4.
| tAC isozyme | KMATP (µM) | Vmax (nmol/min/mg) | Hill coefficient |
|---|---|---|---|
| ESAG4-(P897–R1269) | 24.7 ± 2.3 | 9.7 ± 1.1 | 0.90 ± 0.0 |
| GRESAG4.3-(A867–V1229) | 24.0 ± 2.0 | 8.3 ± 1.0 | 0.94 ± 0.2 |
| GRESAG4.1-(A884–Y1241) | 16.6 ± 0.1 | 2.9 ± 0.1 | 0.75 ± 0.1 |
| GRESAG4.1-(A884–T1131) | 29.5 ± 0.4 | 2.6 ± 0.2 | 0.78 ± 0.1 |
Only the first 230–240 residues of the cytosolic tAC fragments are homologous to other AC domains including the C1A and C2A domains of mammalian ACs. The remaining C-terminal region of ∼110 residues is only found in the ACs from trypanosomes and Leishmania donovanii and is not homologous to any other protein. Compared with the tAC domains, which are highly conserved in GRESAG4.1–4.3 and ESAG4 (83–87% sequence identity), the C-terminal regions are rather variable (54–78% sequence identity). The function of these variable C-terminal regions is still unknown in tACs. However, proteolytical removal of the variable C-terminal region from the isozyme GRESAG4.1 has only minor effects on the catalytic activity (Table I): the KMATP of the digestion product GRESAG4.1-(A884–T1131) is slightly increased from 16.6 to 29.5 µM and the protein shows the same dependency on manganese and magnesium as GRESAG4.1-(A884–Y1241). Consequently, a regulatory role for the C-terminal region on tAC activity is only feasible if this region recruits additional regulatory cofactors from the trypanosomal cytosol or membrane. In contrast to previous in vivo studies (Rolin et al., 1996), a stimulation of tAC activity by trypanosomal calmodulin and/or calcium was not detected, either for the AC domain of ESAG4, the bloodstream-specific AC or for the AC domains of GRESAG4.1 or GRESAG4.3 (data not shown).
Overall structure of tACs
After removal of the variable C-terminal region, high-quality crystals of the AC domains of the isozymes GRESAG4.1 and GRESAG4.3 were readily obtained under a variety of conditions (see Materials and methods; Bieger and Essen, 2000). Orthorhombic crystals of the catalytic domain of GRESAG4.1 (residues A884–T1131) diffracted to 1.4 Å resolution under cryogenic conditions, while tetragonal crystals of GRESAG4.3-(A867–T1118) similarly attained 1.9 Å resolution. The structure of GRESAG4.1 was solved by multiple isomorphous replacement (MIR) using three heavy-atom derivatives (Table II). The final model of GRESAG4.1-(A884–T1131) refined with data at 1.45 Å resolution includes the residues N888–A1122, one molecule of d-DTT, one sulfate ion and 276 water molecules. Seven, mostly solvent-exposed, residues were observed in multiple conformers. The high quality of the final electron density map can be assessed from Figure 1C. Subsequently, the structure of the AC domain of GRESAG4.3 was determined by molecular replacement using the GRESAG4.1 AC domain as a search model (Table II).
Table II. Summary of crystallographic analysis of tACs.
| Data collection statistics | ||||||||||||
| Dataset |
|
Unit cell and space group |
Resolution (Å) |
Measured, unique reflections |
|
|
Rmergea |
I/σ(I)b |
Completeness (%) |
|
||
| GRESAG4.1, crystal form A | a = 49.9 Å, b = 60.1 Å,c = 79.7 Å (P212121) | 10–1.46 | 498 167, 40 911 | 0.082 (0.283) | 24.7 (4.3) | 98.7 (98.9) | ||||||
| + Pb(Me)3Ac | 10–1.50 | 97 405, 31 193 | 0.047 (0.091) | 22.3 (11.5) | 96.7 (80.8) | |||||||
| + GdCl3 | 10–2.80 | 7230, 3401 | 0.123 (0.277) | 13.6 (5.7) | 76.0 (77.2) | |||||||
| + MeHgOH | 10–2.30 | 38 256, 10 880 | 0.079 (0.177) | 24.0 (11.1) | 99.3 (99.6) | |||||||
| GRESAG4.1, crystal form B | a = 123.8 Å, b = 35.8 Å,c = 59.3 Å (P21212) | 10–2.10 | 42 128, 16 265 | 0.045 (0.250) | 21.6 (3.6) | 88.2 (68.8) | ||||||
| GRESAG4.3 | a = 89.7 Å, b = 89.7 Å,c = 67.4 Å (I41) | 20–1.63 | 175 430, 49 013 | 0.036 (0.084) | 21.1 (11.6) | 98.9 (99.7) | ||||||
| + Mg2+ |
|
|
|
23–1.90 |
136 749, 21 016 |
|
|
0.095 (0.194) |
26.4 (13.1) |
99.5 (99.7) |
|
|
| Phasing statistics | ||||||||||||
| Phasing powere |
||||||||||||
| Derivative | Risoc | Sites | RCullisd | 5.2 Å | 3.8 Å | 3.1 Å | 2.7 Å | 2.4 Å | 2.2 Å | 2.0 Å | 1.9 Å | Total |
| Pb(Me)3Ac, A | 0.186 | 5 | 0.66 | 3.15 | 2.10 | 1.80 | 2.01 | 2.05 | 1.91 | 1.94 | 2.15 | 2.04 |
| MeHgOH, A | 0.257 | 5 | 0.81 | 2.60 | 1.51 | 1.27 | 1.31 | 1.44 | 1.39 | 1.46 | ||
| GdCl3 (I), A | 0.265 | 3 | 0.75 | 3.01 | 1.96 | 1.72 | 1.79 | 2.03 | ||||
| GdCl3 (II), A | 0.258 | 3 | 0.76 | 3.28 | 1.89 | 1.48 | 1.55 | 1.90 | ||||
| Figure of merit | 0.81 | 0.69 | 0.63 | 0.57 | 0.58 | 0.51 | 0.46 | 0.45 | 0.55 | |||
| Refinement statistics | ||||||||||||
| Dataset (PDB code) | Resolution(Å) | Reflections(F > 0) | R-factor, Rfreef | No. of atoms,waters, heteroatoms | r.m.s.d.bonds (Å)g | r.m.s.d.angles (°)g | ||||||
| GRESAG4.1, form A (1FX4) | 10–1.46 | 39 779 | 0.178, 0.205 | 2142, 273, 13 | 0.031 | 2.26 | ||||||
| GRESAG4.1, form B | 20–2.00 | 16 219 | 0.240, 0.290 | 1935, 151, 0 | 0.010 | 1.42 | ||||||
| GRESAG4.3, –Mg2+ | 20–1.63 | 33 441 | 0.210, 0.240 | 2084, 265, 0 | 0.010 | 1.40 | ||||||
| GRESAG4.3, +Mg2+ (1FX2) | 20–1.90 | 21 016 | 0.220, 0.230 | 2073, 269, 1 | 0.008 | 1.28 | ||||||
aRmerge = ΣhklΣi|Ii(hkl) – <I(hkl)> |/ΣhklΣiIi(hkl); values in parentheses correspond to the highest resolution shell.
bAs calculated with the program TRUNCATE (CCP4, 1994).
cRiso = Σ||Fderiv| – |Fnative||/Σ|Fnative|
dRCullis = Σ||FPH ± FP| – FH(calc)|/Σ|FPH ± FP|
eThe phasing power is the ratio of the r.m.s. value of |FH| and the r.m.s. of the lack-of-closure error.
fRfactor = Σ|Fobs – Fcalc|/ΣFobs; Rfree calculated with 5% of the data.
gR.m.s.ds for bond angles and length in regard to Engh and Huber parameters (Engh and Huber, 1991).
