

Abstract
Hsp70 and Hsp90 protein chaperones cooperate in a protein-folding pathway required by many “client” proteins. The co-chaperone Sti1p coordinates functions of Hsp70 and Hsp90 in this pathway. Sti1p has three tetratricopeptide repeat (TPR) domains. TPR1 binds Hsp70, TPR2a binds Hsp90, and the ligand for TPR2b is unknown. Although Sti1p is thought to be dedicated to the client folding pathway, we earlier showed that Sti1p regulated Hsp70, independently of Hsp90, in a way that impairs yeast [PSI+] prion propagation. Using this prion system to monitor Sti1p regulation of Hsp70 and an Hsp90-inhibiting compound to monitor Hsp90 regulation, we identified Sti1p mutations that separately affect Hsp70 and Hsp90. TPR1 mutations impaired Sti1p regulation of Hsp70, but deletion of TPR2a and TPR2b did not. Conversely, TPR2a and TPR2b mutations impaired Sti1p regulation of Hsp90, but deletion of TPR1 did not. All Sti1p mutations variously impaired the client folding pathway, which requires both Hsp70 and Hsp90. Thus, Sti1p regulated Hsp70 and Hsp90 separately, Hsp90 is implicated as a TPR2b ligand, and mutations separately affecting regulation of either chaperone impair a pathway that is dependent upon both. We further demonstrate that client folding depended upon bridging of Hsp70 and Hsp90 by Sti1p and find conservation of the independent regulation of Hsp70 and Hsp90 by human Hop1.
Hsp70 and Hsp90 are abundant, essential, and stress-inducible chaperones that assist protein folding. By binding and releasing hydrophobic surfaces on partially folded proteins, Hsp70 plays a general role in helping proteins adopt and maintain native conformations. It also acts in cellular processes during which proteins are partially folded, such as translation and transport across membranes, and protects cells from stress by preventing protein aggregation. Although Hsp90 might also have a general role as a protein chaperone, it is known to act in a well characterized folding pathway for a large number of “client” proteins in the cell, many of which, such as steroid hormone receptors, kinases, and transcription factors, are involved in signaling (1, 2).
This client folding pathway involves Hsp70 and its co-chaperone Hsp40 as well as many co-chaperones that regulate activity of Hsp90. Client proteins interact first with Hsp40 (3), which helps target them to Hsp70, and then are transferred from Hsp70 to Hsp90 to finish folding. The co-chaperone Sti1p (Hop1 in humans) is thought to facilitate this transfer by binding to both Hsp90 and the substrate-bound Hsp70, forming a physical link between them (4–6). Other co-chaperones then bind to Hsp90 while Hop1 and Hsp70 are released from the complex. In vitro, Sti1p both stimulates ATPase activity of Hsp70 and inhibits ATPase activity of Hsp90 (7, 8). These functions of Sti1p agree with data suggesting that in addition to simply bridging Hsp70 and Hsp90, Sti1p and Hop1 affect the client folding pathway through regulation of the conformations and ATPase cycles of the two chaperones (9, 10).
The domain structure and function of Sti1p are evolutionarily conserved. Tetratricopeptide repeat motifs at the amino terminus (TPR1)2 and middle region (TPR2a) mediate the physical interactions of Sti1p with Hsp70 and Hsp90, respectively. A third TPR region immediately downstream of TPR2a, namely TPR2b, is also important for Sti1p function, but the ligand for this domain is unknown. Sti1p also has smaller regions containing conserved aspartate and proline residues following TPR1 (designated DP1) and following TPR2b (designated DP2). Altering TPR residues of Hop1/Sti1p that are known to be important for making contacts with Hsp70 and Hsp90 predictably reduce the ability of Hop1 to physically interact with them and to function in place of Sti1p in a yeast client folding system (11–13). Mutations in DP2, but not DP1, were also shown to be important for Hop1 function and its interaction with human Hsp70.
Although Sti1p and Hop1 have been found in complexes without Hsp70 and Hsp90, as a co-chaperone Sti1p is thought to be dedicated to the client folding pathway. Our recent work with the yeast [PSI+] prion, however, showed that Sti1p was required for a mutant Hsp70 (Ssa1-21p) to impair [PSI+] propagation and that [PSI+] was unaffected by various conditions that impair Hsp90 function (14). SSA1–21 [PSI+] cells thus provide a unique system for monitoring the ability of Sti1p to specifically regulate Hsp70 function. Specific regulation of Hsp90 function by Sti1p is important for cell growth, as depleting Sti1p makes cells hypersensitive to lethal effects of compounds such as radicicol that specifically inactivate Hsp90. Because SSA1–21 cells have normal sensitivity to radicicol, which has no effect on [PSI+] propagation, these cells are also useful for monitoring the ability of Sti1p to specifically regulate Hsp90 function.
We pursued our hypothesis that Sti1p can independently regulate Hsp70 and Hsp90 by using our system to search for mutations in Sti1p that separately affect [PSI+] and radicicol sensitivity. We identified several such mutations, showing that these phenotypes are dependent upon the ability of Sti1p to regulate Hsp70 and Hsp90 separately. Not unexpectedly, Sti1p mutations that affect Hsp70 function were located in regions known to be important for physical interactions of Sti1p with Hsp70, and those affecting Hsp90 function were in regions important for Sti1p interaction with Hsp90. Further, we have shown conservation of the independent regulation of Hsp70 and Hsp90 between Sti1p and its human homolog Hop1.
EXPERIMENTAL PROCEDURES
Yeast Strain, Media, and Growth Conditions
Strain 1016 (Mat α kar1-1 SUQ5 ade2-1 his3Δ202 leu2Δ1 trp1Δ63 ura3-52 SSA1–21 sti1::KanMX (14)) was used. Media with limiting adenine (~10 mg/l) have enough adenine to allow ade2-1 cells to grow but not enough to repress the adenine biosynthetic pathway. In ade2-1 cells, a pigmented adenine precursor accumulates under these conditions because of a block in the pathway. 1/2 YPD (composed of 0.5% yeast extract, 2% peptone, and 2% glucose) is a complex medium with a limiting but undefined amount of adenine. YPAD (excess adenine) is similar but contains 1% yeast extract and is supplemented with 400 mg/liter adenine. Synthetic media are described (15). Cells were grown at 30 °C unless indicated otherwise.
