

Abstract
The origin of the extent of charging and the mechanism by which multiply charged ions are formed in electrospray ionization have been hotly debated for over a decade. Many factors can affect the number of charges on an analyte ion. Here, we investigate the extent of charging of poly(propyleneimine) dendrimers (generations 3.0 and 5.0), cytochrome c, poly(ethylene glycol)s, and 1,n-diaminoalkanes formed from solutions of different composition. We demonstrate that in the absence of other factors, the surface tension of the electrospray droplet late in the desolvation process is a significant factor in determining the overall analyte charge. For poly(ethylene glycol)s, 1,n-diaminoalkanes, and poly(propyleneimine) dendrimers electrosprayed from single-component solutions, there is a clear relationship between the analyte charge and the solvent surface tension. Addition of m-nitrobenzyl alcohol (m-NBA) into electrospray solutions increases the charging when the original solution has a lower surface tension than m-NBA, but the degree of charging decreases when this compound is added to water, which has a higher surface tension. Similarly, the charging of cytochrome c ions formed from acidified denaturing solutions generally increases with increasing surface tension of the least volatile solvent. For the dendrimers investigated, there is a strong correlation between the average charge state of the dendrimer and the Rayleigh limiting charge calculated for a droplet of the same size as the analyte molecule and with the surface tension of the electrospray solvent. A bimodal charge distribution is observed for larger dendrimers formed from water/m-NBA solutions, suggesting the presence of more than one conformation in solution. A similar correlation is found between the extent of charging for 1,n-diaminoalkanes and the calculated Rayleigh limiting charge. These results provide strong evidence that multiply charged organic ions are formed by the charged residue mechanism. A significantly smaller extent of charging for both dendrimers and 1,n-diaminoalkanes would be expected if the ion evaporation mechanism played a significant role.
Introduction
Electrospray ionization (ESI)1 is well-known for its ability to form intact, extensively charged gas-phase macromolecular ions which are ideally suited for analysis by mass spectrometry (MS). The multiple charging phenomenon makes possible accurate molecular weight measurements of large molecules (up to 108 Da)2 on instruments with upper m/z limits. With ESI coupled to Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR-MS), mass measurements of large (105 Da) biomolecules can be made with unprecedented accuracy (within 1 Da).3 Multiply charged ions dissociate more readily than do singly charged ions during MS/MS experiments, and more useful information on molecular sequence and structure can be deduced from their fragmentation.4–7 Electron capture cross sections increase quadratically with charge, making electron capture dissociation (ECD) significantly more efficient with increasing charge.7 ECD of protein ions provides extensive sequence-specific fragments, which greatly improves information from the “top-down” approach to determining molecular sequence and locations of post-translational modifications.8
As shown by several workers,9–18 the analyte charge observed in the gas phase is not typically the same as that in bulk solution. With ESI, gas-phase ions with a charge opposite to that in bulk solution can be formed. For example, protein cations have been formed from basic solutions in which the protein has a net negative charge,9,10 and protein anions have been formed from acidic solutions in which the protein has a net positive charge.10 ESI mass spectra of octaethylporphyrin obtained by Van Berkel, McLuckey, and co-workers were dominated by singly protonated ions even though doubly protonated ions are the predominant species in solution. Spectroscopic measurements indicated that the conversion from doubly to singly charged analyte occurred late in the electrospray process, during desolvation of electrospray droplets within the electrospray interface.11
Several factors which influence the extent of analyte charging in ESI have been identified, including molecular conformation,12,13 competition for charge between the analyte and other species,9,11,14–17 instrumental factors,18 etc. Molecular conformation dramatically influences charge distributions of proteins,12 with elongated conformations producing charge distributions centered around high charge states, and compact conformations producing narrow distributions at low charge.12,13 Competition for charge between analyte and solvent can dramatically reduce the charge on gas-phase analyte ions.14 The charging of electrosprayed peptides depends weakly on pH,15 however, it decreases strongly with increasing solvent gas-phase basicity (GB).9,14–17 Charge reduction is further promoted by instrumental conditions that promote energetic collisions between analyte ions and neutral species, such as high skimmer voltage or capillary temperature.18 Other factors may also play a role in the observed charge state distribution. For example, Cole and co-workers demonstrated that polar, high dielectric solvents favor the formation of high charge states of phospholipid anions19 and diquaternary ammonium cations.20 This was ascribed to the superior ability of polar solvents to stabilize charge.19,20
The mechanism by which gas-phase ions are formed in ESI has been actively debated, and has been the subject of recent reviews.21,22 The two competing mechanisms are the ion evaporation mechanism (IEM)23 and the charged residue mechanism (CRM).24,25 In either mechanism, highly charged droplets are initially produced. These droplets are resistively and/or conductively heated at atmospheric pressure. As a charged electrospray droplet evaporates and decreases in size, the electric field on the droplet surface increases until it overcomes the surface tension, resulting in droplet fission. The number of charges, zR, that can be sustained by a spherical droplet of radius, R, and surface tension, γ, is given by Rayleigh’s equation26
where e is the elementary charge, and εo is the permittivity of the surrounding medium. In the IEM, gas-phase ions are formed by emission from a droplet surface when the electric field becomes sufficiently high. This is believed to occur at droplet diameter ≤20 nm.23 The charges on an electrospray droplet reside on the droplet surface, equidistant from each other to minimize the electrostatic potential energy. Fenn has proposed that the observed number of charges on a multiply charged ion is the number of charges the molecule spans on the droplet surface as it desorbs into the gas phase, which in turn depends on the surface charge density of the droplet and the size and geometry of the analyte.27 In the CRM, droplet evaporation and successive fission events continue until charged nanodroplets containing only single analyte molecules are formed, which evaporate to dry analyte ions.24,25 From the results of their pioneering experiments with cationized poly(ethylene) glycols, Nohmi and Fenn concluded that only ions larger than 5 MDa are formed by the CRM, and that smaller ions are formed by the IEM.27 Fernandez de la Mora28 recently reported that the observed charging of poly(amidoamine) (PAMAM) dendrimers29 and globular proteins electrosprayed from aqueous solutions coincides with zR for water droplets of the same size as the analyte molecules. The radii of the analyte molecules were approximated by assuming the molecules were spheres with densities of 1 g/mL. The author concluded that globular, native proteins and dendrimers of mass ≥6000 Da are formed as ions by the CRM,28 although it was not explained why the IEM would be unable to produce a similar level of charging.