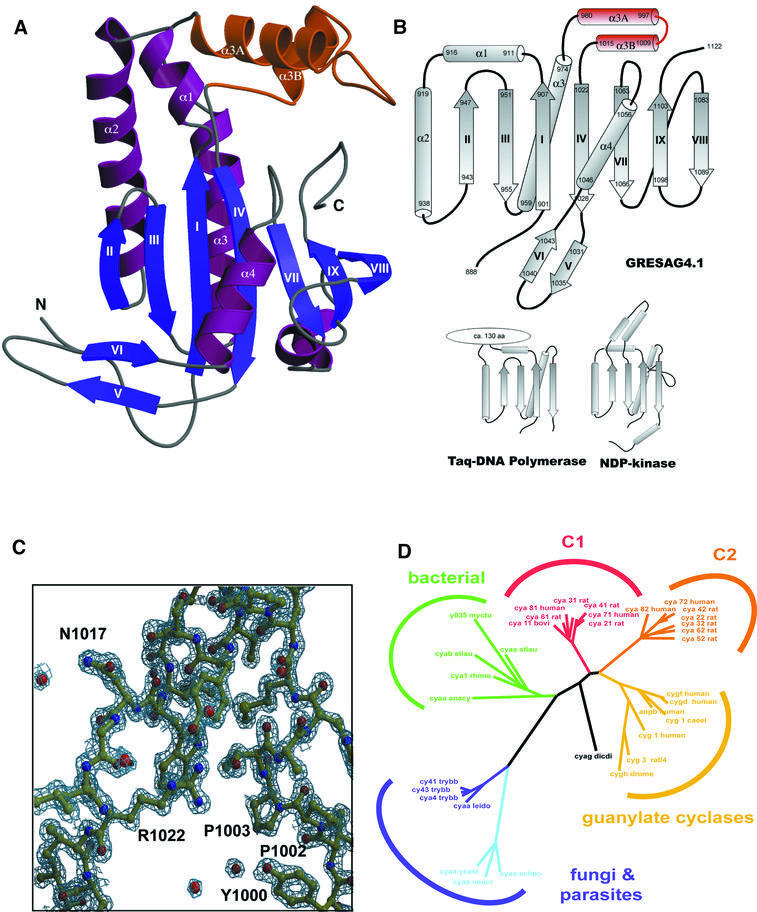
Fig. 1. (A) Overall structure of GRESAG4.1-(A884–T1131). The Δ-subdomain between α3 and β4 that is absent in other ACs is highlighted in orange. (B) Topologies of tACs, DNA polymerase and NDP kinase. The first half of the GRESAG4.1 AC domain (β1–β4, α1, α2) superimposes at 73 Cα positions with Taq DNA polymerase (r.m.s.d. 1.9 Å) and at 42 positions with NDP kinase from D.melanogaster (r.m.s.d. 2.3 Å). The active center is only in ACs and DNA polymerases placed along β1–β4, while NDP kinases harbor the active site in a loop region. (C) σA-weighted 2Fobs – Fcalc electron density map of GRESAG4.1 at 1.46 Å resolution (contouring level 1.7 σ). (D) Dendrogram showing the phylogenetic relationship of ACs and GCs. For its calculation, 40 protein sequences of nucleotidyl cyclases were aligned using the structural alignment between the mammalian C1A and C2A domains and GRESAG4.1 and the multiple and pairwise sequence alignments as computed by PSI-Blast (Altschul et al., 1997) and Clustal_X (Thompson et al., 1997). A non-redundant subset of 22 nucleotidyl cyclase sequences was selected from the resulting multiple sequence alignment to calculate the phylogenetic relationships using PAUPSCRIPT. The sequences of the AC domains of GRESAG4.1 and GRESAG4.3 correspond to the SWISSPROT entries cy41_trybb and cy43_trybb. Figure 1A and Figures 3–5 were made with MOLSCRIPT (Kraulis, 1991) and RASTER3D (Merrit and Murphy, 1994).
GRESAG4.1 and GRESAG4.3 show high structural similarity to each other with a root-mean-square deviation (r.m.s.d.) of only 0.67 Å for 227 equivalent Cα positions. Structural differences are only found at the tip of the β-hairpin motif β5–β6 (residues E1036–K1039), the turn between α3A and α3B (residues C998–D1008) and at the very C-terminus. Both tACs share the overall structure of a seven-stranded β-sheet that is joined at its back by two parallel helices, α2 and α3, and at its front by helix α4 crossing β-strands β1–β4 (Figure 1A and B). Together with the strands β1–β4, the helices α1–α3 form a doubly split βαββαβ sandwich, which is additionally intermitted between α3 and β4 by helices α3A and α3B. This βαββαβ motif, previously coined a palm domain (Artymiuk et al., 1997), forms most of the catalytic surface.
Comparison of tACs with related proteins
Although molecular replacement with the mammalian C1A and C2A domains failed for the structure determination of tACs, the tACs are structurally highly related to the AC domains of class I ACs. A superposition of GRESAG4.1 on the C1A domain of canine ACV gives an r.m.s.d. of 1.45 Å for 127 equivalent Cα positions (Figure 4A), likewise GRESAG4.1 and the C2A domain from rat ACII deviate by 1.64 Å for 134 Cα positions. Hence, the overall structural differences between GRESAG4.1 and the C1A and C2A domains are similar in size to those between the latter two, which superimpose with an r.m.s.d. of 1.3 Å for 153 Cα positions. However, there are two major features that are not present in the structures of mammalian AC domains: first, a 36 amino acid large insertion that forms an α-helical subdomain between α3 and β4; and secondly, an ordered C-terminus (GRESAG4.1, T1105–F1122; GRESAG4.3, A1093– R1106) that extends from the end of β9 along a cleft made between the α4β7 loop and the helical subdomain.
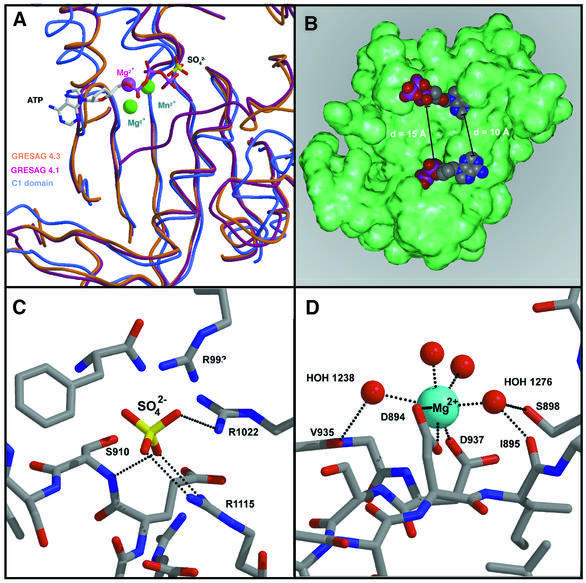
Fig. 4. The catalytic surface of tACs. (A) Superposition of GRESAG4.1 (orange, sulfate), GRESAG4.3 (purple, magnesium) and the mammalian C1A domain (blue, metal centers green, ATPαS) along the active site surface. (B) Model for the binding of ATP (CPK model) to the catalytic surface of a tAC monomer (green). (C) Binding of a sulfate anion (sticks) to the catalytic surface of GRESAG4.1. (D) Magnesium binding site in the monomeric form of GRESAG4.3. The coordination sphere of the magnesium ion (blue) consists of two conserved aspartates and four water ligands. Putative H-bonds are shown as dotted lines.
The secondary structure elements with most of the conserved residues, β1–β4 and α2, are all located at the front side of the palm domain (Figure 1A). Further conservation is observed in β5 and β6, which form the β-hairpin motif in both AC structures. The region from β7 to the C-terminus is located beneath the tAC-specific α-helical subdomain on the opposite site of the catalytic core. In this segment, the sequence similarity between tACs and the C1A domain is as low as that for helices α2 and α3 on the back of the AC domain, as these regions are not involved in catalysis.