Plasmids
Plasmids pRS316STI1 and pRS315STI1 are single copy URA3- and LEU2-based vectors, respectively (16), with STI1 and 500 bp of 5′ and 3′ flanking DNA inserted into the BamHI site. Plasmids pG/N795 and pUCΔSS-26X (17) were used for the glucocorticoid receptor (GR) signaling assay (see below). Plasmids pYDL505 (UGG) and pYDL506 (UAA) (18) were used for the translation read-through assay (see below).
Mutagenesis
Plasmid pRS316STI1 was mutagenized by exposure to hydroxylamine for 1 h at 75 °C as described (19). Site-directed mutagenesis of pRS316STI1 using the QuikChange kit (Stratagene, Burlingame, CA) and appropriate mismatched primers was done to construct Sti1-K75E. Sti1ΔDP2 was made similarly by introducing a stop codon at residue 538. Alleles encoding Sti1ΔTPR1, Sti1ΔDP1, and Sti1ΔTPR2 were generated by using the overlap extension PCR method for deletion mutagenesis (20).
Isolation of Sti1p Mutants
SSA1-21 sti1Δ [PSI+] cells were transformed by the mutagenized pRS316STI1, and 7,000 URA+ transformants were screened for defects in the ability to regulate Hsp70 as allowing growth without adenine at 30 °C and for defects in the ability to regulate Hsp90 as displaying hypersensitivity to radicicol (see “Results”).
Nonsense Suppression (Stop Codon Read-through) Assays
A dual-luciferase assay system (18) was used as described (14). Briefly, early log phase cultures of cells expressing translational fusions of Renilla and firefly luciferase genes, with UGG (from pYDL505) or UAA (from pYDL506) at the sixth codon of the firefly gene, were assayed for luciferase activity using the Promega dual-luciferase assay system in a Zylux FB15 luminometer.
Hormone Induction Assays
Assays were performed as described (21). Briefly, deoxycorticosterone (25 nm final) was added to early log phase cultures, and after 70 min samples were withdrawn for β-galactosidase assays. 100-μl samples were then added to 100 μl of the chemiluminescent β-galactosidase assay reagent Gal-Screen™ (Tropix, Bedford, MA) in 96-well microtiter plates at room temperature. The entire plate was read in a luminometer 1.5 h after the last sample was collected.
Yeast Cell Extracts and Western Analysis
To prepare whole cell extracts, washed cell pellets were suspended in lysis buffer (50 mm Tris-HCl, pH 7.4, 10 mm EDTA, 1 mm dithiothreitol, protease inhibitor (Roche Applied Science)) and broken by agitation with silica beads in a Mini Bead Beater (Biospec Products). Samples containing 20 μg of protein were assayed by Western analysis. The Sti1p antibody used was generated in rabbits using a synthetic peptide corresponding to Sti1p residues 530–544 as antigen. Hsp70 (SPA-822) and Hsp90 (SPA-840) antibodies were from Stressgen, and Hsp104 antibody was a gift from John Glover (University of Toronto).
Quantitative RT-PCR Assays
Quantitative RT-PCR assays were performed using standard methods as described (22).
RESULTS
Identifying Sti1p Mutations That Alter Regulation of Hsp70 and Hsp90
The yeast [PSI+] prion is a self-replicating aggregated form of the translation release factor Sup35p (eRF3) (23–26). [PSI+] propagates in the cytoplasm and is transmitted virtually infallibly between cells as they divide. Aggregation of Sup35p in [PSI+] cells decreases the ability of Sup35p to function in translation termination, which suppresses the ade2-1 nonsense allele in certain yeast strains (27). Nonsuppressed ade2-1 cells require adenine and appear red on limiting adenine because of the accumulation of an Ade2p substrate (see “Experimental Procedures”). Suppression of ade2-1 by [PSI+] confers growth without adenine and a normal white colony color.
We showed earlier that a mutant Hsp70 (Ssa1-21p) impairs [PSI+] propagation, causing a pink coloration on limiting adenine and frequent mitotic loss of [PSI+], which is diagnosed as red [psi−] colonies. Unlike wild type cells, SSA1-21 [PSI+] cells also require adenine at 30 °C (28). Depleting the Hsp90 co-chaperone Sti1p in SSA1-21 cells restores a normal [PSI+] phenotype, so SSA1-21 sti1Δ [PSI+] cells are white and grow without adenine at 30 °C. Thus, impairment of [PSI+] by Ssa1-21p depends upon the ability of Sti1p to regulate Hsp70 (14). We also showed that this ability of Sti1p to regulate Hsp70 with regard to [PSI+] is independent of Hsp90. Furthermore, SSA1-21 cells have normal sensitivity to the Hsp90-inhibiting drugs geldanamycin and radicicol, and deleting STI1 in wild type and SSA1-21 cells makes them similarly hypersensitive to growth inhibition caused by both drugs, indicating that Ssa1-21p does not affect the ability of Sti1p to regulate Hsp90. Therefore, SSA1–21 sti1Δ cells can be used to identify mutations in Sti1p that separately affect its ability to regulate Hsp70 and Hsp90.
We used these cells to screen for such mutations. As expected, SSA1-21 sti1Δ [PSI+] cells transformed by the control plasmid carrying wild type Sti1p do not grow without adenine at 30 °C and have normal radicicol sensitivity, whereas those with the empty vector grow without adenine and are hypersensitive to radicicol. To identify Sti1p mutations, SSA1-21 sti1Δ [PSI+] cells were first transformed by a library of randomly mutagenized plasmids carrying STI1. Transformants that grew without adenine at 30 °C yet displayed normal radicicol sensitivity were selected as expressing Sti1p having impaired ability to regulate Hsp70 but retaining the ability to regulate Hsp90. Transformants hypersensitive to radicicol but remaining unable to grow without adenine at 30 °C were selected as expressing Sti1p having impaired ability to regulate Hsp90 but retaining the ability to regulate Hsp70.
