

Abstract
The G protein-coupled receptor kinase 2 (GRK2) phosphorylates and desensitizes ligand-activated G protein-coupled-receptors. Here, evidence is shown for a novel role of GRK2 in regulating chemokine-mediated signals. The presence of increased levels of GRK2 in human embryonic kidney (HEK) 293 cells produced a significant reduction of the extracellular signal-regulated kinase (ERK) response to CCL2. This effect is independent of its role in receptor phosphorylation because the kinase-deficient mutant GRK2K220R was able to reduce this response, and ERK activation by CCR2BIX, a phosphorylation-defective receptor mutant, was also inhibited by GRK2. Constructs containing the Gαq-binding RGS-like RH domain of GRK2 or its Gβγ-binding domain could not reproduce the inhibition, thus revealing that GRK2 acts downstream of G proteins. Interestingly, chemokine-driven mitogen-activated protein kinase kinase (MEK) stimulation is not affected in cells overexpressing GRK2 or GRK2K220R or in splenocytes from heterozygous GRK2 mice, where reduced kinase levels correlate with enhanced ERK activation by chemokines. We find GRK2 and MEK in the same multimolecular complex, thus suggesting a mechanism for GRK2 regulation of ERK activity that involves a direct or coordinate interaction with MEK. These results suggest an important role for GRK2 in the control of chemokine induction of ERK activation at the level of the MEK–ERK interface.
INTRODUCTION
G protein-coupled receptor kinases (GRKs) are serine/threonine protein kinases that control the desensitization process of the G protein-coupled receptor family (Penela et al., 2003; Willets et al., 2003). GRK2 in particular is one of the best characterized GRK isoforms and has been shown to phosphorylate and desensitize the agonist-bound form of many G protein-coupled receptors (GPCRs) (Aragay et al., 1998b). Receptor phosphorylation is followed by high-affinity binding of arrestins to the receptor, which sterically inhibits further G protein activation. Although it is now clear that arrestins serve additional roles as multifunctional adaptor molecules, emerging new evidence also unveils unexpected roles for GRKs, including the phosphorylation of nonreceptor substrates, of non-GPCR receptors, and the association to different proteins such as phosphoinositide 3-kinase, GIT, or clathrin (Penela et al., 2003; Willets et al., 2003). Moreover, the GRK2/3 subfamily members contain a C-terminal Gβγ-binding region (Carman et al., 2000; Willets et al., 2003) and are able to inactivate G protein-dependent pathways in a phosphorylation-independent manner involving their avid interaction with Gαq family members via their RGS-like RH N-terminal domain (Carman et al., 1999; Sallese et al., 2000).
Chemokines bind to a variety of GPCRs, thus mediating chemotactic and proadhesion effects in leukocytes and other cell types (Rossi and Zlotnik, 2000). The CC chemokine receptor, CCR2, exclusively binds CCL2 produced by endothelial cells, smooth muscle cells, and macrophages in response to a variety of mediators. Disruption of the mouse CCR2 gene diminishes leukocyte adhesion to microvasculature and monocyte extravasation in response to proinflammatory stimuli (Kuziel et al., 1997). CCL2 has also been implicated in a variety of inflammatory diseases, including rheumatoid arthritis, atherosclerosis, and alveolitis as well as in macrophage infiltration of tumors (Gerard and Rollins, 2001). In this line, emerging evidence has proven that the mitogen-activated protein kinase (MAPK) pathway is activated in response to chemokine stimulation and plays an important role in chemotaxis and integrin activation mediated by chemokines (Ashida et al., 2001; Ge et al., 2003; Jiménez-Sainz et al., 2003).
Several recent studies have confirmed a role for GRKs and arrestins in the phosphorylation and desensitization of chemokine receptors (see references in (Aragay et al., 1998a,b). In fact, it has been demonstrated that chemokines can differ in their ability to induce receptor phosphorylation and desensitization, thus providing a molecular mechanism for agonist-induced attenuation of receptor signaling (Oppermann et al., 1999). Interestingly, GRK2 is highly expressed in peripheral blood leukocytes and in some myeloid and lymphoid cell lines. Alterations in GRK activity have been demonstrated upon T-cell activation (De Blasi et al., 1995), and patients with rheumatoid arthritis show decreased GRK2 expression in peripheral blood mononuclear cells (Lombardi et al., 1999).