Although class II ACs are topologically related to membrane-bound GCs, structure-based sequence comparisons and calculation of their evolutionary relationships indicate a divergence between GC and AC families that preceded the speciation into different AC classes (Figure 1D). Furthermore, these data suggest a close relationship between tACs and bacterial ACs such as those from mycobacteria or Stigmatella aurantiaca. All of them presumably act as homodimers. These results suggest that the topology of membrane association is not indicative of the evolutionary relationships between cyclase families, as membrane association occurs in phylogenetically distant cyclase families, like the bacterial and mammalian ACs.
Monomeric versus dimeric state of tACs
The catalytic activity of nucleotidyl cyclases was generally found to depend on the dimerization of the AC/GC domains. In both crystal structures of the C2A domain (Zhang et al., 1997b) and the chimeric complex between bovine Gsα, C1A from canine ACV and C2A from rat ACII (Tesmer et al., 1997), these AC domains were only observed as homo- and heterodimers. Furthermore, various studies on the guanylate cyclases and class I ACs demonstrated an association of the AC/GC domains that preceded stimulation of these cyclases by effectors (Garbers, 1989; Harteneck et al., 1990; Buechler et al., 1991; Yan et al., 1996). In contrast, all three crystal forms obtained for the tACs GRESAG4.1 and GRESAG4.3 comprise the AC domains in a monomeric instead of a dimeric state. Likewise, none of the crystal contacts that are made by the tAC domains is conserved among the crystal forms.
While the monomeric state is apparently catalytically inactive, biochemical data indicate that in solution the monomeric state of the tAC domains equilibrates dynamically with a dimeric, catalytically active species. Analytical gel filtration experiments with GRESAG4.1-(A884–T1131) demonstrated the predominance of a dimeric species at low ionic strength (Figure 2A). However, at higher ionic strength, the elution profile was progressively shifted towards the monomeric species, which indicates the occurrence of monomeric and dimeric states at varying ratios in solution. This sensitivity towards salt suggests that the dimerization of tACs significantly depends on polar interactions in the dimer interface. Furthermore, in tAC homodimers, two catalytic sites are postulated along the 2-fold symmetric dimer interface (Liu et al., 1997), while in class I ACs only one site is formed in the interface due to the asymmetry of the C1C2 heterodimer. The existence of two catalytic sites per tAC homodimer was proven by enforcing heterodimerization between the wild-type enzyme of GRESAG4.1 and several catalytically inactive mutants that should selectively destroy one or both active sites upon association with a wild-type AC domain. If added in stoichiometric excess, the addition of the single site mutants D949A or E943A to 2.0 nM GRESAG4.1-(A884–Y1241) causes a 60% loss of activity, which is consistent with the loss of one active site per wt–mutant heterodimer (Figure 2B). Consequently, when the double mutant D949A/R1053A is added, the activity of GRESAG4.1 drops to <10%, indicating that both active sites of the dimer are functionally impaired (Figure 2B). Finally, if complementary pairs of single site mutants are mixed, a restoration of one active site can be observed per mutant–mutant heterodimer. For example, an equimolar mixture of the D949A and R1053A mutants exhibits a 4-fold increased specific activity above the expected background level (Figure 2C).
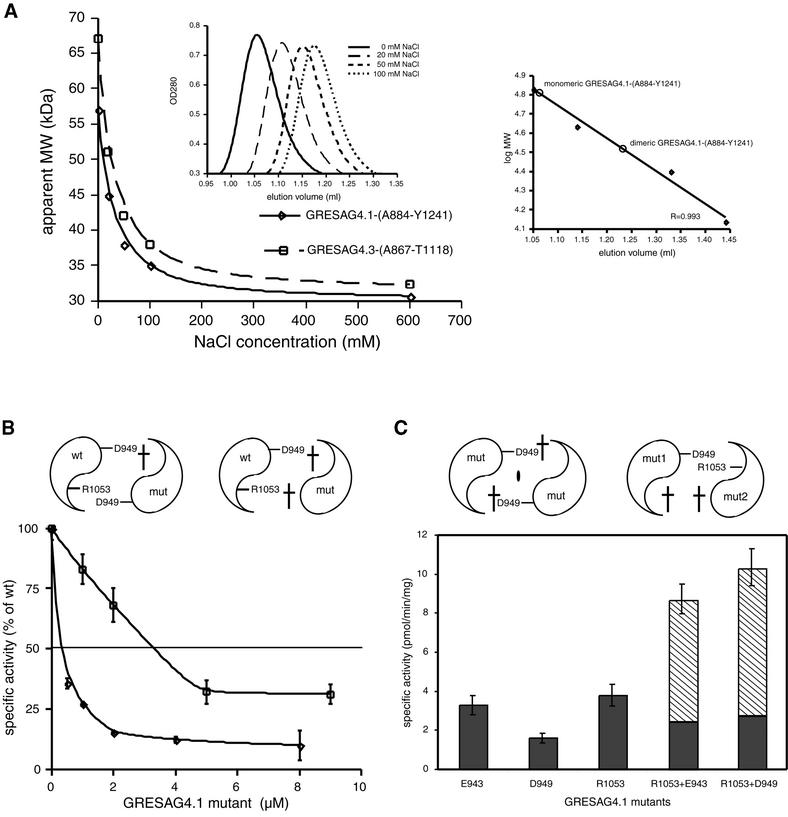
Fig. 2. The monomer–dimer equilibrium of tAC domains. (A) Analytical gel filtrations were carried out in the presence of increasing NaCl concentrations on a SMART chromatography station (Pharmacia). Each run was made with 50 µg of purified protein in 20 mM Tris–HCl pH 8.0, flow rate 100 µl/min. The continuous shift of the retention factor (Rf) (left inlay) indicates a dynamic equilibrium between a monomeric and a dimeric species. At low salt, the Rf value corresponds to the molecular weight of a tAC dimer while in the presence of 600 mM NaCl only the monomeric species is present. The gel filtration column S75 PC 3/20 was calibrated (right inlay) using bovine serum albumin (67.0 kDa), ovalbumin (43.0 kDa), chymotrypsinogen A (25 kDa) and ribonuclease A (13.7 kDa). (B) Inhibition of wild-type GRESAG4.1 activity by addition of the mutant D949A (squares) and the double mutant D949A/R1053A (diamonds). AC assays were carried out at a protein concentration of 2 nM (4 nM) for the D949A (D949A/R1053A) mutant as previously described (Bieger and Essen, 2000). (C) Specific activities of the mutants E943A, D949A and R1053A and the equimolar mixtures of E943A with R1053A and D949A with R1053A.
Dimerization is fundamental to AC activity and might be of regulatory importance if the association of intact tACs in the membrane is promoted by effector binding either to the putative extracellular receptor domains or to the cytosolic regions. Such a cytosolic mode of activation was previously shown for the retinal guanylyl cyclase-1 (retGC-1) (Olshevskaya et al., 1999; Yu et al., 1999). In this case, activation of retGC-1 coincides with a transition from a monomeric to a dimeric retGC-1 species in the membrane when retGC-1 binds to GC activating proteins (GCAP) at calcium concentrations that are sufficiently low to drive dimerization of GCAP. The weak dimerization tendency of isolated tAC domains as compared with the pronounced pairing of the mammalian C1A and C2A domains makes a similar activation mechanism also attractive for tACs in the membrane context, although suitable activating ligands have yet to be identified in T.brucei. The absence of steric overlaps in a model of the tAC dimer that was assembled on the basis of the C1C2 heterodimer (Figure 3A) indicates that the dimerization of tAC domains proceeds without a requirement for major conformational changes. However, most of the twenty polar interactions that were observed in the C1C2 heterodimer are missing in the putative tAC dimer due to sequence differences. Despite the lack of a conserved dimer interface, the structural element that contributes together with helix α2 most to the dimer interface in class I enzymes, the β5–β6 hairpin (residues D1031–Y1043), is structurally well preserved between tACs and the C1A and C2A domains (Figure 3B). In both tAC structures, this hairpin, also coined an arm region, is well ordered, as indicated by its low B-factors. The orthogonal kink between the arm region and the adjacent β4 strand and α4 helix is made by two highly conserved glycines, G1027 and G1045, at the N- and C-terminal ends of the arm region. The backbone conformations of the glycines occupy regions in torsional space (G1027, Φ = –176°/Ψ = 170°; G1045, Φ = 106°/Ψ = –163°), which are unfavorable for other amino acids. A further interaction that is conserved between evolutionary distant ACs is the salt bridge between K945 on β2 and D1042 residing on β6 of the arm region (2.8 Å). In class I ACs, this salt bridge becomes occluded in the ventral cavity upon dimerization of the C1A and C2A domains. Here, the corresponding Lys/Glu pair on the C2 domain (K938, D1018) forms hydrogen bonds to the N1 and N6 atoms of the adenine ring of bound substrate (Tesmer et al., 1997). Mutations at these positions relax the substrate specificity of class I ACs (Beuve, 1999), although it is not clear to what extent the precise backbone conformation of the arm domain contributes to substrate specificity. Accordingly, a disruption of the K945–D1042 salt bridge in the K945A mutant of GRESAG4.1 leads to a dramatic loss of activity (Table III). In the absence of other dominant interactions, it might be the binding of the arm region towards the base of the substrate that additionally stabilizes a transient tAC dimerization.