Many clones both required adenine at 30 °C and were hypersensitive to radicicol, and therefore they were defective for Sti1p regulation of both Hsp70 and Hsp90. Among these, we expected to find sti1 alleles with early termination codons. We also considered that single missense mutations might completely inactivate Sti1p, and therefore we sequenced 35 alleles that failed to complement Sti1p in both screens. All contained either early termination codons or multiple missense substitutions (data not shown), indicating that single missense mutations that disrupt Sti1p regulation of both Hsp70 and Hsp90, if they exist, are rare.
We also constructed STI1 alleles lacking domains known to be important for specific interactions of Sti1p with Hsp70 and Hsp90 (6, 13, 29). Lastly, we assessed the abilities of human Hop1, a specific Hop1 mutant (K73E) known to be weakened in its interaction with human Hsp70 (13), and the homologous Sti1p mutant (K75E), to complement Sti1p function in [PSI+] propagation. The STI1 deletion alleles and the locations of mutations identified in the screens are diagrammed in Fig. 1.
FIGURE 1. Sti1p domain structure and location of mutations that impair regulation of Hsp70 and Hsp90.

TPR repeat and aspartate-proline (DP) repeat regions are indicated. Amino acid residue numbers at the borders of the domains are shown along the top of the diagram (scale is approximate). Amino acid substitutions impairing the ability of Sti1p to regulate Hsp70 are indicated above the diagram of the intact gene, and those that impair Hsp90 regulation are shown below. Alleles lacking specific domains are shown with the deleted domain absent or spanned by a dotted line.
Sti1p Mutations That Affect Regulation of Hsp70
The degree of red coloration of [PSI+] colonies and the frequency of appearance of red [psi−] colonies, which are readily observed on plates with limiting adenine, qualitatively reflect the degree to which Sti1p functions in the impairment of [PSI+] in SSA1-21 cells. Cells expressing Sti1ΔTPR1 and Sti1-C49Y were stably [PSI+] and lacked pigment accumulation (Fig. 2). These phenotypes are similar to that of cells lacking Sti1p (Fig. 2, sti1Δ), which shows that these alterations essentially abolished the ability of Sti1p to regulate Hsp70 with regard to [PSI+] propagation. Cells expressing Sti1p with the other substitutions in TPR1 (A31T, S44F, A63V, K75E, and G76N) displayed very faint pink coloration (Fig. 2). Thus, these mutations considerably impaired the ability of Sti1p to weaken [PSI+] propagation, showing that these residues also are important for Sti1p regulation of Hsp70. Together, these data show that TPR1, which is known to mediate the physical interaction of Sti1p with the C terminus of Hsp70 (11), is critical for the ability of Sti1p to regulate Ssa1-21p in a way that impairs [PSI+] propagation and reveal novel residues of Sti1p that are crucial for this regulation.
FIGURE 2. [PSI+] phenotypes of SSA1–21 sti1Δ cells expressing various Sti1 and Hop1 proteins.
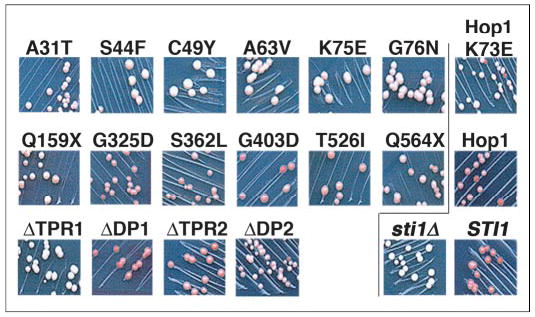
Cells with plasmids carrying the STI1 or HOP1 alleles were streaked for colonies onto plates maintaining selection for the plasmid and containing limiting adenine. To the left of the line, the top panels show cells expressing Sti1p mutants selected as affecting Hsp70 regulation, the middle panels are those selected as affecting Hsp90 regulation, and the bottom panels are those with engineered deletions (see Fig. 1). To the right of the line, the top and middle panels show cells expressing Hop1 variants in place of Sti1p, and the bottom panels show Sti1p wild type (STI1) and null (sti1Δ) controls. Weakening of [PSI+] by Ssa1-21p requires Sti1p, so sti1Δ cells are white and uniformly [PSI+], whereas STI1 cells are pink and have spontaneously arising [psi−] colonies (red) in the streak. The degree to which Sti1p mutations affect the ability of Sti1p to weaken [PSI+] propagation can be estimated by the extent to which the phenotypes resemble these controls. For example, Hop1 regulates Ssa1-21p essentially as well as Sti1p with regard to [PSI+] propagation, whereas Sti1ΔTPR1 (lower left) is essentially unable to regulate Ssa1-21p in a way that weakens [PSI+], and ΔDP2 has partial activity in this regard.
To quantify the degree to which the mutations affected Sti1p function with regard to [PSI+], we measured the extent of read-through of a nonsense codon in a luciferase reporter transcript (Fig. 3). There was a general correlation between the overt [PSI+] phenotype and the degree to which [PSI+] was capable of causing nonsense suppression. It is readily apparent from the plot of the data that substitutions spanning TPR1, in particular between Ser44 and Ala63, most significantly reduced the ability of Sti1p to regulate Ssa1-21p with regard to [PSI+]. As anticipated, Sti1ΔTPR1 was phenotypically Sti1-null regarding its effects on [PSI+]. As the substitutions that were identified in the screen for defects in the ability of Sti1p to regulate Hsp90 were selected to retain the ability to regulate Hsp70, they predictably had modest if any effects on [PSI+].
FIGURE 3. Relative ability of Sti1p and Hop1 variants to weaken [PSI+] as measured by nonsense suppression.