In this context, we have studied the impact of altering GRK2 protein levels and activity on chemokine-induced extracellular signal-regulated kinase (ERK)1/2 activation. We have analyzed the MAPK response to CCL2 and CXCL12 in GRK2-transfected cells and in splenocytes from hemizygous GRK2 mice. We show here that elevated levels of GRK2 can inhibit chemokine-mediated induction of ERK activity. Conversely, in heterozygous GRK2 (+/–) mice, with decreased levels of GRK2, chemokine activation promotes a more robust ERK stimulation. Interestingly, such effects seem to be independent of the ability of GRK2 to phosphorylate GPCR or to interact with G protein subunits. Furthermore, we report that mitogen-activated protein kinase kinase (MEK) and GRK2 are present in the same multimolecular complex, and postulate a new mode of regulation of ERK activation by GRK2 occurring at the interface between MEK and ERK.
MATERIALS AND METHODS
Materials and Plasmids
EBNA-HEK293 cells were purchased from the American Type Culture Collection (Manassas, VA). Culture media and LipofectAMINE plus were from Invitrogen (Carlsbad, CA). Epidermal growth factor (EGF) was obtained from Upstate Biotechnology (Lake Placid, NY), and CCL2 was purchased from PeproTech (Rocky Hill, NJ). The cDNAs encoding hemagglutinin (HA)-ERK1 and CXCR4 were provided by Dr. J. Moscat and Dr. M. Fresno, respectively (Centro de Biología Molecular, Madrid, Spain). The pCDNA3-CCR2BIX was generously provided by Dr. I. F. Charo (California University, San Francisco, CA). The pCDNA3-HA-MEK1 plasmid was kindly provided by Dr. P. Crespo (CSIC-University of Cantabria, Cantabria, Spain). The pcDNA3-D110A-GRK2 mutant was donated by Dr. J. L. Benovic (Thomas Jefferson University, Philadelphia, PA). Other plasmids were constructed in our laboratory. The minigene construct containing GRK2 amino acids 438–689 was generated by PCR and then subcloned via NcoI-BclI sites into the pBSSK plasmid engineered to contain the 3′ and 5′ untranslated regions of the pRK5-α1B-AR minigene construct (Luttrell et al., 1993) kindly donated by Dr. S. Cotecchia (Lausanne University, Lausanne, Switzerland). The resulting pBSSK-minigene GRK2 438–689 was verified by sequencing and subcloned into the pREP4 vector (Invitrogen). For the construction of the pREP4-GRK2 1-546, a stop codon together with a BamHI restriction site was introduced by PCR following the triplet for Gly 546 of bovine GRK2. After subcloning into pBSSK, and rescuing the N-terminal region with the ScaI-RsrI fragment of pBSSK-full-length GRK2, the sequence between RsrI and the introduced stop codon was verified by sequencing. The resulting GRK2 1-546 coding region was then introduced into pREP4.
Polyclonal C-16 and C-14 antibodies that recognize ERK1 and ERK2 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Polyclonal antibodies anti-phospho-ERK1/2 and phospho MEK1/2 were purchased from Cell Signaling Technology (Beverly, MA). The polyclonal antibody Ab9 against full-length GRK2 was kindly provided by Dr. J. L. Benovic (Thomas Jefferson University). The monoclonal anti-HA antibody was obtained from the Microscopy facility at the Centro de Biologia Molecular (Madrid, Spain). The anti-MEK antibody was obtained from Santa Cruz Biotechnology (C-18). Anti-GRK2 antibodies against glutathione S-transferase (GST)-fusion proteins containing regions 436-689 and 50-145 of bovine GRK2 were generated in our laboratory (Murga et al., 1996), as was the anti-β-arrestin antibody Ab186.
Cells and Animals
HEK293 and Mono Mac cells were maintained essentially as described previously (Jiménez-Sainz et al., 2003). Spleen cells were obtained from 3-mo-old heterozygous GRK2 (+/–) and wild-type littermates. The heterozygous mice (Jaber et al., 1996) were kindly provided by Drs. R. J. Lefkowitz and M. G. Caron (Duke University, Durham, NC) and backcrossed with C57BL6 and 129SV mice. Spleens were collected from mice, and cell suspensions were obtained by dispersion with a syringe in the presence of RPMI 1640 medium containing nonessential amino acids and 10% fetal bovine serum (FBS). Cells were washed twice in RPMI 1640 medium with 10% FBS, and erythrocytes were lysed in medium containing 150 mM NH4Cl, 10 mM HKCO3, and 1 mM EDTA, pH 7.4, at 37°C for 5 min. Then, splenocytes were washed in RPMI 1640 medium with 10% FBS and counted by trypan blue exclusion. For MEK and mitogen-activated protein kinase (MAPK) assays, the splenocytes were serum starved before ligand addition.