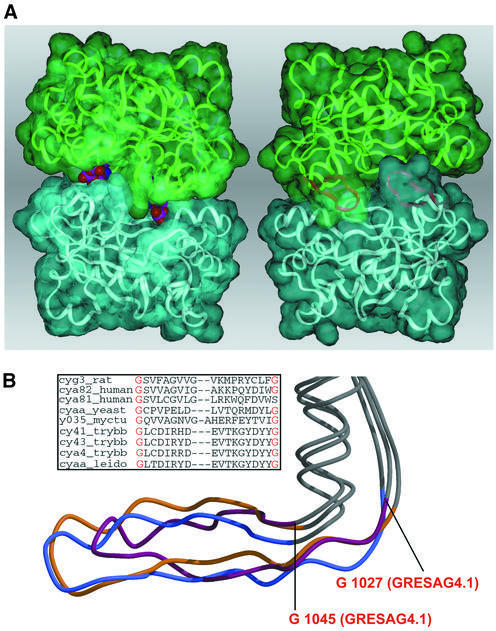
Fig. 3. The dimerization of tACs. (A) Model of the tAC dimer as shown from the dorsal (left) and the ventral site (right). Whereas the ventral site is blocked by the pairing of the arm regions (purple), the dorsal site allows substrate access as indicated by ATP molecules occupying both active sites (CPK models). (B) Structural comparison between the arm region of GRESAG4.1 (purple), the C1 (yellow) and C2 domain (blue). The inlay shows the sequence divergence between tACs and other nucleotidyl cyclases in this region.
Table III. Mutagenesis of catalytically important residues in GRESAG4.1.
| Mutation | Specific activity(% wild type) | Correlate inmammalian ACs | Context inmammalian ACs(Tesmer et al., 1997) | Vmax |
|---|---|---|---|---|
| D906A | 0.005 | Mg2+ coordination | D396 VC1 | – |
| E943A | 0.08 | salt bridge to R934 | K936 IIC2 (salt bridge to D923; C1–C2 interface) | wild type 10-fold decreased (Tang et al., 1995) |
| K945A | 0.1 | binds to N1 of adenine | K938 IIC2 (salt bridge to D1018 IIC2) | wild type (Tang et al., 1995) |
| D949A | 0.008 | Mg2+ coordination | D440 VC1 | <10-fold inactive (Tang et al., 1995) |
| R1022A | 60 | γ-phosphate binding | R484 VC1 | wild type10-fold decreased (Tang et al., 1995) |
| N1049A | 0.03 | active site structure,water coordination | N1025 IIC2 | <10-fold-inactive (Yan et al., 1997) |
| R1053A | 0.1 | stabilization of transition state, binds to α- and/orβ-phosphate | R1029 IIC2 | <10-fold- inactive (Tang et al., 1995; Yan et al., 1997) |
| E1055A | 54 | (salt bridge to R1022 inGRESAG4.1) | E518 VC1 (salt bridge to R484) | wild type (Tang et al., 1995; Dessauer et al., 1997) |
The active site surfaces of tACs
The surface regions that were identified in class I ACs to constitute the catalytic site cover most of the front side of the palm region. In the tAC crystals, these rather polar and flat surfaces are differently engaged in crystal contacts. In crystals of GRESAG4.1-(A884–T1131), the C-terminus of the tAC domain (residues T1105–F1122) runs along the active site surface by making several polar interactions with catalytic residues. The binding of the C-terminal stretch towards the active site is obviously dictated by crystal packing and not due to a putative autoinhibitory function, as the shortened tAC domain of GRESAG4.1-(A884–D1117) exhibits similar specific activities to that of GRESAG4.1-(A884–T1131) (data not shown).
Attempts to cocrystallize tACs with nucleotide analogs were unsuccessful due to the strong propensity of tACs to crystallize in inactive, monomeric states. However, a previous model of the substrate ATP in the catalytic site of the C1C2 dimer (Tesmer et al., 1997, 1999) fits nicely into the two catalytic sites of the modeled GRESAG4.1 dimer. The binding of two ATP molecules per tAC dimer occurs without steric interference; the closest approach is 10 Å between the adenine rings while the α-phosphoryls at which the cyclization reaction occurs are 15 Å distant from each other (Figure 4B). Likewise, the Hill coefficients that correspond only to a slight anticooperativity in GRESAG4.1, GRESAG4.3 and ESAG4 (Table I) indicate almost independent substrate binding by the two catalytic sites.
In the model of the tAC–ATP complex, the terminal γ-phosphoryl of ATP is stabilized as part of the leaving group by salt bridges to R1022 and R1115. Mutagenesis of R1022, which resides on strand β4, moderately affects the catalytic efficiency of GRESAG4.1; the KM value of the R1022A mutant increases ∼3-fold (45 µM) while Vmax drops by 59% (1.2 nmol/min/mg) when compared with the Michaelis–Menten parameters of the wild-type enzyme. A similar contribution to catalysis was shown for the homologous residue R484 in the mammalian C1A domain (Tesmer et al., 1997, 1999). In contrast, the second arginine, R1115, contributes to the stabilization of the γ-phosphoryl only in tACs. Here, the ordered C-terminal stretch approaches the active site in such a way that the side-chain of R1115 is closely positioned at the γ-phosphoryl binding site while the same region is completely disordered in crystal structures of the mammalian ACs. Interestingly, in the GRESAG4.1 structure, the catalytic site surface harbors a sulfate ion from the crystallization buffer that forms ionic interactions with R1022 (NH2, 3.0 Å; NH1, 3.0 Å) and R1115 (NH1, 3.0 Å; NH1, 3.1 Å) as well (Figure 4C). This sulfate on the surface of monomeric GRESAG4.1 is only 2.0 Å away from the predicted binding site of the γ-phosphoryl of ATP. Magnesium-independent binding of sulfate to the active site surface correlates with the observed sensitivities of tAC activities towards dianions like phosphate and sulfate (IC50 for GRESAG4.1-(A884–T1131): 21 mM for Pi, 53 mM for sulfate). The apparently preformed anion binding site also recruits the dipole moment of helix α1, whose N-terminus, only 6.3 Å distant, is directed towards the sulfate or γ-phosphoryl, respectively. The precise alignment of helix α1 is controlled by a highly conserved β-bulge I907–S910, which connects the C-terminus of β1 with α1. Interestingly, in DNA polymerases the corresponding helix is likewise orientated by a preceding β-bulge. Therefore, it is likely that ACs and DNA polymerases share the way by which the leaving group is stabilized during phosphoryl transfer.