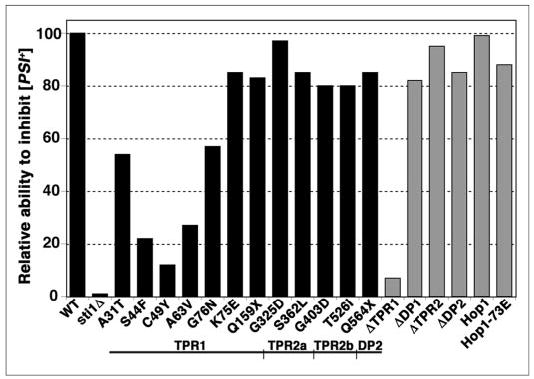
A read-through of nonsense codons caused by [PSI+] was quantified using a bicistronic mRNA encoding different forms of luciferase separated by a linker, which contains or lacks a stop codon. In SSA1-21 sti1Δ [PSI+] cells expressing the transcript with the intervening stop codon, the ratio of expression of the luciferase downstream of the linker to that of the luciferase upstream of the linker provides a measurement of stop codon read-through, which is related to [PSI+] “strength.” This ratio is then normalized to the same ratio from the same cells expressing the mRNA without the stop codon. The height of the bars reflects the extent to which the Sti1 proteins retain the ability to regulate Ssa1-21p in a way that weakens [PSI+], relative to wild type Sti1p, which is set at 100. Data for the randomly isolated mutations are plotted as black bars; those for the Sti1p deletions and Hop1 derivatives are plotted as gray bars. Below the graph is a linear diagram of the Sti1p domain structure (not to scale) aligned to show the region of the protein where the indicated mutations are located.
Sti1p Mutations That Affect Regulation of Hsp90
Sti1p regulation of Hsp90 is reflected in the hypersensitivity of cells lacking Sti1p to growth inhibition by the small compound radicicol, which specifically inactivates Hsp90. The degree of sensitivity to such growth inhibition of cells expressing the Sti1p mutants in place of Sti1p is shown in Fig. 4. It is clear from these data that the C-terminal portion of Sti1p is critical for the ability of Sti1p to regulate Hsp90 with regard to its essential function. All of the mutations isolated as causing hypersensitivity to radicicol were in residues within TPR2a, TPR2b, and DP2 or were predicted to cause deletions of these regions by introducing premature termination codons. The TPR2b mutations provide the first in vivo evidence that Hsp90 is a ligand for this Sti1p domain. Also, cells expressing Sti1-Q564stop (Q564X) and the engineered Sti1ΔDP2 proteins remained completely hypersensitive to the lethal effects of radicicol, suggesting that an intact DP2 region is necessary for Sti1p regulation of Hsp90. In contrast, the Sti1ΔTPR1 and Sti1ΔDP1 proteins conferred nearly wild type sensitivity to radicicol, which indicates that TPR1 and DP1 are dispensable for Sti1p regulation of Hsp90 in this context.
FIGURE 4. Sensitivity of SSA1–21 sti1Δ [psi−] cells expressing mutant Sti1 proteins to the Hsp90 inhibitor radicicol.

Upper panel, 5-fold serial dilutions of cells expressing Sti1 or Hop1 proteins (indicated above the panel) were grown on rich medium containing 25 μg/ml radicicol for 7 days at 33 °C. Lower panel, identical aliquots of the three highest dilutions of the same cells were grown on similar plates without radicicol for 2 days at 30 °C followed by 2 days at 25 °C. Between the panels is the Sti1p diagram described in the legend for Fig. 3.
Effects of Sti1p Mutations on Steroid Hormone Signaling
We next tested the function of the Sti1p mutants in a process that requires simultaneous regulation of Hsp70 and Hsp90 by Sti1p. The folding pathway for Hsp90 client proteins in which Sti1p acts by coordinating Hsp70 and Hsp90 functions is highly conserved. Among client proteins in humans are steroid hormone receptors. Although yeast lack such receptors, a system consisting of hormone receptors and reporter genes in which expression is regulated by hormone-induced activation of the receptor has been developed for monitoring steroid hormone signaling in yeast (30, 31). Proper folding of the receptor, which allows it to bind ligand and activate expression of the reporter, requires its interaction with the Hsp70/Sti1p/Hsp90 machinery. This system has provided valuable insight into functions of these and other mammalian and yeast factors that are involved in this folding and signaling process (32–34).
We monitored Sti1p function in this client folding pathway by quantifying activity of β-galactosidase expressed from a promoter that is transcriptionally activated by glucocorticoid receptor after induction by exposure to the GR ligand deoxycorticosterone. Efficient expression of β-galactosidase in our strains was dependent upon both deoxycorticosterone and the presence of Sti1p, which demonstrates the requirement for Sti1p in GR folding and activation (Fig. 5A). The only Sti1p mutations that abolished function in this pathway were Sti1ΔTPR1, C49Y, Q159stop (Q159X), and Sti1ΔTPR2 (Fig. 5B). Thus, the simultaneous presence of unmodified TPR1 and TPR2 was necessary for GR activation. Although structural data suggest that Cys49 is not involved in contacting Hsp70 directly (11), it apparently is critical for Hsp70 interaction or regulation in GR activation. Sti1-Q159stop and Sti1ΔTPR2 both lack regions known to be required for binding to Hsp90. These results show that Sti1p regulation of both Hsp70 and Hsp90 is necessary for GR signaling in yeast and suggest that simultaneous interaction of Sti1p with Hsp70 and Hsp90 is required for GR activation.
FIGURE 5. Stimulation of GR activity by Sti1 and Hop1 proteins.
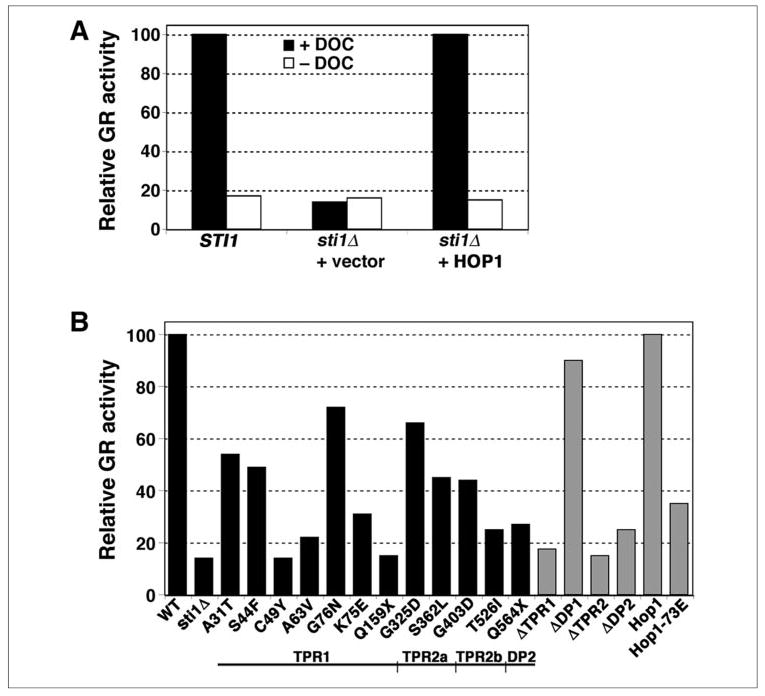
A, deoxycorticosterone (DOC) was added to one-half of the split cultures of SSA1-21 sti1Δ [psi−] cells expressing GR and carrying a GR-regulated β-galactosidase gene. The β-galactosidase activities of the cultures were measured and normalized to that from cells expressing wild type Sti1p, which was set at 100. B, the β-galactosidase activities of deoxycorticosterone-treated cultures expressing the various Sti1p and Hop1 proteins, normalized as in A, are shown. Below the plot is the Sti1p diagram described in the legend for Fig. 3.