Transfections
Human embryonic kidney (HEK) 293 cells were transiently transfected with LipofectAMINE (Invitrogen) as described previously (Jiménez-Sainz et al., 2003). Transient expression was confirmed by immunoblot analysis of whole-cell lysates using specific antisera. CCL2, CXCL12, and epidermal growth factor (EGF) stimulation of HEK293 cells was performed 48 h after transfection at 37°C in culture medium without serum, during the indicated time periods. The cells were serum starved for 16 h before ligand addition to minimize basal kinase activity.
Lysis, Immunoprecipitation, and Western Blotting
MAPK and MEK activation were assessed as described previously (Jiménez-Sainz et al., 2003) using anti-phospho-ERK and anti-phospho-MEK antibodies, respectively. For testing GRK and β-arrestin expression, splenocytes obtained from wild-type and GRK2 (+/–) mice were lysed in ice-cold n-dodecyl-β-d-maltoside buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 20 mM MgCl2, 1% n-d-maltoside, 10 mM NaF, 1 mM Na3VO4, and a cocktail of protease inhibitors), and equal amounts of lysates were resolved by SDS-PAGE. For MEK-GRK2 coimmunoprecipitation, a buffer containing 1% Triton X-100, 10% glycerol, 135 mM NaCl, 20 mM Tris-HCl, pH 7.5, 0.5 mM EDTA, 10 mM NaF, and 1 mM Na3VO4 was used both for cell lysis and subsequent washes. For endogenous MEK-GRK2 coimmunoprecipitation, 60 × 106 HEK293 or Monomac cells and 2 μg anti-MEK (sc-219) were used per lane for these experiments, and radioimmunoprecipitation assay buffer was used for both lysis and washes. The presence of MAPK, β-arrestin, GRK6, and GRK2 peptides in the lysates was determined by using anti-ERK1/ERK2, Ab186, anti-GRK6, Ab9, and anti-GRK2 436-689 and 50-145 antibodies, respectively. Blots were developed using a chemiluminescent method (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom). Quantitation of bands was performed as described previously (Jiménez-Sainz et al., 2003). All data are expressed as the mean ± SEM. Student's t test was used to compare numerical data, and p values <0.05 were considered to represent significant differences.
Fluorescence-activated Cell Sorting (FACS) Analysis
Splenocytes from GRK2 (+/+) and (+/–) mice were incubated with phycoerythrin-labeled anti-mouse CXCR4 antibody (BD Biosciences PharMingen, San Diego, CA) using the appropriate PE-conjugated isotype control (IgG2b) to determine background labeling. FACS detection was carried out using FACScalibur (BD Biosciences PharMingen) and subsequent analysis using CellQuest software.
RESULTS
In previous studies, we have analyzed the pathway of activation of MAPKs by CCL2 in monocytes and HEK293-CCR2B cells, which involves heterotrimeric G proteins, protein kinase C, phosphoinositide 3-kinase, and Ras (Aragay et al., 1998a; Jiménez-Sainz et al., 2003). We have also reported that activation of CCR2B by CCL2 leads to its rapid phosphorylation by GRK2. To further analyze the role of GRK2 in chemokine signaling, we have studied the effect of altering kinase levels in the activation of ERK by CCL2. Coexpression of GRK2 and CCR2B produces a robust decrease in CCL2-induced ERK phosphorylation (65–70% reduction) compared with that observed in cells expressing the receptor alone (Figure 1A). The reduction is dose dependent, because gradually increasing the expression levels of GRK2 (up to 4.5-fold the endogenous amount of protein) produced an enhanced inhibitory effect on CCL2-stimulated ERK activity (Figure 1B). EGF activation of ERK phosphorylation is not affected by the expression of GRK2 (Figure 1C), thus demonstrating that the inhibitory effect of GRK2 is specific for the activated CCR2B receptor.
Figure 1.

Inhibition of CCL2-mediated ERK stimulation in the presence of GRK2. HEK293 cells transiently transfected with pcDNA3-FLAG-CCR2B, pcDNA3, or pcDNA3-GRK2, and pcDNA1-HA-ERK1 were stimulated with 20 nM CCL2 (A) for different time periods. Lysates were subjected to immunoblotting with anti-phosphorylated ERK (top) and reprobed with anti-ERK and anti-GRK2 antibodies (bottom). Data normalized for total ERK levels are expressed as the average fold-increase ± SEM of HA-ERK1 phosphorylation of five independent experiments. *p < 0.05 versus controls activated at 5 min. Arrowheads indicate the migration of HA-ERK protein. (B) The indicated amounts of pcDNA3-GRK2 or pcDNA-GRK2K220R DNA were transfected into HEK293 cells, and phospho-ERKs, ERKs, and GRK2 were analyzed as described in A. (C) Cells were stimulated with 100 ng/ml EGF and processed as described above.