Another hallmark in class I ACs and DNA polymerases is two-metal ion catalysis. According to recent structural and enzymological data, two magnesium ions coordinate both to the active site and the triphosphate moiety of the substrate molecule ATP (Park et al., 1997; Tesmer et al., 1999). In the structure of monomeric GRESAG4.3, a single magnesium ion is bound to two acidic, surface-exposed residues of the catalytic site, D894 (OD1, 2.5 Å; OD2, 2.9 Å) and D937 (OD1, 2.3 Å); four additional water ligands complete the slightly disturbed octahedral coordination shell around the magnesium ion in the absence of bound substrate (Figure 4D). These two aspartates are highly conserved among ACs and were suggested to support the stabilization of the pentavalent transition state at the α-phosphate. Mutagenesis of each of these metal ligands in GRESAG4.1 (D906 and D949; see Table III) and class I ACs (Tang et al., 1995) leads to a profound loss of catalytic activity. Accordingly, when superimposing the GRESAG4.3 structure on the C1C2 heterodimer, this magnesium ion is found at the same position as the magnesium ions in the structure of the C1C2 dimer liganded to Mg2+-pyrophosphate and the product analog 2′-d-3′-AMP.
Taken together, the similar structural features of the active site surfaces (Figure 4A) and the common mutagenesis profile (Table III) indicate that tACs follow an almost identical catalytic mechanism to the related class I ACs. However, despite the appealing similarities in the nucleotide binding sites, tACs and class I ACs strongly differ in their response to so-called P-site inhibitors. Mammalian ACs are non-competitively inhibited by 2′-deoxy-adenosine and its 3′-mono- or polyphosphate derivatives. These P-site inhibitors (Desaubry et al., 1996) synergistically inactivate mammalian ACs by dead-end inhibition only in the presence of pyrophosphate. Consequently, the apparent affinities of mammalian class I ACs towards P-site inhibitors are exceptionally high with IC50 values being in the nanomolar range. When GRESAG4.1-(A884–T1131) was assayed with 2′-deoxyadenosine and its 3′-monophosphate analog at concentrations up to 100 µM no significant inhibition was observed, either in the presence or in the absence of PPi. However, PPi alone inhibited GRESAG4.1-(A884–T1131) with an apparent IC50 value of <1 mM. This observation corroborates an earlier study on intact tACs in T.brucei membrane preparations (Martin et al., 1978), which reported an IC50 value of 135 µM. Insensitivities towards P-site inhibitors were also described for the bacterial AC from Bordella pertussis and the soluble ACs from bovine sperm. According to structural and biochemical studies on mammalian ACs, P-site inhibitors occupy the active site together with Mg2+-pyrophospate (Tesmer et al., 1997) and impair substrate release from the C1C2 heterodimer (Dessauer and Gilman, 1997). Interestingly, the residues bordering the adenine binding site of the tAC homodimer are well conserved. Therefore, the different behavior towards this class of substrate-like inhibitors is presumably not linked to structural differences in the active site itself, but due to the looser association of tACs.
The Δ-subdomain
During the activation of class I ACs, most interactions between the C1C2 heterodimer and Gsα occur in a groove formed by α2 and the α3–β4 loop of the C2 domain. Similarly, in the GCAP-mediated activation of membrane-bound GCs, the α3–β4 loop was shown to interact with GCAP (Sokal et al., 1999). In tACs, this region, or Δ-subdomain due to its deltoid shape, bears the most striking topological difference to other ACs and GCs: 36 amino acids are inserted in the α3–β4 loop (Figures 1A and 5A) where they form two additional helical segments, the 26 Å long helix α3A (N980–E997) and the shorter segment α3B (P1009–L1015). The multiple sequence alignment in Figure 5A shows that this Δ-subdomain is uniquely conserved in the class II ACs from the parasites T.brucei and L.donovanii, but missing in other class II ACs like the ACG from Dictyostelium discoideum. Parts of the α3A helix are tightly associated with the α1 helix and the preceding β-bulge, e.g. the side chains of L982 and F989 form a hydrophobic cluster with A912 and L913. Thereby, R1020 on the N-terminus of strand β4 organizes major parts of the Δ-subdomain on top of the globular tAC domain by making H-bonds to Y986 (OH, 3.1 Å), E990 (OE2, 3.0 Å), T1004 (OG1, 3.0 Å) and Y1012 (OH, 2.8 Å). Consequently, when comparing the structures of GRESAG4.1 and GRESAG4.3, the Δ-subdomain is structurally almost invariant due to its packing on the globular core, several stabilizing interactions inside the Δ-subdomain and overall low mobility as reflected by low B-factors. Furthermore, the proximity of the transmembrane segment of GRESAG4.1 (residues F863–P883) and the N-terminus of the tAC domain sets topological constraints on how membrane-bound and dimerized tACs position their Δ-subdomains relative to the membrane context. According to such a model (Figure 5E), the Δ-subdomain points towards the cytosolic bulk, ∼45 Å away from the membrane surface, where it is easily accessible for interactions with other, regulatory proteins.
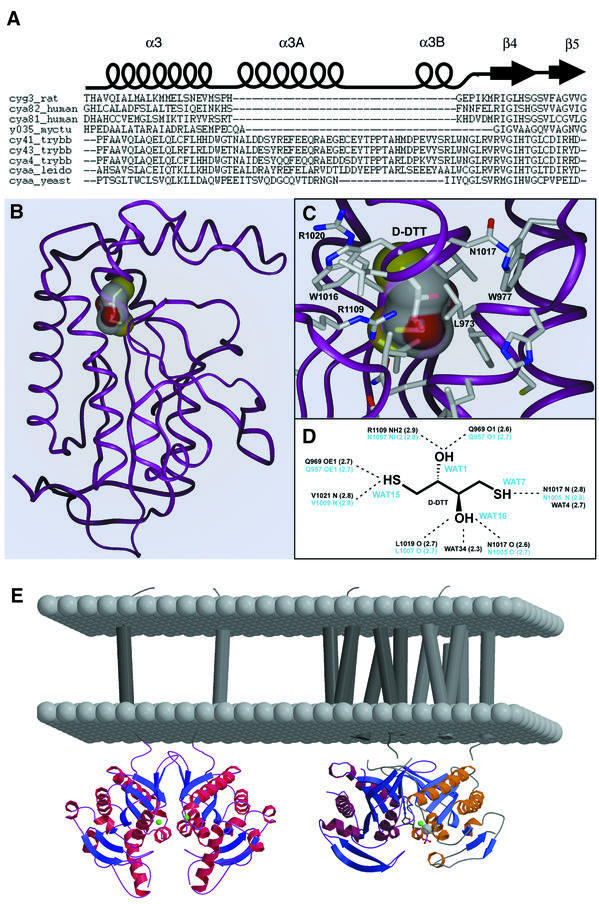
Fig. 5. The Δ-subdomain. (A) Multiple sequence alignment of nucleotidyl cyclases along the β4–α3 insertion. (B) Stereospecific binding of d-DTT in GRESAG4.1. (C and D) Interactions between d-DTT and its binding site below the Δ-subdomain. All residues that make contacts with d-DTT are strictly conserved among other tACs. (E) Models of tAC dimers (left) and mammalian C1C2 heterodimers (right) in their native membrane context. The Δ-subdomain of tACs is distally located from the membrane.
d-DTT binding and allosteric regulation
Together with the α4–β7 loop and parts of β4 and β8, the two helices α3A and α3B enclose an internal cavity that has an overall volume of 95 Å3 as calculated by GRASP (Nicholls, 1992). This cavity (Figure 5B) is mostly hydrophilically walled by backbone atoms; only the side chains of R1109, located on β8, and Q969 on α3 point directly into the center of the cavity. During the refinement of the 1.45 Å structure of GRESAG4.1, electron density came up in the cavity that could be assigned as a single d-DTT molecule (Figure 5C). The interaction between the tAC domain and DTT is stereospecific, as racemic dl-DTT was used as reductant during protein purification and crystallization. Most interactions between the hydroxyl and sulfhydryl groups of d-DTT and the cavity involve main chain atoms: Q969 (O–O1, 2.6 Å; OE1–S1, 2.7 Å), N1017 (O–O2, 2.6 Å; N–S2, 2.8 Å), L1019 (O–O2, 2.7 Å) and V1021 (N–S1, 2.8 Å). Only two H-bonds are made between d-DTT and side chains, O1–R1109 NH2 (2.9 Å) and S1–Q969 OE1 (2.7 Å). Furthermore, two water molecules H-bond to O2 (2.3 Å) and S2 (2.7 Å). In both tAC structures the cavity is completely occluded and without access to bulk solvent. However, in the GRESAG4.3 structure four water molecules are located instead of d-DTT inside the cavity and H-bond to R1109, Q969, V1021 and N1017 (Figure 5D). Similarly to structurally well characterized carbohydrate binding sites (Quiocho, 1986, 1993), these waters occupy in this d-DTT-free structure almost the same positions as the hydroxyls and sulfhydryls of d-DTT in the GRESAG4.1–d-DTT complex.