We recently reported a novel phenomenon in which the absolute abundances of high charge states of protein, peptide, and synthetic polymer ions are increased, relative to those obtained from conventional “denaturing” solutions, by adding certain solvents, such as glycerol and m-nitrobenzyl alcohol, into electrospray solutions.14,30,31 For example, adding m-NBA at a level of 1% to a 47% water/50% methanol/3% acetic acid solution of cytochrome c (10−5 M) results in an increase in the maximum charge state from 21+ to 24+ and an increase in the average charge state from 17.3+ to 20.8+.31 Preliminary results31 indicated that the increased charging is due to an increase in the surface tension of the electrospray droplets. The usefulness of this method was recently demonstrated by McLafferty and co-workers,8 who performed ECD on highly charged analyte ions formed from glycerol-containing electro-spray solutions, as well as other solutions, to obtain extensive sequence information deduced from cleavage at 97% of the interresidue bonds of a 29 kDa protein. Here, we present additional evidence that the increased analyte charging is brought about by increasing the surface tension of the charged droplets from which the ions are formed. We also show that the degree of multiple charging observed for relatively small organic ions (100–7200 Da), electrosprayed from various solutions, is inconsistent with these small ions being formed by the IEM.
Experimental Section
Experiments are performed on a quadrupole mass spectrometer with an in-house-built electrospray source. This instrument is described elsewhere.32 Ions are generated by nanoelectrospray using needles that are made from 1.0 mm o.d./0.78 mm i.d. borosilicate capillaries. These capillaries are pulled to a tip with an inner diameter of ~4 μm using a Flaming/Brown micropipet puller (Model P-87, Sutter Instruments, Novato, CA). The electrospray is initiated by applying a potential of ~1000 V to a Pt wire (0.127 mm diameter, Aldrich, Milwaukee, WI) inserted into the nanoelectrospray needle to within ~2 mm of the tip. The wire and nanoelectrospray needle are held in place with a patch clamp holder (WPI Instruments, Sarasota, FL). The flow rates are between 60 and 200 nL/min. No pneumatic assistance or back pressure is used. The ions and droplets generated by electrospray are sampled from atmospheric pressure through a 12 cm long stainless steel capillary (0.50 mm i.d.) which is heated to 195 °C. The voltages on the heated metal capillary, the first and second skimmers are 12, 15, and 7 V, respectively. The voltage on the tube lens was 80 V for experiments with cytochrome c and poly(ethylene glycol)s (PEGs), 75 V for dendrimers, and 55 V for diaminoalkanes.
Methanol (99.99%) was obtained from EM Science (Gibbs-town, NJ). Glycerol (99.5+%), 2-methoxyethanol (99%), 1,n-diaminoalkanes (n = 4, 5, 7, 8, 10, 12), m-nitrobenzyl alcohol (m-NBA; 98%), poly(propyleneimine) hexadecaamine dendrimer, generation 3.0 (DAB-16; 16 peripheral amino groups; 1687 Da), poly(propyleneimine) tetrahexacontaamine dendrimer, generation 5.0 (DAB-64; 64 peripheral amino groups; 7168 Da), and PEG with an average molecular weight of 1500 Da were obtained from Aldrich Chemical Co. (Milwaukee, WI). Glacial acetic acid (99.9%), dimethyl sulfoxide (DMSO; 99.9%), 2-propanol (99.9%), sodium acetate monohydrate (99.5%), and formamide (99.5%) were obtained from Fisher Scientific (Fair Lawn, NJ). m-Chlorophenol was obtained from Matheson, Coleman, and Bell (East Rutherford, NJ), and n-butyl acetate (98%) was obtained from MCB Manufacturing Chemists (Cincinnati, OH). Equine cytochrome c (>95%) was purchased from Sigma Chemical Co. (St. Louis, MO) and was used without further purification. Electrospray solutions of cytochrome c, PEG 1500, diaminoalkanes, and dendrimers were prepared with analyte concentrations of ~10 μM, ~0.1% weight/volume, ~100 μM, and ~50 μM, respectively. The reported solution compositions are on a volume/volume basis. All cytochrome c containing electrospray solutions used in this study contained 3% acetic acid. Addition of small amounts of acid to water/methanol solutions results in higher charge states due to denaturation of proteins in solution.12 All PEG 1500-containing solutions contained 28 mM sodium acetate to promote formation of sodiated cations.
One parameter used to describe a given charge state distribution is the average charge state (zavg). This parameter is calculated as follows
where N is the number of observed analyte charge states in a given mass spectrum, zi is the net charge of the ith charge state, and wi is the signal intensity of the ith charge state. For PEGs, which are polydisperse, wi is the sum of the intensities of the various PEG oligomers of the ith charge state. Other parameters are zbase, the analyte charge state of highest abundance in a mass spectrum, and zmax, the maximum analyte charge state in a mass spectrum. The surface tension of m-NBA was measured at UC Berkeley, as described previously.31 Surface tension data for other solvents were obtained from refs 33 and 34. Boiling point data are from ref 33. Surface tension values are at 25 °C, unless otherwise noted. Molecular radii from Scherrenberg et al.,35 who characterized poly(propyleneimine) dendrimers by viscometry and small-angle neutron scattering, are used for our calculations of zR for DAB-16 and DAB-64. Reported experimental errors correspond to one standard deviation from three replicate measurements, with a fresh nanoelectrospray tip used for each measurement, unless otherwise noted. The simplifying assumption of charges spaced in a hexagonal array is used to calculate the distance between charges on the surfaces of charged droplets.36 This produces values that are slightly high for small droplets (< ~60 Å). To calculate the distance between charges on the smallest multiply charged droplets, i.e., droplets with a Rayleigh limiting charge of two, three, or four charges, the charges are assumed to lie on a spherical droplet in such a way as to maximize the distance between charges.
Results and Discussion
Dendrimers
Electrospray mass spectra of DAB-16 formed from both single and two-component solutions are shown in Figure 1. For the single-component solutions, the average charge state, zavg, decreases in the order water (4.7+; Figure 1a), acetic acid (3.6+; Figure 1f), methanol (3.3+; Figure 1c), and 2-propanol (2.6+; Figure 1e). The surface tension of water, acetic acid, methanol, and 2-propanol is 72, 27, 22, and 21 mN/m, respectively. Thus, the extent of charging follows the order of solvent surface tension.
Figure 1.