All of the other Sti1p mutations, except ΔDP1, caused substantial reduction in GR activation. Although Sti1ΔDP2 had only a modest effect on Hsp70 regulation as measured by its affects on [PSI+], it only slightly restored GR activation, suggesting that its defect in GR activation was related to its inability to complement Sti1p regarding Hsp90 regulation. The G403D and T526I mutations in TPR2b also reduced GR activation considerably, further showing the importance of TPR2b for Sti1p function, presumably through regulation of Hsp90 in this pathway. Overall, the degree to which the Sti1p mutations reduced GR activation correlated roughly with the degree to which they reduced the ability to regulate Hsp70 or Hsp90 independently, which suggests that the basis for Sti1p regulation of the individual and cooperative Hsp70 and Hsp90 functions is similar.
GR Activation Requires Bridging of Hsp70 and Hsp90 by Sti1p
In addition to regulating the enzymatic activities of Hsp70 and Hsp90, Sti1p is thought to act in the Hsp90 client folding pathway by simultaneously binding the two chaperones to bridge them and facilitate transfer of substrate from Hsp70 to Hsp90. We used our system to test this hypothesis by simultaneously expressing Sti1ΔTPR1 and Sti1ΔTPR2 in SSA1-21 sti1Δ cells. As anticipated, although independent regulation of both Hsp70 and Hsp90 functions regarding [PSI+] and radicicol sensitivity were largely restored, GR activation was only marginally above background values (Fig. 6). These results indicate that although the truncated proteins were able to regulate Hsp70 and Hsp90 separately in the same cell, the client folding pathway depended on the physical linking of the two chaperones by intact Sti1p.
FIGURE 6. Sti1Δ TPR1 and Sti1Δ TPR2 independently regulate Hsp70 and Hsp90 in the same cell, but GR activation requires intact Sti1p.

Upper panels, SSA1-21 sti1Δ [PSI+] cells lacking Sti1p (sti1Δ) or expressing wild type (STI1) or truncated forms of Sti1p as indicated were streaked onto medium with limiting adenine and grown as described in the legend for Fig. 2. Lower panels, a dilution series of the cells shown in A were grown as described for Fig. 4 with (+) or without (−) radicicol. Relative levels of GR activation for the same transformants, measured as described in the legend for Fig. 5 and normalized to wild type set at 100, are indicated at the bottom.
Human Hop1 Regulates [PSI+] Propagation Like Sti1p
Others have shown that the loss of GR signaling in a yeast strain lacking Sti1p is restored by human Hop1 and that a mutation in Hop1 (K73E) that reduces Hop1 interaction with human Hsp70 reduces Hop1 function in this GR signaling (13, 29). Thus, Hop1 and Sti1p are functionally conserved with regard to GR activation. We transformed SSA1-21 sti1Δ [PSI+] cells with plasmids containing intact Hop1, Hop1 with the K73E mutation, and Sti1p with the homologous mutation (K75E) to test the functional conservation between Hop1 and Sti1p with regard to [PSI+] propagation.
Hop1 was capable of weakening the [PSI+] phenotype (Fig. 2, center right) and inhibiting [PSI+]-mediated nonsense suppression (Fig. 3) to levels similar to that of Sti1p, indicating that Hop1 functions like Sti1p with regard to [PSI+] propagation. Additionally, the K73E mutation reduced the ability of Hop1 to inhibit [PSI+], suggesting that these Hop1 effects on [PSI+] were mediated through regulation of Hsp70. The K75E mutation of Sti1p also reduced the ability of Sti1p to weaken [PSI+] (Figs. 2 and 3), and both Hop1-K73E and Sti1-K75E were impaired to a similar degree for GR signaling (Fig. 5B). These data demonstrate functional conservation between Sti1p and Hop1 in both [PSI+] propagation and GR activation. Additionally, neither Hop1-K73E nor Sti1-K75E was altered in regulating sensitivity to radicicol compared with their wild type counterparts, which shows that the distinct regulation of Hsp70 and Hsp90 by Sti1p and Hop1 is evolutionarily conserved.
Abundance of Sti1 and Heat Shock Proteins
To assess the possibility that effects caused by the mutations were due to altered protein production or stability, we used Western analysis to determine overall abundance of the Sti1 proteins (Fig. 7A). Except for Sti1-Q564stop (Q564X) and Sti1ΔDP1, which were elevated, the amount of the different Sti1 proteins did not vary significantly from that of wild type Sti1p, suggesting that the mutations reduced function of Sti1p rather than its abundance.
FIGURE 7. Relative expression of Sti proteins and protein chaperones.