Conclusion
Functional studies on the precise roles and regulation of membrane-bound ACs in trypanosomatids have been severely hampered by the presence of multiple, highly homologous class II ACs in these parasitic organisms. For example, at least six orthologous gene copies per cell were found for ESAG4, which is specifically expressed in the bloodstream form by its association with the variable surface glycoprotein (VSG) expression sites. Likewise, the tAC GRESAG4.1 is encoded by nine orthologs. These numbers represent only lower limits until the complete genome sequence of T.brucei has been revealed. Together with the mutagenesis data, the structural analysis of two isozymes, GRESAG4.1 and GRESAG4.3, confirmed that tACs structurally and mechanistically resemble class I enzymes but comprise an additional, unique structural motif, the Δ-subdomain. The topological location of this subdomain in a region where other nucleotidyl cyclases like the mammalian class I ACs or the membrane-bound GCs bind their regulatory cofactors suggests a regulatory role for the Δ-subdomain. Furthermore, the observed structural invariance of the Δ-subdomain and the strict conservation of residues that line the binding pocket for d-DTT indicate that this domain serves equivalent functions in the different isozymes of trypanosomatids and leishmanial species. As the observed binding of d-DTT might be a case of molecular mimicry, one might speculate that the binding pocket recognizes other, small molecule compounds that exert a unique allosteric control on tAC activity. For example, the cAMP-triggered transition between the proliferative, long slender form and the non-proliferative short stumpy form was found to be induced by a soluble activity released by bloodstream trypanosomes, which was termed SIF (stumpy induction factor) (Vassella et al., 1997). Alternatively, the Δ-subdomain could bind in trypanosomes a yet unidentified proteinaceous cofactor that affects dimerization of the tAC domains similarly to the retGC-1/GCAP system.
Interestingly, product inhibition of tAC activity by PPi itself might be a primary regulatory pathway for these ACs. Recent 31P-NMR studies showed that PPi is more abundant than ATP in T.cruzi (Urbina et al., 1999) where it apparently partitions both to the cytosol and the acidocalcisomes, a specialized vacuolous compartment of trypanosomatids. At least some trypanosomal enzymes use PPi instead of ATP as an energy source (Bringaud et al., 1998; Rodrigues et al., 1999), and it might be falling cytosolic PPi concentrations that cause transient tAC activation during the different metabolic states of the parasite. In this scenario, the high propensity of tACs to adopt a monomeric state might make them especially sensitive to PPi-mediated inhibition. AC activity that is solely under metabolic control would not be unique to tACs and has been described for several other ACs: soluble ACs from mammalian testis and cyanobacteria are activated by bicarbonate (Chen et al., 2000), while a bacterial AC from Brevibacterium liquefaciens was found to be stimulated by the glycolysis product pyruvate (Peters et al., 1991).
Materials and methods
Plasmid construction, site-directed mutagenesis and protein expression
Gene fragments that code for the cytosolic regions of GRESAG4.3-(L867–V1229) and ESAG4-(P897–R1269) were amplified from genomic DNA of the T.brucei strain 927 using Takara Taq polymerase (BioWhittaker Europe) and 30 cycles at 30s/94°C, 2 min/58°C and 5 min/72°C. For amplification, the primer pairs 5′-CTCGCTCGAGCA CAAATAAAGGGG and 5′-GCGGATCCGCTAGCGGACTTGCGTGC TTCACTCTC were used for the GRESAG4.3 gene and 5′-CTC GCTCGAGCTATTATCACGTAGTGGCGCTGTCATTTTCCAAGTC and 5′-GCGGATCCCATATGTGACACCGTCAATGACTTTCAG for ESAG4 (restriction sites underlined). The DNA fragment of GRESAG4.3 (1.09 kbp) was digested with NheI and XhoI, and the 1.11 kbp fragment of ESAG4 with NdeI and XhoI and ligated into the expression vector pET-28a (Invitrogen) using standard cloning techniques. In comparison with the SWISSPROT entries CY43_TRYBB and CYA4_TRYBB from T.brucei strain AnTat1.3, several sequence differences were found for the coding sequences that were confirmed by repeated PCR amplifications and sequencing of both strands. The GRESAG4.3 sequence (SWISSPROT entry CY43_TRYBB) shows the sequence exchanges R959C, H994S, M995L, N1098S, C1164W and G1165R, while the cloned ESAG4 gene (CYA4_TRYBB) has the exchanges K937M, C969R, Q990R, S1047P and R1101Q. The exchanges probably reflect sequence heterogeneity for these multigenic isozymes in T.brucei; none of the exchanges is observed at positions that are crucial for catalytic activity.
Site-directed mutagenesis of the GRESAG4.1-(A884–Y1241) gene fragment was carried out by fusion PCR (Ho et al., 1989) using Takara Taq polymerase (BioWhittaker Europe) and 30 cycles at 30s/94°C, 2 min/58°C and 5 min/72°C with pET-gres4.1cyto (Bieger and Essen, 2000) as template. Accordingly, the double mutant D949A/R1053A was generated with the plasmid pET-gres4.1cyto(D949A) as template. The mutated gene fragments of GRESAG4.1 were digested with NheI and XhoI and cloned into pET-28a.
The plasmids pET-gres4.1cyto, pET-gres4.3cyto and pET-es4cyto were transformed into E.coli BL21(DE3) cells. Protein expression, renaturation of the tAC fragments from inclusion bodies by dialysis and further purification were carried out using the previously described protocol for GRESAG4.1-(A884–Y1241) (Bieger and Essen, 2000).
Limited proteolysis and mass spectrometry
To digest the cytosolic fragments of GRESAG4.1, GRESAG4.3 and ESAG4 down to the AC domains the recombinant isozymes were incubated for 30 min at 4°C at a concentration of 0.5 mg/ml with 0.05 mg/ml subtilisin [GRESAG4.1-(A884–Y1241)] and 0.05 mg/ml proteinase K [GRESAG4.3-(A867–V1229) and ESAG4-(P897–R1269)]. The cleavage reaction was carried out in 20 mM Tris–HCl pH 8.0 as reaction buffer and stopped by adding 10 mM phenylmethylsulfonyl fluoride. The digestion reaction yielded for all three tACs a predominant product of ∼30 kDa mass. The digestion products were loaded onto a pre-equilibrated Resource Q column (Pharmacia). The column was washed with 5 vols of the reaction buffer and the AC domains of the three tAC isozymes were eluted with 60 ml of a linear NaCl gradient (0–600 mM). The eluate was concentrated and loaded onto a Superdex 75 gel filtration column (Pharmacia) in 20 mM Tris–HCl pH 8.0, 0.6 M NaCl.
The purified AC domain of GRESAG4.1 was N-terminally sequenced by Edman degradation and analyzed on an electrospray mass spectrometer (ABI) to determine its molecular mass and the location of its N- and C-termini.
Crystallization, data collection and structure determination
The tAC GRESAG4.1-(A884–T1131) was crystallized in two forms, A and B, as previously described (Bieger and Essen, 2000). Crystals of GRESAG4.3-(A867–T1118) were grown in hanging drops using vapor diffusion at 18°C by mixing 1 µl of protein solution (6 mg/ml) in 5 mM Tris–HCl pH 8.0 with 1 µl of 25% (w/v) PEG4000, 0.2 M MgCl2 pH 7.0 as crystallization buffer. After 2 days, the tetragonal crystals were grown to final size and the PEG4000 concentration was increased to 35% (w/v) in the reservoir.