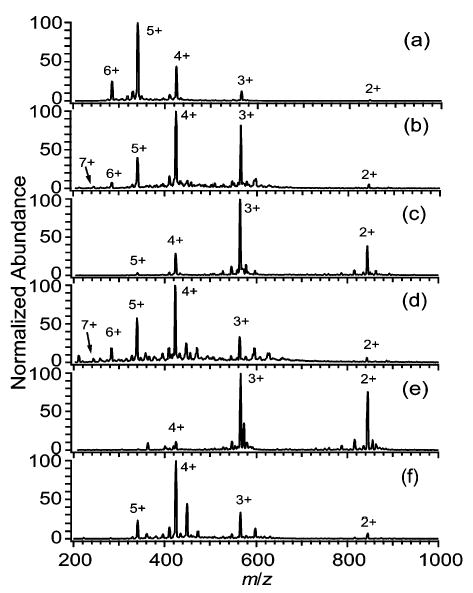
Representative mass spectra of DAB-16 (50 μM) electrosprayed from solutions of (a) water, (b) 99% water/1% m-NBA, (c) methanol, (d) 95% methanol/5% m-NBA, (e) 2-propanol, and (f) acetic acid. Singly charged ions (at m/z 1688) were detected only in spectrum (e) at 4% relative abundance.
Addition of 1% m-NBA (50 mN/m) into the aqueous electrospray solution decreases zavg by 19%, and reduces the charge state of highest abundance, zbase, from 5+ to 4+ (Figure 1b). (The maximum charge, zmax, appears to increase, however, with 7+ present at 2.6% relative abundance in Figure 1b. This will be discussed with the results for DAB-64). In contrast to the results obtained with aqueous solutions, adding 5% m-NBA to methanol solutions increases zavg, zbase, and zmax, by 27%, 33%, and 40%, respectively (Figure 1c, d). Addition of m-NBA to water lowers the surface tension, particularly later in the droplet lifetime since water has a higher vapor pressure and will preferentially evaporate. In contrast, m-NBA added to methanol increases the surface tension, with a larger increase occurring later in the droplet lifetime due to preferential evaporation of methanol. The charge state distributions of spectra obtained from water or methanol to which small amounts of m-NBA have been added are remarkably similar. This result indicates that the ions are formed late in the droplet lifetime when most of the more volatile components (water and methanol) have evaporated. Thus, addition of m-NBA to electrospray solutions of dendrimers produces a reduction or an enhancement in analyte charging in a manner consistent with surface tension dependent charging.
A similar trend in charge with surface tension of the single component solutions is observed for the larger dendrimer, DAB-64 (Figure 2a, c, e, f), although subtle differences, i.e., Figure 2c vs 2e, may be obscured by the presence of impurities or possibly by the occurrence of adduction, or by other factors. Addition of m-NBA to methanol solutions of DAB-64 produces an increase of zavg, zbase, and zmax by 31%, 33%, and 44%, respectively (Figure 2c, d), a result similar to that obtained for the smaller dendrimer. The highest observed values of zbase (11+) and zavg (9.9+) are obtained from aqueous solutions (Figure 2a). Unlike with DAB-16, addition of m-NBA to aqueous solutions of DAB-64 results in a bimodal charge distribution, with one distribution centered around 13+ and another centered around 9+ (Figure 2b). A bimodal distribution indicates that more than one conformation of the analyte is present, with more compact conformations giving rise to distributions of lower charge.12,13 Others have also presented evidence that dendrimers are not completely rigid. For example, several experimental35,37,38 and theoretical39,40 studies indicate that dendrimers assume open, extended structures in solvents in which the peripheral groups of the dendrimer interact favorably with the solvent, and assume compact structures in solvents in which the interactions are less favorable. Larger dendrimers have more degrees of conformational freedom, which may be the reason a bimodal distribution is observed upon addition of m-NBA to aqueous solutions of DAB-64 (Figure 2b), while only a subtler broadening of the charge state distribution, i.e., the appearance of 7+, occurs for DAB-16 (Figure 1b).
Figure 2.
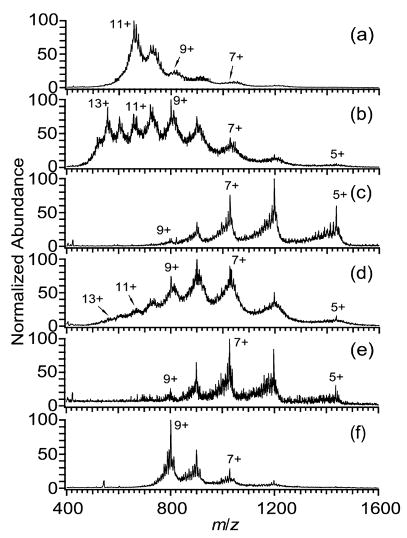
Representative mass spectra of DAB-64 (50 μM) electrosprayed from solutions of (a) water, (b) 99% water/1% m-NBA, (c) methanol, (d) 95% methanol/5% m-NBA, (e) 2-propanol, and (f) acetic acid.
The values of zbase and zavg are all within 10% of the Rayleigh limiting charge, zR, for DAB-16 electrosprayed from water, methanol, and 2-propanol except for zavg obtained from methanol, which is within 17% of zR. Electrospray spectra from acetic acid solutions have a slightly greater degree of charging than that calculated for zR, with zavg and zbase greater than zR by 16% and 28%, respectively. Interestingly, the value of zavg obtained when electrospraying from 5% m-NBA/95% methanol is equal to zR for droplets composed only of m-NBA. This suggests that the analyte ions may be formed from electrospray droplets that have evaporated to nearly 100% m-NBA. When the larger dendrimer, DAB-64, is electrosprayed from single component solutions, values of zavg and zbase are within 10% of zR, except in the cases of acetic acid and 2-propanol, for which zavg and zbase are within 33% of zR. The discrepancies between the calculated and observed charging may be due in part to the dendrimers assuming different molecular sizes in nonaqueous solvents,35,37–40 or to impurities that could have a significant effect on the surface tension late in the droplet lifetime. The molecular sizes used in the calculation of zR were measured only in aqueous solutions.35 As expected, there is very good agreement between the observed and calculated charging for both DAB-16 and DAB-64 electrosprayed from aqueous solutions (Tables 1, 2).
Table 1.