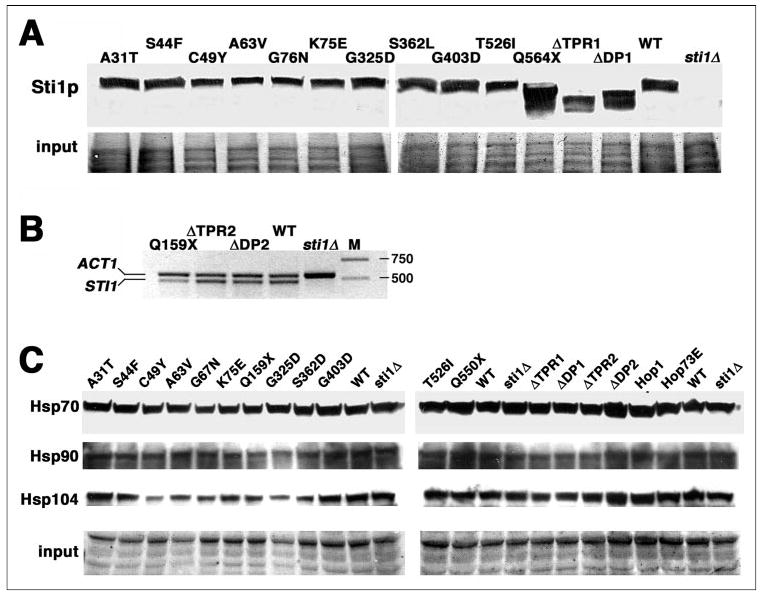
A, whole cell lysates of cells expressing the various Sti1 proteins, indicated at the top, were subjected to Western analysis using antibody to a Sti1p epitope spanning amino acid residues 530–544. B, abundance of mRNA from cells expressing indicated Sti1p variants that lack the epitope for the Sti1p antibody was quantified by RT-PCR and compared with mRNA from cells expressing wild type Sti1p (WT) and lacking Sti1p (sti1Δ). Abundance of actin mRNA was measured as a control in all reactions. Lane M shows molecular mass markers of 750 and 500 bp. The expected sizes of PCR products are 567 bp for actin and 496 bp for STI1. C, lysates of cells used for A and B were subjected to Western analysis probing for Hsp70, Hsp90, and Hsp104 as indicated. For A and C, portions of membranes stained by Amido Black are shown as loading and transfer controls (Input).
Our Sti1p antibody recognizes an epitope spanning residues 530–544, which overlaps the border between TPR2b and DP2. Because this epitope is absent in the Sti1-Q159stop (Q159X), Sti1ΔTPR2, and Sti1ΔDP2 proteins, we used RT-PCR to quantify the relative abundance of STI1 mRNA as a measure of expression of these three proteins (Fig. 7B). Although the ΔTPR2 and ΔDP2 transcripts were as abundant as wild type, the amount of Q159stop mRNA was noticeably reduced, which was expected because transcripts with early nonsense mutations are targeted for degradation by the nonsense-mediated mRNA decay pathway (35). The abundance of the Q159stop protein could be expected to be reduced correspondingly, which probably contributed to its reduced ability to cause [PSI+] inhibition.
It was also possible that the Sti1p mutations were exerting their effects by altering expression of Hsp70, Hsp90, or Hsp104. Hsp104 is essential for [PSI+] propagation and can interact with Sti1p under certain non-optimal growth conditions (36). We therefore repeated the Western analysis to measure abundance of these chaperones. As shown in Fig. 7C, there were modest variations in steady-state abundance of the three chaperones in cells expressing the different Sti1 proteins, but the small relative differences were not likely to contribute significantly to the effects caused by the Sti1p mutations. Rather, the effects of the mutations are more likely due to disruptions in the ability of Sti1p to interact physically with the chaperones or to regulate their enzymatic activities.
DISCUSSION
Although the role for Sti1p in coordinating action of Hsp70 and Hsp90 in the client protein folding pathway is well established, we now demonstrate that Sti1p can independently regulate the individual functions of Hsp70 and Hsp90. We also show that physical linking of Hsp70 and Hsp90 by Sti1p is required for efficient function of the client folding pathway. Our results delineate Sti1p domains that specify differences in Sti1p regulation of the two chaperones and identify novel amino acid residues that are critical for these Sti1p functions. We further identify Hsp90 as a ligand for TPR2b and provide evidence that the independent regulation of Hsp70 and Hsp90 is evolutionarily conserved by showing complementary functions of the human Sti1p homolog Hop1.
Previous in vitro data defines TPR1 and TPR2 as mediating Hop1/Sti1p interactions with Hsp70 and Hsp90, respectively (5, 37). In agreement with these studies, we identified TPR1 as largely sufficient for Sti1p regulation of Hsp70 with regard to [PSI+] and TPR2 as essential for regulation of Hsp90. Moreover we show that TPR1 was dispensable for regulation of Hsp90 with regard to the essential function of Hsp90 and that TPR2 was dispensable for Sti1p regulation of Hsp70. Nevertheless, as both Hsp70 and Hsp90 are important for client protein folding, mutations affecting regulation of either chaperone predictably impaired steroid receptor activation, and we found that regulation of this activation required both TPR1 and TPR2 to be present on the same polypeptide.
Although the ligand for TPR2b is unknown, others have found that point mutations in TPR2b of Hop1, selected by sequence alignment as in residues possibly involved in chaperone interactions, reduce Hop1 binding to Hsp70 but not to Hsp90 (13). Although these data were stated to contrast with a report that deletions within TPR2b inhibit Hsp90 binding (5, 13) they pointed to Hsp70 as a putative ligand for TPR2b. Unlike all of our Sti1p mutations, the Hop1 TPR2b substitutions did not affect Hop1 function in GR activation in yeast. Our mutations in TPR2b, identified in a screen of random Sti1p mutants, caused radicicol hypersensitivity and impaired GR activation without affecting [PSI+]. These physiological effects clearly reveal the importance of TPR2b for Hsp90 regulation and suggest that TPR2b does not influence Hsp70 activity directly. They also uncover limitations of in vitro interaction studies in predicting the effects of mutations in vivo.
Aside from some of the effects of mutating DP2, our data agree with earlier reports showing that TPR1 and TPR2a of Hop1/Sti1p are important for physical interactions with Hsp70 and Hsp90, respectively, and that DP2 is more important than DP1 for steroid receptor activation (5, 6, 11–13, 37). The Sti1p mutant lacking DP2 was nonfunctional with regard to radicicol sensitivity but was affected only modestly with regard to [PSI+]. Thus, although DP2 played some role in Hsp70 regulation, it was more important for the regulation of Hsp90. Earlier work showing that point mutations in DP2 reduced both Hsp70 binding in vitro and GR activation in yeast led to the proposal that DP2 and TPR1 cooperate to bind Hsp70 (13). Although our data do not rule out this possibility, they show that any loss of Hsp70 interaction that might be caused by truncation or complete deletion of DP2 has only small effects on Hsp70 regulation in vivo and that the major cause of the reduced GR activation by DP2 mutation might be the impaired ability of Hop1 to regulate Hsp90. It is possible that DP2 function either is affected differentially by deletion and substitution mutations or has diverged between Hop1 and Sti1p. Whatever the reason, the discrepancies between the two studies remain to be resolved experimentally.