X-ray data were collected at 100 K from GRESAG4.1-(A884–T1131) frozen in 2.0 M ammonium sulfate pH 7.0, 20% (v/v) glycerol. For the GRESAG4.3-(A867–T1118) crystals, the mother liquor was used as cryo-protection buffer. Heavy metal derivatives of crystal form A of GRESAG4.1-(A884–T1131) were prepared by incubating the crystals in the cryo buffer with 10 mM of the heavy metal salt for 4–8 h. Diffraction data were collected on a Rigaku RU-200 CuKα rotating anode with focusing mirrors (Charles Supper) and a 30 cm MAR research imaging plate. Synchrotron data were collected from frozen crystals at beamlines BW6 and BW7B, HASYLAB, Hamburg, using a MAR CCD detector and wavelengths of 1.07 and 1.00 Å, respectively.
Data for crystal form A of GRESAG4.1-(A884–T1131) were indexed and integrated with DENZO (HKL research) in space group P212121 (a = 49.9 Å, b = 60.1 Å, c = 79.7 Å) up to 1.45 Å resolution. The crystals have one monomer per asymmetric unit with 30% solvent content. Likewise, crystallographic data of GRESAG4.3-(A867–T1118) were collected at 1.9 Å resolution and indexed and integrated with MOSFLM in space group I41 (a = 89.7 Å, c = 67.1 Å, solvent content 35%, with one monomer per asymmetric unit).
The structure of GRESAG4.1-(A884–T1131) was solved by MIR using an initial native 1.9 Å dataset collected at beamline BW6. After phase refinement with SHARP (La Fortelle and Bricogne, 1997) and SOLOMON (Abrahams and Leslie, 1996), a 1.9 Å MIR map was obtained that allowed semi-automatic tracing and model building by WARP (Lamzin and Wilson, 1997). Further refinement was carried out using CNS 0.9 (Brunger et al., 1997) with a native 1.46 Å dataset that was recollected at beamline BW7B. The refinement converged at an R-factor/R-free of 0.178/0.205 for data between 10.0 and 1.45 Å (Table I). The structure of the GRESAG4.3 AC domain was solved by molecular replacement (MR) using AMoRe (CCP4, 1994) and the structure of GRESAG4.1-(A884–T1131) as search model. Refinement with the native data set proceeded to a final R-factor/R-free of 0.200/0.229 for data between 10 and 1.9 Å.
All tAC models exhibited good stereochemistry as analyzed by PROCHECK (Laskowski et al., 1993) while the following side chains were missing or only partially defined in electron density maps: E1036, V1037, R1092 (CD-NH2), D1116 (CB-OD2) and E1119 (CB-OE2) for GRESAG4.1, and E985 (CG-OE2), E987 (CB-OE2), K1027 (CG-NZ) and R1080 (CG-NH2) for GRESAG4.3. Coordinates and structure factors of GRESAG4.1 and GRESAG4.3 have been deposited in the RCSB Protein Data Bank under accession codes 1FX2 and 1FX4.
Acknowledgments
Acknowledgements
The authors are very grateful for the encouragement and enduring support of D.Oesterhelt, the valuable comments of D.Griffith, the mutagenesis work of U.Heider, and the assistance of H.Bartunik at synchrotron beamline BW6, MPG-ASMB, Hamburg, and W.Burmeister at ID14-3, ESRF, Grenoble. This work is supported by a predoctoral fellowship (B.B.) of the Fonds der Chemischen Industrie.
References
- Abrahams J.P. and Leslie,A.G.W. (1996) Methods used in the structure determination of bovine mitochondrial F1-ATPase. Acta Crystallogr. D, 52, 30–42. [DOI] [PubMed] [Google Scholar]
- Alexandre S., Paindavoine,P., Tebabi,P., Pays,A., Halleux,S., Steinert,M. and Pays,E. (1990) Differential expression of a family of putative adenylate/guanylate cyclase genes in Trypanosoma brucei. Mol. Biochem. Parasitol., 43, 279–288. [DOI] [PubMed] [Google Scholar]
- Altschul S.F., Madden,T.L., Schäffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artymiuk P.J., Poirrette,A.R., Rice,D.W. and Willett,P. (1997) A polymerase I palm in adenylyl cyclase? Nature, 388, 33–34. [DOI] [PubMed] [Google Scholar]
- Barzu O. and Danchin,A. (1994) Adenylyl cyclases: a heterogeneous class of ATP-utilizing enzymes. Prog. Nucleic Acid Res. Mol. Biol., 49, 241–283. [DOI] [PubMed] [Google Scholar]
- Beuve A. (1999) Conversion of a guanylyl cyclase to an adenylyl cyclase. Methods, 19, 545–550. [DOI] [PubMed] [Google Scholar]
- Bieger B. and Essen,L.-O. (2000) Crystallization and preliminary X-ray analysis of the catalytic domain of the adenylate cyclase GRESAG4.1 from Trypanosoma brucei. Acta Crystallogr. D, 56, 359–362. [DOI] [PubMed] [Google Scholar]
- Bringaud F., Baltz,D. and Baltz,T. (1998) Functional and molecular characterization of a glycosomal PPi-dependent enzyme in trypanosomatids: pyruvate, phosphate dikinase. Proc. Natl Acad. Sci. USA, 95, 7963–7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger A.T., Adams,P.D. and Rice,L.M. (1997) New applications of simulated annealing in X-ray crystallography and solution NMR. Structure, 5, 325–336. [DOI] [PubMed] [Google Scholar]
- Buechler W.A., Nakane,M. and Murad,F. (1991) Expression of soluble guanylate cyclase activity requires both enzyme subunits. Biochem. Biophys. Res. Commun., 174, 351–357. [DOI] [PubMed] [Google Scholar]
- Chen Y., Cann,M.J., Litvin,T.N., Iourgenko,V., Sinclair,M.L., Levin,L.R. and Buck,J. (2000) Soluble adenylyl cyclase as an evolutionary conserved bicarbonate sensor. Science, 289, 625–628. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Desaubry L., Shoshani,I. and Johnson,R.A. (1996) 2′,5′-Dideoxyadenosine 3′-polyphosphates are potent inhibitors of adenylyl cyclases. J. Biol. Chem., 271, 2380–2382. [DOI] [PubMed] [Google Scholar]
- Dessauer C.W. and Gilman,A.G. (1996) Purification and characterization of a soluble form of mammalian adenylyl cyclase. J. Biol. Chem., 271, 16967–16974. [DOI] [PubMed] [Google Scholar]
- Dessauer C.W. and Gilman,A.G. (1997) The catalytic mechanism of mammalian adenylyl cyclase: equilibrium binding and kinetic analysis of P-site inhibition. J. Biol. Chem., 272, 27787–27795. [DOI] [PubMed] [Google Scholar]
- Dessauer C.W., Scully,T.T. and Gilman,A.G. (1997) Interactions of forskolin and ATP with the cytosolic domains of mammalian adenylyl cyclase. J. Biol. Chem., 272, 22272–22277. [DOI] [PubMed] [Google Scholar]
- Engh R.A. and Huber,R. (1991) Accurate bond and angle parameters for X-ray protein-structure refinement. Acta Crystallogr. A, 47, 392–400. [Google Scholar]
- Garbers D.L. (1989) Guanylate cyclase, a cell surface receptor. J. Biol. Chem., 264, 9103–9116. [PubMed] [Google Scholar]
- Harteneck C., Koesling,D., Soling,A., Schultz,G. and Bohme,E. (1990) Expression of soluble guanylyl cyclase. Catalytic activity requires two enzyme subunits. FEBS Lett., 272, 221–223. [DOI] [PubMed] [Google Scholar]
- Ho S.N., Hunt,H.D., Horton,R.M., Pullen,J.K. and Pease,L.R. (1989) Site-directed mutagenesis by overlap extension using polymerase chain reaction. Gene, 77, 51–59. [DOI] [PubMed] [Google Scholar]
- Hurley J.H. (1998) The adenylyl and guanylyl cyclase superfamily. Curr. Opin. Struct. Biol., 8, 770–777. [DOI] [PubMed] [Google Scholar]
- Kraulis P.J. (1991) MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- La Fortelle E. and Bricogne,G. (1997) Maximum-likelihood heavy-atom parameter refinement in the MIR and MAD methods. Methods Enzymol., 276, 472–494. [DOI] [PubMed] [Google Scholar]
- Lamzin V.S. and Wilson,K.S. (1997) Automated refinement for protein crystallography. Methods Enzymol., 277, 269–305. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur,M.W., Moss,D.S. and Thornton,J.M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr., 26, 283–291. [Google Scholar]
- Liu Y., Ruoho,A.E., Rao,V.D. and Hurley,J.H. (1997) Catalytic mechanism of the adenylyl and guanylyl cyclases: modeling and mutational analysis. Proc. Natl Acad. Sci. USA, 94, 13414–13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin B.R., Voorheis,H.P. and Kennedy,E.L. (1978) Adenylate cyclase in bloodstream forms of Trypanosoma brucei sp. Biochem. J., 175, 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews K.R. (1999) Developments in the differentiation of Trypanosonma brucei. Parasitol. Today, 15, 76–80. [DOI] [PubMed] [Google Scholar]
- Merrit E.A. and Murphy,M.E.P. (1994) Raster3D version 2.0. A program for photorealistic molecular graphics. Acta Crystallogr. D, 50, 869–873. [DOI] [PubMed] [Google Scholar]
- Nicholls A. (1992) GRASP: Graphical representation and analysis of surface properties. Columbia University, New York, NY.