GRK2 has been shown to phosphorylate the CCR2B receptor and desensitize CCL2-induced calcium signaling (Aragay et al., 1998a). To analyze whether the reduction observed in CCL2-mediated ERK activation in the presence of excess GRK2 involved receptor phosphorylation, similar experiments were performed using a kinase-defective mutant, GRK2K220R (Kong et al., 1994). Unexpectedly, the expression at similar levels of GRK2K220R produces a robust decrease in CCL2-induced ERK phosphorylation (52% mean reduction) (Figure 2A). These results suggest that the inhibitory effect produced by GRK2 is not only due to the phosphorylation of the ligand-activated CCR2B receptor. This point was further addressed by analyzing the effect of GRK2 on the activation of ERK by a mutated form of the receptor, CCR2BIX (Franci et al., 1996). This receptor has serine/threonine residues in the C-terminal tail of the protein mutated to alanines, is poorly phosphorylated by GRK2, and consequently, overexpression of GRK2 does not block CCR2BIX-induced calcium response (Aragay et al., 1998a). In cells transfected with CCR2BIX, CCL2 triggered a marked increase of ERK phosphorylation (Figure 2B) that was potently impaired by GRK2 (70% inhibition). Together, these results suggest that expression of GRK2 negatively regulates ERK activation induced by CCL2 and that this effect is mostly independent of receptor phosphorylation.
Figure 2.
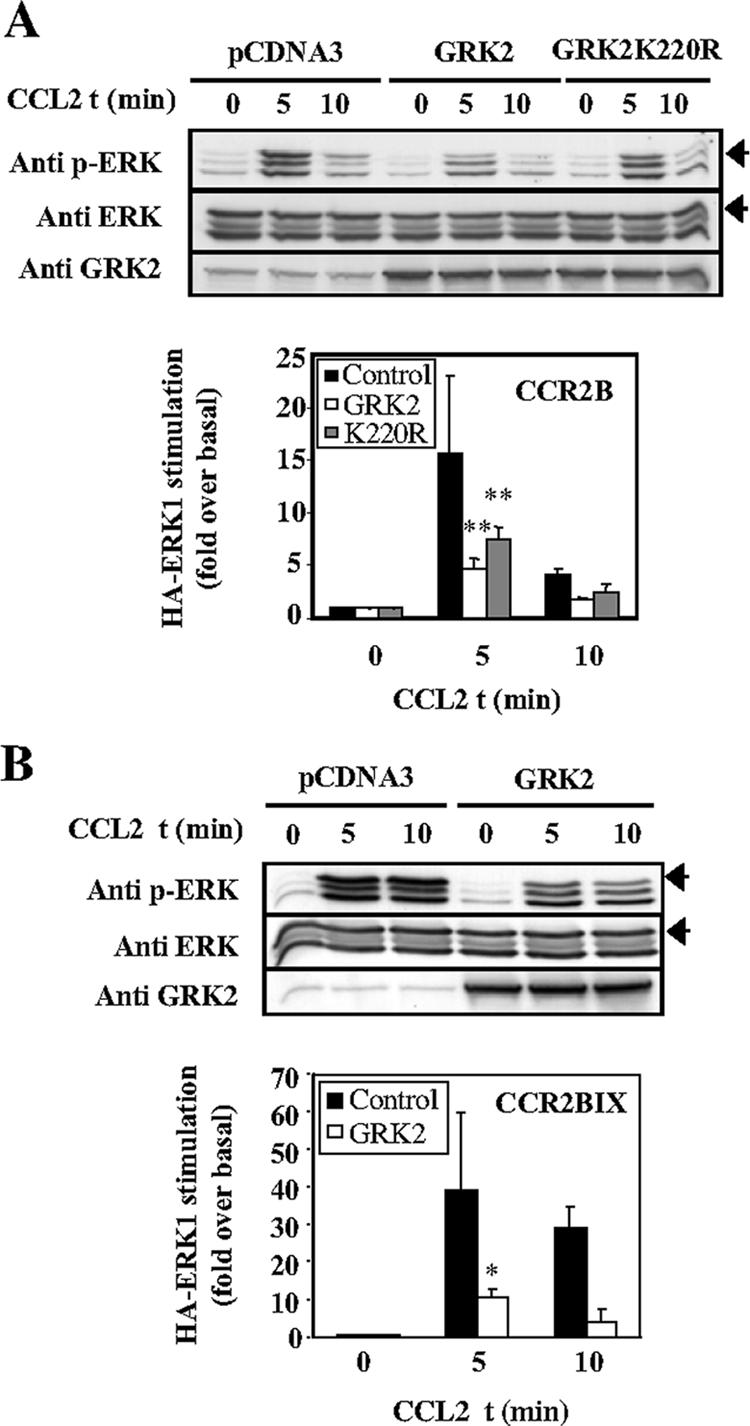
The inhibitory effect of GRK2 on CCL2-stimulated ERK activation is independent of receptor phosphorylation. (A) Cells were transfected as in Figure 1 also using pcDNA-GRK2K220R. Analysis of HA-ERK1 phosphorylation and quantitation was performed as in Figure 1 (mean ± SEM of four independent experiments). **p < 0.01 versus controls activated at 5 min. (B) ERK stimulation by the Ser/Thr mutant receptor CCR2BIX is also reduced in presence of GRK2. Cells were transfected as in Figure 1 using pcDNA3-FLAG-CCR2BIX. Analysis was performed as described above, and mean fold-increase represented. *p < 0.01 versus controls activated at 5 min. Arrowheads indicate the migration of HA-ERK.
GRK2 is a multidomain molecule that has been shown to interact with different proteins (Figure 3A): 1) the N-terminal region holds an RGS-like RH domain (aa 51–173), which binds to Gαq (Carman et al., 1999; Sallese et al., 2000); 2) the central catalytic domain is responsible for the kinase activity of the protein; and 3) the C-terminal domain contains a pleckstrin homology domain (PH), which