Comparison of the Experimentally Measured Average Charge State (zavg) and Charge State of Highest Abundance (zbase) of DAB-16 Dendrimer (5 × 10−5 M), with the Rayleigh Charge (zR) of a Spherical Droplet of the Same Sizea as the Dendrimer, for Electrospray Solutions with Various Surface Tensions (Errors reported as one standard deviation from three replicate measurements)
| electrospray solventb | surface tension (mN/m) | zR | zbase | zavg | zbase/zR | zavg/zR |
|---|---|---|---|---|---|---|
| water | 72 | 5.1 | 5 | 4.72 ± 0.01 | 0.98 | 0.93 |
| m-NBAc | 50d | 4.2e | 4 | 4.2 ± 0.1 | 0.94 | 1.00 |
| acetic acid | 27 | 3.1 | 4 | 3.6 ± 0.1 | 1.28 | 1.16 |
| methanol | 22 | 2.8 | 3 | 3.3 ± 0.1 | 1.07 | 1.17 |
| 2-propanol | 21 | 2.7 | 3 | 2.63 ± 0.02 | 1.09 | 0.96 |
Radius of 11.8 Å, from ref 35.
Single-component electrospray solutions were used, unless otherwise noted.
The electrospray solution was 5% m-NBA/95% methanol.
Surface tension of m-NBA (98% pure material).
Rayleigh charge for a droplet composed of only m-NBA.
Table 2.
Comparison of the Experimentally Measured Average Charge State (zavg) and Charge State of Highest Abundance (zbase) of DAB-64 Dendrimer (5 × 10−5 M), with the Rayleigh Charge (zR) of a Spherical Droplet of the Same Sizea as the Dendrimer, for Electrospray Solutions with Various Surface Tensions (Errors are reported as one standard deviation from three replicate measurements)
| electrospray solventb | surface tension (mN/m) | zR | zbase | zavg | zbase/zR | zavg/zR |
|---|---|---|---|---|---|---|
| water | 72 | 11.0 | 11 | 9.9 ± 0.1 | 1.00 | 0.90 |
| m-NBAc | 50d | 9.2e | 8 | 8.8 ± 0.4 | 0.87 | 0.96 |
| acetic acid | 27 | 6.8 | 9 | 8.4 ± 0.2 | 1.33 | 1.24 |
| methanol | 22 | 6.1 | 6 | 6.7 ± 0.3 | 0.98 | 1.10 |
| 2-propanol | 21 | 6.0 | 7 | 7.6 ± 0.5 | 1.17 | 1.27 |
Radius of 19.8 Å, from ref 35.
Single-component electrospray solutions were used, unless otherwise noted.
The electrospray solution was 5% m-NBA/95% methanol.
Surface tension of m-NBA (98% pure material).
Rayleigh charge for a droplet composed of only m-NBA.
The observation that the extent of charging of dendrimers is approximately equal to the Rayleigh limiting charge of a solvent droplet of the same size as the analyte is consistent with these ions being formed by the charged residue mechanism (CRM).24,25,28 In this mechanism, the sequence of evaporation and fission events of electrospray droplets eventually results in charged nanodroplets that each contain one analyte molecule. Charge is partitioned between the analyte molecule and the solvent molecules as the final few solvent molecules evaporate, resulting in the formation of an unsolvated gas-phase analyte ion. The number of charges available to the analyte ion is limited by the number of charges on the nanodroplet, which is a function of the surface tension of the solvent.26
In the ion evaporation mechanism (IEM), in which the analyte ion desorbs from the surface of a charged droplet that is larger than the analyte molecule, the analyte charge is determined by how many charges the analyte molecule spans on the surface of the charged droplet as it desorbs into the gas phase, i.e., by the surface charge density of the droplet and the size of the analyte molecule.27 Figure 3 shows the surface charge density, the charge–charge distance, and the number of charges of a water droplet charged at the Rayleigh limit as a function of droplet radius. The charge density at the Rayleigh limit increases significantly with decreasing droplet size. This leads to a corresponding decrease in the distance between charges with decreasing droplet size. The reason for this is that there are fewer overall charges on smaller droplets and, hence, fewer pairwise interactions between charges. Although the Rayleigh formula might not apply rigorously for very small droplets, the trend in charge density with droplet size is clear. An ion desorbing from a droplet which is much larger than the analyte will not carry away as many charges as it would if it were formed by the CRM. For example, if DAB-16 ions desorbed from the surface of a 10 nm water droplet charged at the Rayleigh limit, the ions would each span only one charge as they left the droplet surface. This value is only one-fifth the observed zbase for this analyte electrosprayed from water (Table 1). As described by Fenn,27 the IEM charge is calculated as the number of charges on the droplet surface that are spanned by the cross section of the molecule. The largest area the analyte molecule could imaginably present to the droplet surface is the total surface area of the molecule. Even if the entire molecular surface area were exposed to charges on the droplet surface, then DAB-16 ions would each span only 2.5 charges on average as they left the surface of a 10 nm water droplet. This calculated value is only half the observed zbase for this analyte electrosprayed from water (Table 1). Thus, only the CRM can produce the extent of dendrimer charging observed in these experiments.
Figure 3.
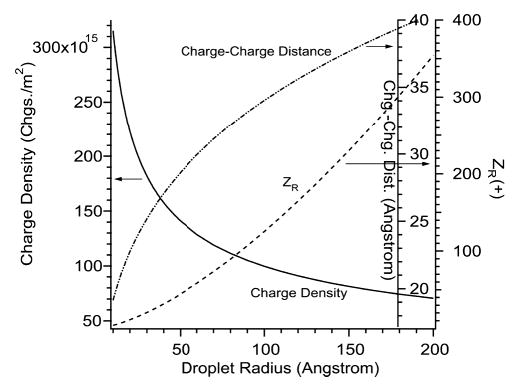
Effects of droplet radius on the surface charge density, the distance between charges, and the total number of charges (zR) for a spherical water droplet charged at the Rayleigh limit. Accurate charge–charge distances for the smallest charged droplets are given in Figure 10.
It has been proposed that a protonating agent, such as ammonium acetate, is needed for optimum production of protonated gas-phase analyte ions.41 In these experiments, no such agent was added to the electrospray solutions, yet highly protonated molecular ions are formed (Figure 1). This indicates that solvated protons (H3O+) are sufficient by themselves to cationize the analyte.
Cytochrome c
Electrospray mass spectra of cytochrome c ions formed from denaturing solutions (97% water/3% acetic acid or 47% water/50% methanol/3% acetic acid) containing varying levels of glycerol, m-NBA, DMSO, formamide, m-chlorophenol, and 2-methoxyethanol were obtained. Increasing the level of each of these solvents up to a certain level increases analyte charging. Beyond this level a reduction in charge and/or loss of stable electrospray signal occurs (Figure 4). For example, increasing the level of DMSO from 0% to 50% increases zavg by 17%, but zavg decreases by 15% as the DMSO level is increased from 50% to 87%. Figure 5 shows a plot of zavg obtained using optimum charge enhancing concentrations of additives as a function of the surface tension of the least volatile solvent component. There is a rough correlation between the extent of analyte charging and the surface tension of the least volatile solvent component, with charge enhancement decreasing in the order m-NBA > glycerol > DMSO > m-chlorophenol ≈ formamide ≈ 2-methoxyethanol. These solvents all increase zavg, but only glycerol and m-NBA increase zMax of cytochrome c ions, to 23+ and to 24+, respectively, from 21+ (Figure 4b).