We earlier showed that Ssa1-21p impairs [PSI+] propagation through enhanced substrate binding because of increased ATP hydrolysis, which promotes substrate binding, or decreased ADP release, which stabilizes the substrate-bound state (14, 38). As Sti1p is a potent activator of Ssa1 ATPase in vitro (7), its depletion, which should reduce Ssa1-21p ATPase activity, has the expected effect of improving [PSI+] in SSA1-21 cells. Hop1 does not stimulate ATPase of Ssa1p in vitro, however, so its ability to restore inhibition of [PSI+] in SSA1-21 sti1Δ cells was somewhat surprising. Possibly, Hop1 does stimulate Ssa1p ATPase in vivo in a reaction that might involve other components of the chaperone machinery. Alternatively, the interaction of Ssa1p with the TPR1 domain of Hop1 is enough to confer regulation of Ssa1-21p in a way that influences [PSI+]. A possible explanation for such regulation would be that Hop1, which has a higher affinity for Ssa1p than Sti1p and preferentially binds the ADP-bound form of Hsp70 (7, 9), stabilizes this state of Ssa1-21p.
There was a correlation between the extent of impairment of either Hsp70 or Hsp90 and the extent to which the client folding pathway, which depends on both chaperones, was affected. This observation holds even if regulation of one of the chaperones was essentially unaffected (TABLE ONE). These results suggest that the way Sti1p regulates Hsp70 and Hsp90 both independently and as cooperative chaperones is similar. Moreover, the ability to separate lethal effects of Hsp90-inhibiting compounds from effects on [PSI+] propagation suggests that the essential function of Hsp90 is unrelated to the ability of Sti1p to regulate Hsp70 and, therefore, is separate from the client folding pathway.
TABLE ONE. Summary of effects of Sti1p mutations on Hsp70 and Hsp90 function.
[PSI+] values reflect data from Figs. 2 and 3, radicicol resistance from data in Fig. 4, and GR activity from data in Fig. 5. Numbers in parentheses indicate the chaperone (70, Hsp70; 90, Hsp90) affected by Sti1p mutations.
|
Assaya |
||||
|---|---|---|---|---|
| Stilp | Corresponding Hop1 residues | [PSI+] inhibition (70) | Radicicolres(90) | GR activity (70/90) |
| None | +++ | +++ | +++ | |
| Null | − | − | − | |
| A31T | A30 | + | +++ | + |
| S44F | S42 | +/− | ++ | +/− |
| C49Y | A47 | − | ++ | − |
| A63V | G61 | +/− | +++ | +/− |
| K75E | K73 | +/−b | +++ | +/− |
| G76N | G74 | + | +++ | + |
| Q159stop | T151 | ++ | − | − |
| G325D | G288 | +++ | +/− | + |
| S362L | S326 | ++ | − | +/− |
| G403D | G367 | ++ | − | +/− |
| T526I | S481 | ++ | − | +/− |
| Q564stop | S519 | ++ | − | +/− |
| ΔTpr1-(1–147) | 1–139 | − | +++ | +/− |
| ΔDP1-(148–200) | 140–187 | +++ | +++ | ++ |
| ΔTpr2-(201–536) | 188–491 | +++ | − | − |
| ΔDP2-(537–589) | 492–543 | ++ | − | +/− |
| Hop1-73E | +/−b | ++ | +/− | |
| Hop1 | +++ | ++ | +++ | |
Subjective values: +++, ≥90% of wild type Sti1p activity; ++, 75–90% Sti1p activity; +, 50–75% Sti1p activity; +/−, 20–50% Sti1p activity; −, <20% Sti1p activity.
Impairment of [PSI+] as measured by colony phenotype and nonsense suppression did not correlate. The values shown reflect the more significant effect.
Our inability to identify single missense mutations in Sti1p that impaired regulation of both Hsp70 and Hsp90 is consistent with our findings that the separate domains of Sti1p confer the ability to regulate Hsp70 and Hsp90, suggesting that at least two mutations might be required to eliminate the ability of Sti1p to regulate both chaperones. These observations suggest that Sti1p does not have a specific activity essential for regulation of both Hsp70 and Hsp90.
Aside from bridging Hsp70 and Hsp90, Sti1/Hop1 can regulate enzymatic activities of these chaperones (9, 10). The mechanisms underlying this regulation are not well understood, and it is possible that simply binding to the chaperones is enough for Sti1p to influence ATPase cycles or interactions of other regulatory factors. Further work with our novel mutants, in particular those altered in residues predicted to be outside of chaperone interaction surfaces, will aid in elucidating these regulatory mechanisms.
Acknowledgments
We thank David Smith and Jonathan Dinman for plasmids and Andy Golden for critical review of the manuscript.
Footnotes
The abbreviations used are: TPR, tetratricopeptide repeat; RT, reverse transcription; GR, glucocorticoid receptor.