- Olshevskaya E.V., Ermilov,A.N. and Dizhoor,A.M. (1999) Dimerization of guanylyl cyclase-activating protein and a mechanism of photoreceptor guanylyl cyclase activation. J. Biol. Chem., 274, 25583–25587. [DOI] [PubMed] [Google Scholar]
- Park Y., Choi,H., Lee,D.S. and Kim,Y. (1997) Improvement of the 3′-5′ exonuclease activity of Taq DNA polymerase by protein engineering in the active site. Mol. Cells, 7, 419–424. [PubMed] [Google Scholar]
- Peters E.P., Wilderspin,A.F., Wood,S.P., Zvelebil,M.J., Sezer,O. and Danchin,A. (1991) A pyruvate-stimulated adenylate cyclase has a sequence related to the fes/fps oncogenes and to eukaryotic cyclases. Mol. Microbiol., 5, 1175–1181. [DOI] [PubMed] [Google Scholar]
- Quiocho F.A. (1986) Carbohydrate-binding proteins: tertiary structures and protein–sugar interactions. Annu. Rev. Biochem., 55, 287–315. [DOI] [PubMed] [Google Scholar]
- Quiocho F.A. (1993) Probing the atomic interactions between proteins and carbohydrates. Biochem. Soc. Trans, 21, 442–448. [DOI] [PubMed] [Google Scholar]
- Rodrigues C.O., Scott,D.A. and Docampo,R. (1999) Characterization of a vacuolar pyrophosphatase in Trypanosoma brucei and its localization to acidocalcisomes. Mol. Cell. Biol., 19, 7712–7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolin S., Hanocq-Quertier,J., Paturiaux-Hanocq,F., Nolan,D., Salmon,D., Webb,H., Carrington,M., Voorheis,P. and Pays,E. (1996) Simultaneous but independent activation of adenylate cyclase and glycosylphosphatidylinositol-phospholipase C under stress conditions in Trypanosoma brucei. J. Biol. Chem., 271, 10844–10852. [DOI] [PubMed] [Google Scholar]
- Sokal I., Haeseleer,F., Arendt,A., Adman,E.T., Hargrave,P.A. and Palczewski,K. (1999) Identification of a guanylyl cyclase-activating protein-binding site within the catalytic domain of retinal guanylyl cyclase 1. Biochemistry, 38, 1387–1393. [DOI] [PubMed] [Google Scholar]
- Sunahara R.K., Dessauer,C.W. and Gilman,A.G. (1996) Complexity and diversity of mammalian adenylyl cyclases. Annu. Rev. Pharmacol. Toxicol., 36, 461–480. [DOI] [PubMed] [Google Scholar]
- Tang W.J. and Gilman,A.G. (1992) Adenylyl cyclases. Cell, 70, 869–872. [DOI] [PubMed] [Google Scholar]
- Tang W.J. and Gilman,A.G. (1995) Construction of a soluble adenylyl cyclase activated by Gsα and forskolin. Science, 268, 1769–1772. [DOI] [PubMed] [Google Scholar]
- Tang W.J., Stanzel,M. and Gilman,A.G. (1995) Truncation and alanine-scanning mutants of type I adenylyl cyclase. Biochemistry, 34, 14563–14572. [DOI] [PubMed] [Google Scholar]
- Tesmer J.J., Sunahara,R.K., Gilman,A.G. and Sprang,S.R. (1997) Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsα.GTPγS. Science, 278, 1907–1916. [DOI] [PubMed] [Google Scholar]
- Tesmer J.J., Sunahara,R.K., Johnson,R.A., Gosselin,G., Gilman,A.G. and Sprang,S.R. (1999) Two-metal-ion catalysis in adenylyl cyclase. Science, 285, 756–760. [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Gibson,T.J., Plewniak,F., Jeanmougin,F. and Higgins,D.G. (1997) The Clustal_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res., 25, 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tielens A.G.M. and Van Hellemond,J.J. (1998) Differences in energy metabolism between Trypanosomatidae. Parasitol. Today, 14, 265–271. [DOI] [PubMed] [Google Scholar]
- Urbina J.A. et al. (1999) Trypanosoma cruzi contains major pyrophosphate stores and its growth in vitro and in vivo is blocked by pyrophosphate analogs. J. Biol. Chem., 274, 33609–33615. [DOI] [PubMed] [Google Scholar]
- Vassella E., Reuner,B., Yutzy,B. and Boshart,M. (1997) Differentiation of African trypanosomes is controlled by a density sensing mechanism which signals cell cycle arrest via the cAMP pathway. J. Cell Sci., 110, 2661–2671. [DOI] [PubMed] [Google Scholar]
- Whisnant R.E., Gilman,A.G. and Dessauer,C.W. (1996) Interaction of the two cytosolic domains of mammalian adenylyl cyclase. Proc. Natl Acad. Sci. USA, 93, 6621–6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S.Z., Hahn,D., Huang,Z.H. and Tang,W.J. (1996) Two cytoplasmic domains of mammalian adenylyl cyclase form a Gsα- and forskolin-activated enzyme in vitro. J. Biol. Chem., 271, 10941–10945. [DOI] [PubMed] [Google Scholar]
- Yan S.Z., Huang,Z.W., Shaw,R.S. and Tang,W.J. (1997) The conserved asparagine and arginine are essential for catalysis of mammalian adenylyl cyclase. J. Biol. Chem., 272, 12342–12349. [DOI] [PubMed] [Google Scholar]
- Yu H., Olshevskaya,E., Duda,T., Seno,K., Hayashi,F., Sharma,R.K., Dizhoor,A.M. and Yamazaki,A. (1999) Activation of retinal guanylyl cyclase-1 by Ca2+-binding proteins involves its dimerization. J. Biol. Chem., 274, 15547–15555. [DOI] [PubMed] [Google Scholar]
- Zhang G., Liu,Y., Qin,J., Vo,B., Tang,W.J., Ruoho,A.E. and Hurley,J.H. (1997a) Characterization and crystallization of a minimal catalytic core domain from mammalian type II adenylyl cyclase. Protein Sci., 6, 903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G., Liu,Y., Ruoho,A.E. and Hurley,J.H. (1997b) Structure of the adenylyl cyclase catalytic core. Nature, 386, 247–253. [DOI] [PubMed] [Google Scholar]