Figure 4.
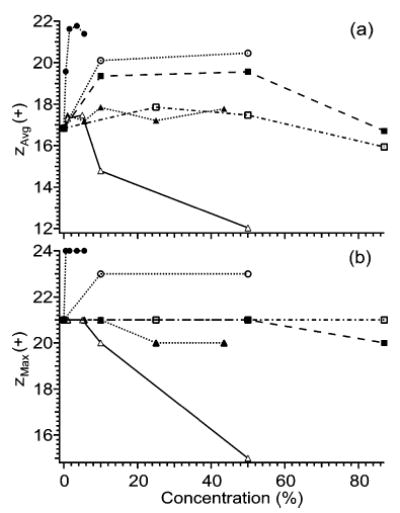
Effects of adding m-NBA (closed circles), glycerol (open circles), DMSO (closed squares), 2-methoxyethanol (open squares), m-chlorophenol (closed triangles), and formamide (open triangles) to electrospray solutions of cytochrome c (10−5 M), on (a) average and (b) maximum charge state. For m-NBA and m-chlorophenol, the starting solution is 47% methanol/50% water/3% acetic acid. For all other compounds, the starting solution is 97% water/3% acetic acid.
Figure 5.
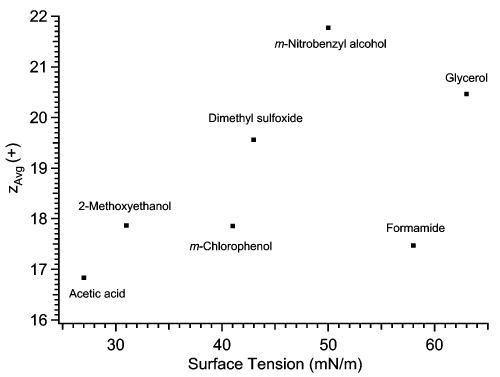
Effects of the surface tension of the least volatile solvent component on the highest values for zavg achieved in the experiments described in the text. Each point is labeled with the name of the corresponding least volatile solvent component.
DMSO and formamide have the additional effect of promoting bimodal charge distributions. For the standard denaturing solution, only a single charge distribution centered around 17+ is observed. This distribution shifts toward higher charge as DMSO or formamide is added into the electrospray solution resulting in an increasing average charge state. As the level of formamide surpasses ~10% or the level of DMSO surpasses ~87%, a second distribution centered around 12+ or 14+, respectively, is observed. The appearance of the distribution at lower charge has the effect of reducing the average charge state. It was noted previously that glycerol also induces bimodal protein charge distributions.31 Polyols, such as glycerol, stabilize folded conformations of proteins,42 which give rise to charge distributions centered at lower charge.12
Low vapor pressure alone is not sufficient for a solvent to promote high charge states. Addition of up to 48% n-butyl acetate, a solvent with a relatively high boiling temperature (126 °C) and low surface tension (25 mN/m), to electrospray solutions (47% water/50% methanol/3% acetic acid; n-butyl acetate displacing water and methanol) of cytochrome c decreases zavg by up to 6.5%. Similarly, as the level of acetic acid (bp 118 °C, surface tension = 27 mN/m) in an aqueous cytochrome c solution is increased from 3% to 90%, the average charge decreases by 6%.
Electrospray solutions of cytochrome c contain acetic acid to promote denaturation. Increasing the level of acetic acid in a water/acetic acid solution decreases the surface tension (Figure 6).43 Evaporation of a water/acetic acid solution results in an enrichment in acetic acid due to the low vapor pressure of acetic acid relative to that of water.44 Therefore, when such solutions are used in ESI, the surface tension of the droplets is continuously changing with time. For ions formed by the CRM, the surface tension of the droplets from which ions are formed is closer to that of acetic acid (27 mN/m) than to that of water (72 mN/m). Adding low vapor pressure solvents with surface tensions higher than that of acetic acid then results in “mature” electrospray droplets with increased surface tension, and hence an improved ability to sustain charges. As expected, the charge-enhancing solvents identified thus far all have higher surface tensions and boiling points than acetic acid.
Figure 6.
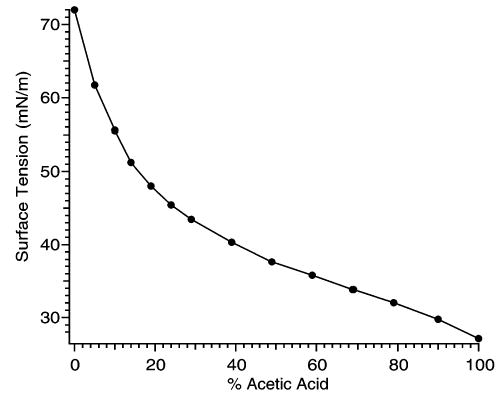
Effects of increasing the percentage acetic acid (v/v) on the surface tension of water/acetic solutions. Data are experimentally measured values from ref 43.
PEGs
Synthetic polymers, such as PEGs, are typically cationized in ESI by metal cation adduction, e.g., Na+ (Figure 7).45 A higher value of zavg is observed when PEG 1500 is electrosprayed from aqueous solutions (2.61 ± 0.09; Figure 7c) than when it is electrosprayed from methanol solutions (2.39 ± 0.08; Figure 7a). Adding m-NBA at a level of 20% to methanol solutions of PEG 1500 results in a dramatic charge enhancement, increasing zavg by 40% (Figure 7b). Adding m-NBA at a level of 1% to aqueous solutions of PEG 1500 decreases zavg by 4% (Figure 7d). As described previously,31 the addition of m-NBA also shifts the observed molecular weight distributions slightly but reproducibly toward lower molecular weight.
Figure 7.