References
- 1.Pratt WB, Toft DO. Exp Biol Med. 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 2.Pratt WB, Galigniana MD, Morishima Y, Murphy PJ. Essays Biochem. 2004;40:41–58. doi: 10.1042/bse0400041. [DOI] [PubMed] [Google Scholar]
- 3.Hernandez MP, Chadli A, Toft DO. J Biol Chem. 2002;277:11873–11881. doi: 10.1074/jbc.M111445200. [DOI] [PubMed] [Google Scholar]
- 4.Smith DF, Sullivan WP, Marion TN, Zaitsu K, Madden B, McCormick DJ, Toft DO. Mol Cell Biol. 1993;13:869–876. doi: 10.1128/mcb.13.2.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen S, Prapapanich V, Rimerman RA, Honore B, Smith DF. Mol Endocrinol. 1996;10:682–693. doi: 10.1210/mend.10.6.8776728. [DOI] [PubMed] [Google Scholar]
- 6.Chen S, Smith DF. J Biol Chem. 1998;273:35194–35200. doi: 10.1074/jbc.273.52.35194. [DOI] [PubMed] [Google Scholar]
- 7.Wegele H, Haslbeck M, Reinstein J, Buchner J. J Biol Chem. 2003;278:25970–25976. doi: 10.1074/jbc.M301548200. [DOI] [PubMed] [Google Scholar]
- 8.Richter K, Muschler P, Hainzl O, Reinstein J, Buchner J. J Biol Chem. 2003;278:10328–10333. doi: 10.1074/jbc.M213094200. [DOI] [PubMed] [Google Scholar]
- 9.Johnson BD, Schumacher RJ, Ross ED, Toft DO. J Biol Chem. 1998;273:3679–3686. doi: 10.1074/jbc.273.6.3679. [DOI] [PubMed] [Google Scholar]
- 10.Hernandez MP, Sullivan WP, Toft DO. J Biol Chem. 2002;277:38294–38304. doi: 10.1074/jbc.M206566200. [DOI] [PubMed] [Google Scholar]
- 11.Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, Hartl FU, Moarefi I. Cell. 2000;101:199–210. doi: 10.1016/S0092-8674(00)80830-2. [DOI] [PubMed] [Google Scholar]
- 12.Odunuga OO, Hornby JA, Bies C, Zimmermann R, Pugh DJ, Blatch GL. J Biol Chem. 2003;278:6896–68904. doi: 10.1074/jbc.M206867200. [DOI] [PubMed] [Google Scholar]
- 13.Carrigan PE, Nelson GM, Roberts PJ, Stoffer J, Riggs DL, Smith DF. J Biol Chem. 2004;279:16185–16193. doi: 10.1074/jbc.M314130200. [DOI] [PubMed] [Google Scholar]
- 14.Jones GW, Song Y, Chung S, Masison DC. Mol Cell Biol. 2004;24:3928–3937. doi: 10.1128/MCB.24.9.3928-3937.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sherman F. Methods Enzymol. 1994;194:3–21. doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- 16.Sikorski RS, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Louvion JF, Warth R, Picard D. Proc Natl Acad Sci U S A. 1996;93:13937–13942. doi: 10.1073/pnas.93.24.13937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harger JW, Dinman JD. RNA (NY) 2003;9:1019–1024. doi: 10.1261/rna.5930803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schatz PJ, Solomon F, Botstein D. Genetics. 1988;120:681–695. doi: 10.1093/genetics/120.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee J, Lee HJ, Shin MK, Ryu WS. BioTechniques. 2004;36:398–400. doi: 10.2144/04363BM04. [DOI] [PubMed] [Google Scholar]
- 21.Riggs DL, Roberts PJ, Chirillo SC, Cheung-Flynn J, Prapapanich V, Ratajczak T, Gaber R, Picard D, Smith DF. EMBO J. 2003;22:1158–1167. doi: 10.1093/emboj/cdg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song Y, Azakami H, Shamima B, He J, Kato A. FEBS Lett. 2002;512:213–217. doi: 10.1016/s0014-5793(02)02258-5. [DOI] [PubMed] [Google Scholar]
- 23.Wickner RB. Science. 1994;264:566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 24.Stansfield I, Jones KM, Kushnirov VV, Dagkesamanskaya AR, Poznyakovski AI, Paushkin SV, Nierras CR, Cox BS, Ter-Avanesyan MD, Tuite MF. EMBO J. 1995;14:4365–4373. doi: 10.1002/j.1460-2075.1995.tb00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhouravleva G, Frolova L, Le Goff X, Le Guellec R, Inge-Vechtomov S, Kisselev L, Philippe M. EMBO J. 1995;14:4065–4072. doi: 10.1002/j.1460-2075.1995.tb00078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wickner RB, Taylor KL, Edskes HK, Maddelein ML, Moriyama H, Roberts BT. J Struct Biol. 2000;130:310–322. doi: 10.1006/jsbi.2000.4250. [DOI] [PubMed] [Google Scholar]
- 27.Cox BS. Heredity. 1965;20:505–521. [Google Scholar]
- 28.Jung G, Jones G, Wegrzyn RD, Masison DC. Genetics. 2000;156:559–570. doi: 10.1093/genetics/156.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carrigan PE, Riggs DL, Chinkers M, Smith DF. J Biol Chem. 2005;280:8906–8911. doi: 10.1074/jbc.M414245200. [DOI] [PubMed] [Google Scholar]
- 30.Schena M, Yamamoto KR. Science. 1988;241:965–967. doi: 10.1126/science.3043665. [DOI] [PubMed] [Google Scholar]
- 31.Schena M, Picard D, Yamamoto KR. Methods Enzymol. 1991;194:389–398. doi: 10.1016/0076-6879(91)94029-c. [DOI] [PubMed] [Google Scholar]
- 32.Picard D, Khursheed B, Garabedian MJ, Fortin MG, Lindquist S, Yamamoto KR. Nature. 1990;348:166–168. doi: 10.1038/348166a0. [DOI] [PubMed] [Google Scholar]
- 33.Nathan DF, Lindquist S. Mol Cell Biol. 1995;15:3917–3925. doi: 10.1128/mcb.15.7.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duina AA, Chang HC, Marsh JA, Lindquist S, Gaber RF. Science. 1996;274:1713–1715. doi: 10.1126/science.274.5293.1713. [DOI] [PubMed] [Google Scholar]
- 35.Hentze MW, Kulozik AE. Cell. 1999;96:307–310. doi: 10.1016/s0092-8674(00)80542-5. [DOI] [PubMed] [Google Scholar]
- 36.Abbas-Terki T, Donze O, Briand PA, Picard D. Mol Cell Biol. 2001;21:7569–7575. doi: 10.1128/MCB.21.22.7569-7575.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lassle M, Blatch GL, Kundra V, Takatori T, Zetter BR. J Biol Chem. 1997;272:1876–1884. doi: 10.1074/jbc.272.3.1876. [DOI] [PubMed] [Google Scholar]
- 38.Jones GW, Masison DC. Genetics. 2003;163:495–506. doi: 10.1093/genetics/163.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]