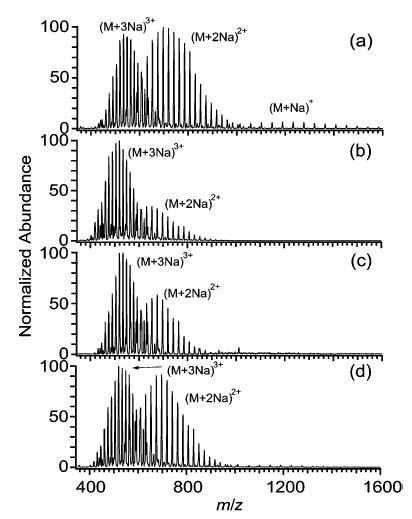
Electrospray mass spectra of PEG 1500 (0.1% w/v) from solutions containing (a) methanol, (b) 80% methanol/20% m-NBA, (c) water, and (d) 99% water/1% m-NBA. All solutions contained 28 mM sodium acetate to facilitate the formation of Na+ adducts.
These trends are consistent with surface tension dependent charging, but it is noteworthy that the highest value of zavg is achieved not from the aqueous solution, but from the methanol/m-NBA solution (2.79 ± 0.01). This may be due to PEG molecules assuming more extended conformations in methanol/m-NBA solutions than in aqueous solutions.
Diaminoalkanes
Conformational effects can clearly contribute to the observed charge state distribution. To avoid conformational effects, the dependence of charging of 1,n-diaminoalkanes as a function of solvent was investigated. Both (M + 2H)2+ and (M + H)+ of 1,12-diaminododecane (DADD) are typically formed in ESI. The charging of this diamine follows the same basic trends as dendrimers and PEGs (Figure 8). A higher zavg (1.56 ± 0.01) is observed from aqueous solutions (Figure 8a) than from methanol solutions (1.14 ± 0.03; Figure 8c). The addition of 1% m-NBA to aqueous solutions reduces zavg by 17% (to 1.30 ± 0.03; Figure 8b), whereas the addition of 5% m-NBA to methanol solutions increases zavg by 5.3% (to 1.20 ± 0.02; Figure 8d). m-NBA was not added into aqueous solutions at levels greater than 1% due to solubility limitations. Gas-phase proton transfer46 from doubly protonated diamines to solvent molecules does not appear to be responsible for these trends in charging. The GB of m-NBA is not known, however, the GBs of nitrobenzene and benzyl alcohol (184 and 179 kcal/mol, respectively) are both higher than that of methanol (173 kcal/mol)47 indicating that m-NBA likely also has a GB higher than that of methanol. Thus, adding m-NBA to methanol solutions should have produced a reduction in charging, instead of an enhancement, if proton transfer was responsible for the observed trends in charging.
Figure 8.
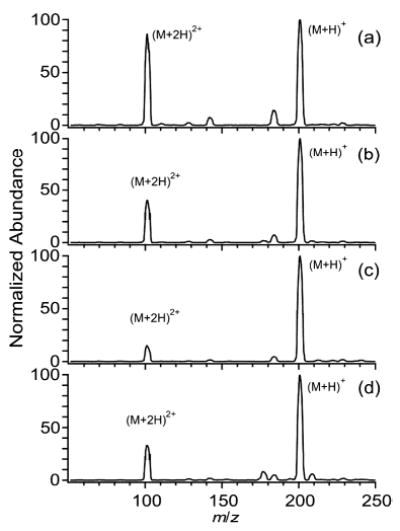
Electrospray mass spectra of 1,12-diaminododecane (10−4 M) from solutions of (a) water, (b) 99% water/1% m-NBA, (c) methanol, and (d) 95% methanol/5% m-NBA.
1,n-Diaminoalkanes (H3N(CH2)nNH2) of varying chain length were electrosprayed from water, methanol, and 5% m-NBA/95% methanol solutions. For aqueous solutions, zavg rises slightly from n = 4, for which only singly charged ions are detected, to n = 5, for which the doubly charged ion is detected at 6% relative abundance. A dramatic increase is then observed from n = 5 to 7, with zavg increasing by nearly 50% (Figure 9). A gradual increase in zavg of 11% is observed from n = 7 to 12. In contrast to these results, zavg gradually increases by only 24% from n = 4 to 12 with methanol solutions. The data of 5% m-NBA/95% methanol solutions closely follow the data of methanol solutions, except for an abrupt rise of 19% from n = 10 to 12 (the corresponding increase for methanol solutions is only 3%).
Figure 9.
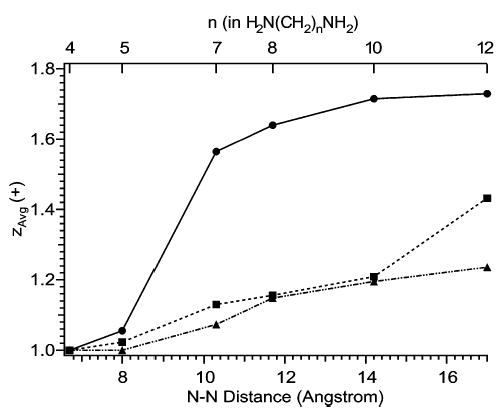
Effects of varying the chain length on the average charge state of 1,n-diaminoalkanes electrosprayed from solutions of water (circles), methanol (triangles), and 95% methanol/5% m-NBA (squares).
For diamines electrosprayed from water, a dramatic drop in dication abundance occurs for diamines with N–N distances below that of 1,7-diaminoalkane, for which the N–N distance is 10.1 Å. This distance is 2.7 Å less than the diameter of a water droplet for which zR = 2, the shortest distance between a pair of charges on a water droplet permitted by the Rayleigh equation (Figure 10). The distance between charges on droplets charged at the Rayleigh limit increases with increasing droplet size and charge (Figures 3, 10). For example, the distance between charges (measured along a line passing through the droplet) on water droplets for which zR = 2, 3, and 4, is 12.8, 14.4, and 16.3 Å, respectively (Figure 10). A large drop in dication abundance is expected when the N–N distance is smaller than the distance between charges on the droplet. Thus, the observed charging of diamines n = 7 and 8 is greater than predicted from Rayleigh calculations. This may be due to the analyte molecule stabilizing the charged nanodroplet. Also, in the final stages of evaporation, water molecules would be expected to solvate the charged ends of the dication, forming something closer to a “dumbbell” structure (Figure 11b) than to a sphere with the diamine running through the center (Figure 11a). Such a structure would have a slightly larger diameter than that of a sphere.
Figure 10.
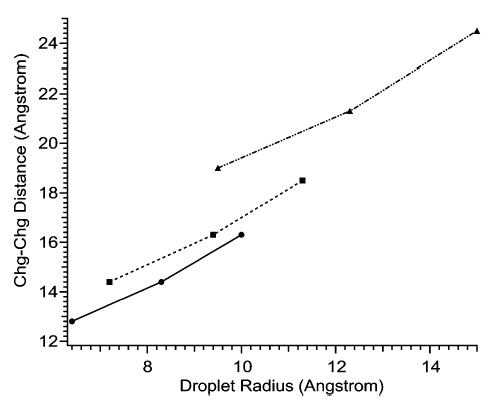
Effects of varying the droplet radius on the distance between charges on spherical droplets of water (circles), methanol (triangles), and m-NBA (squares), charged at the Rayleigh limit, for droplets for which zR = 2, 3, and 4 elementary charges. The distance between charges increases with increasing zR.
Figure 11.
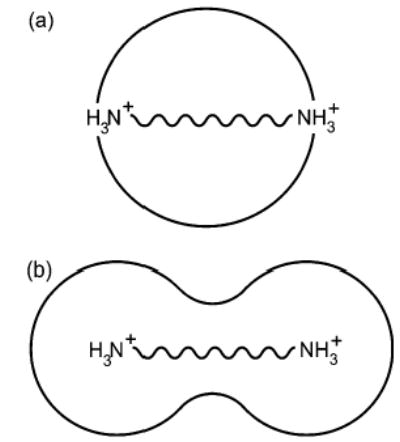
Cartoon representations of a diprotonated diamine in a spherical water droplet with (a) its charged ends at the surface, and (b) of the same dication with approximately the same volume of water distributed between two spheres which solvate the charges.
No dramatic increase in zavg is observed with methanol solutions. The distance between charges on a methanol droplet for which zR = 2 is 19 Å (Figure 10). This value is 2 Å longer than the N–N distance of the largest diamine (n = 12) used in this study. Extending the chain length to some point beyond n = 12 would presumably result in a large increase in charging similar to the one observed for aqueous solutions. Unfortunately, we are not aware of larger diaminoalkanes that are commercially available. The gradual increase in relative abundance of (M + 2H)2+ with increasing chain length is unexpected, based solely on the Rayleigh equation, because two charges are not supposed to approach each other closely enough in methanol droplets to be spanned by the diamines. The appearance of the small fraction of doubly protonated ions could be due to a number of factors, including fluctuation of charges around their equilibrium positions on the droplets, occasionally allowing doubly protonated diamines to be formed, or stabilization of nanodroplets by the analyte molecule, or nonspherical droplets (Figure 11b).
In the case of 5% m-NBA/95% methanol, the exact composition of droplets from which analyte ions are formed is not known due to differential evaporation rates of methanol and m-NBA. It is noteworthy, however, that zavg drops abruptly as the N–N distance decreases below 14.2–17 Å (n = 10–12), which is similar to the distance between charges for a droplet containing only m-NBA for which zR = 2 (14.4 Å).
As with dendrimers, the observation of doubly protonated diamines is inconsistent with the IEM. Droplets charged at the Rayleigh limit with charge–charge distances capable of being spanned by the diamines are on the same order of size as the molecules themselves (Figure 10). Diamines at the surfaces of larger droplets would each be able to reach only a single charge. For example, the distance between charges on a 20 nm water droplet, charged at the Rayleigh limit (zR = 125) is 34 Å, twice the N–N distance of the largest diamine (n = 12) used in these experiments. It is possible that higher charge states could be produced by a dynamic ion evaporation mechanism whereby one end of the diaminoalkane is desorbed and charge reorganization at the droplet surface results in protonating the other end of the diaminoalkane as well. This process would be analogous to the asymmetric droplet fission process observed for large electrospray droplets in which tiny droplets are emitted from a larger droplet but these tiny droplets carry away a disproportionate fraction of the charge.
Conclusions
The extent of multiple charging observed in electrospray ionization depends on many factors. While charge reduction of an analyte is common, charge enhancement is not. We demonstrate for the first time a clear relationship between the analyte charge and the surface tension of the electrospray droplets for a variety of protonated and cationized species. Droplets with a higher surface tension can hold more charges before reaching the Rayleigh limit. The higher charge density on the surface of such droplets makes available more charges for analyte ions that are formed from the droplets. This phenomenon is the origin of the dramatic charge enhancement or supercharging brought about by adding solvents, such as m-NBA, into denaturing electrospray solutions of proteins. In the absence of other factors, there is a good correlation between the number of charges on an analyte ion and the Rayleigh limiting charge of a solvent droplet of the same size as the analyte molecule. This result is consistent with multiply charged organic ions as small as one hundred daltons being formed by the charged residue mechanism. If these ions were formed by the ion evaporation mechanism, the mechanism that is widely believed to be responsible for the formation of small multiply charged organic ions, then the observed extent of charge would be lower by a factor of ~5.
Although these results provide strong evidence that multiply protonated or cationized organic molecules are produced by the charged residue model, we cannot rule out the possibility that the singly charged ions or multivalent cations/anions are formed by the ion evaporation mechanism. However, it is clear that the transition from solution-phase to gas-phase structure for multiply charged organic ions is determined by the solvent evaporation process.
Acknowledgments
Generous financial support for this research was provided by the National Institutes of Health (Grant No. R01-GM64712-01). We also acknowledge Hewlett-Packard Co. for their contribution of mass spectrometer components.
References
- 1.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 2.Chen R, Cheng X, Mitchell DW, Hofstadler SA, Wu Q, Rockwood AL, Sherman MG, Smith RD. Anal Chem. 1995;67:1159–1163. [Google Scholar]
- 3.Kelleher NL, Senko MW, Siegel MM, McLafferty FW. J Am Soc Mass Spectrom. 1997;8:380–383. [Google Scholar]
- 4.McLafferty FW. Acc Chem Res. 1994;27:379–386. [Google Scholar]
- 5.Jockusch RA, Schnier PD, Price WD, Strittmatter EF, Demirev PA, Williams ER. Anal Chem. 1997;69:1119–1126. doi: 10.1021/ac960804q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reid GE, Wu J, Chrisman PA, Wells JM, McLuckey SA. Anal Chem. 2001;73:3274–3281. doi: 10.1021/ac0101095. [DOI] [PubMed] [Google Scholar]
- 7.Zubarev RA, Horn DM, Fridriksson EK, Kelleher NL, Kruger NA, Lewis MA, Carpenter BK, McLafferty FW. Anal Chem. 2000;72:563–573. doi: 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
- 8.Sze SK, Ge Y, Oh H, McLafferty FW. Proc Natl Acad Sci USA. 2002;99:1774–1779. doi: 10.1073/pnas.251691898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le Blanc JCY, Wang J, Guevremont R, Siu KWM. Org Mass Spectrom. 1994;29:587–593. [Google Scholar]
- 10.Kelly MA, Vestling MM, Fenselau CC, Smith PB. Org Mass Spectrom. 1992;27:1143–1147. [Google Scholar]
- 11.Chillier XFD, Monnier A, Bill H, Gülacüar FO, Buchs A, McLuckey SA, Van Berkel GJ. Rapid Commun Mass Spectrom. 1996;10:229–304. [Google Scholar]
- 12.Chowdhury SK, Katta V, Chait BT. J Am Chem Soc. 1990;112:9012–9013. [Google Scholar]
- 13.Konermann L, Douglas DJ. Biochemistry. 1997;36:12 296–12 302. doi: 10.1021/bi971266u. [DOI] [PubMed] [Google Scholar]
- 14.Iavarone AT, Jurchen JC, Williams ER. J Am Soc Mass Spectrom. 2000;11:976–985. doi: 10.1016/S1044-0305(00)00169-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang G, Cole RB. Org Mass Spectrom. 1994;29:419–427. [Google Scholar]
- 16.Schnier PD, Gross DS, Williams ER. J Am Soc Mass Spectrom. 1995;6:1086–1097. doi: 10.1016/1044-0305(95)00532-3. [DOI] [PubMed] [Google Scholar]
- 17.Mirza UA, Chait BT. Anal Chem. 1994;66:2898–2904. doi: 10.1021/ac00090a017. [DOI] [PubMed] [Google Scholar]
- 18.Thomson BA. J Am Soc Mass Spectrom. 1997;8:1053–1058. [Google Scholar]
- 19.Cole RB, Harrata AK. J Am Soc Mass Spectrom. 1993;4:546–556. doi: 10.1016/1044-0305(93)85016-Q. [DOI] [PubMed] [Google Scholar]
- 20.Wang G, Cole RB. J Am Soc Mass Spectrom. 1996;7:1050–1058. doi: 10.1016/1044-0305(96)00051-7. [DOI] [PubMed] [Google Scholar]
- 21.Kebarle P, Peschke M. Anal Chim Acta. 2000;406:11–35. [Google Scholar]
- 22.Cole RB. J Mass Spectrom. 2000;35:763–772. doi: 10.1002/1096-9888(200007)35:7<763::AID-JMS16>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 23.Iribarne JV, Thomson BA. J Chem Phys. 1976;64:2287–2294. [Google Scholar]
- 24.Dole M, Mach LL, Hine RL, Mobley RC, Ferguson LP, Alice MP. J Chem Phys. 1968;49:2240–2249. [Google Scholar]
- 25.Schmelzeisen-Redeker G, Bütfering L, Röllgen FW. Int J Mass Spectrom Ion Processes. 1989;90:139–150. [Google Scholar]
- 26.Lord Rayleigh. Philos Mag. 1882;14:184–186. [Google Scholar]
- 27.a Fenn JB. J Am Soc Mass Spectrom. 1993;4:524–535. doi: 10.1016/1044-0305(93)85014-O. [DOI] [PubMed] [Google Scholar]; b Nohmi T, Fenn JB. J Am Chem Soc. 1991;114:3241–3246. [Google Scholar]
- 28.Fernandez de la Mora J. Anal Chim Acta. 2000;406:93–104. [Google Scholar]
- 29.Tolic LP, Anderson GA, Smith RD, Brothers HM, Spindler R, Tomalia DA. Int J Mass Spectrom Ion Processes. 1997;165:405–418. [Google Scholar]
- 30.Iavarone AT, Jurchen JC, Williams ER. Anal Chem. 2001;73:1455–1460. doi: 10.1021/ac001251t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iavarone AT, Williams ER. Int J Mass Spectrom. 2002;219:63–72. [Google Scholar]
- 32.Rodriguez-Cruz SE, Klassen JS, Williams ER. J Am Soc Mass Spectrom. 1999;10:958–968. doi: 10.1016/S1044-0305(99)00068-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lide, D. R., Ed. CRC Handbook of Chemistry and Physics; CRC Press: Boca Raton, 1996.
- 34.Daubert, T. E.; Danner, R. P. Physical and Thermodynamic Properties of Pure Chemicals Data Compilation; Hemisphere Publishing Group: New York, 1989.
- 35.Scherrenberg R, Cousseens B, Van Vliet P, Edouard G, Brackman J, De Brabander E. Macromolecules. 1998;31:456–461. [Google Scholar]
- 36.Fenn JB, Rosell J, Meng CK. J Am Soc Mass Spectrom. 1997;8:1147–1157. [Google Scholar]
- 37.Chai M, Niu Y, Youngs PL, Rinaldi J. J Am Chem Soc. 2001;123:4670–4678. doi: 10.1021/ja002824m. [DOI] [PubMed] [Google Scholar]
- 38.De Backer S, Prinzie Y, Verheijen W, Smet M, Desmedt K, Dehaen W, De Schryver FC. J Phys Chem A. 1998;102:5451–5455. [Google Scholar]
- 39.Murat M, Grest GS. Macromolecules. 1996;29:1278–1292. [Google Scholar]
- 40.Welch P, Muthukumar M. Macromolecules. 1998;31:5892–5897. [Google Scholar]
- 41.Felitsyn N, Peschke M, Kebarle P. Int J Mass Spectrom. 2002;219:39–62. [Google Scholar]
- 42.Back JF, Oakenfull D, Smith MB. Biochemistry. 1979;18:5191–5196. doi: 10.1021/bi00590a025. [DOI] [PubMed] [Google Scholar]
- 43.Álvarez E, Vázquez G, Sánchez-Vilas M, Sanjurjo B, Navaza JM. J Chem Eng Data. 1997;42:957–960. [Google Scholar]
- 44.Siu KWM, Guevremont R, Le Blanc JCY, O’Brien RT, Berman SS. Org Mass Spectrom. 1993;28:579–584. [Google Scholar]
- 45.Wong SF, Meng CK, Fenn JB. J Phys Chem. 1988;92:546–550. [Google Scholar]
- 46.Williams ER. J Mass Spectrom. 1996;31:831–842. doi: 10.1002/(SICI)1096-9888(199608)31:8<831::AID-JMS392>3.0.CO;2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hunter, E. P.; Lias, S. G. In NIST Chemistry Webbook, NIST Standard Reference Database Number 69; Mallard, W. G.; Lindstrom, P. J., Eds., National Institute of Standards and Technology: Gaithersburg, MD 29899, March 1998.