binds to Gβγ and lipids (Carman et al., 2000). To study the GRK2 inhibitory mechanism in more detail, experiments were performed expressing the CCR2B receptor together with full-length GRK2, the GRK2 C-terminal domain (aa 438–689), or a construct including the RH and the catalytic domain (aa 1–546). Only the full-length GRK2 protein was able to produce an inhibitory effect on CCL2 activation of ERK (Figure 3B), thus supporting the notion that GRK2-mediated inhibition of ERK stimulation cannot be explained by Gβγ or Gα sequestration. Interestingly, a GRK2 point mutation, D110A, with impaired binding to Gαq proteins (Sterne-Marr et al., 2003), also showed a similar effect in down-regulating ERK phosphorylation as that observed with GRK2 (Figure 3C), as does the D110A-K220R double mutant (our unpublished data). This result demonstrates that Gαq binding is not required for GRK2 to exert its inhibition of ERK stimulation.
Figure 3.

The independent expression of different GRK2 domains does not block ERK activation by CCL2. (A) GRK2 domain structure. Full-length GRK2 plus constructs GRK2 1-546 and GRK2 438-689 are represented schematically. The RH, which binds to Gαq subunits, the catalytic region, and the PH subdomain, which binds to Gβγ, are indicated. (B) HEK293 cells were transiently transfected with pcDNA3-FLAG-CCR2B and pcCDNA1-HA-ERK1 in the presence of the indicated GRK2 constructs. Analysis of ERK activation was performed and quantified as in Figure 1. Expression levels of GRK2 and GRK2 domains were analyzed by Western Blot as detailed in Materials and Methods (bottom). (C) Cells transfected with the Gαq-binding-defective mutant pcDNA3-D110A-GRK2 or the same amount of pcDNA3-GRK2 or pcDNA3 as controls were processed as described above. This experiment was repeated three times.
To better define the level at which GRK2 is affecting ERK activation, we analyzed the stimulation of MEK (an ERK1/2 upstream activator) by chemokines with specific anti-phospho-MEK antibodies. CCL2 induces an increase in MEK phosphorylation that is maximal at 5 min (Figure 4A). Nevertheless, the expression of high levels of GRK2 does not result in any apparent reduction on CCL2-induced MEK phosphorylation even under conditions where a prominent reduction of ERK activation was observed. Similar results were obtained with the kinase-deficient GRK2K220R mutant (Figure 4B) and when using cotransfected HA-MEK (our unpublished data). Interestingly, a similar pattern of both MEK stimulation and ERK modulation was observed in cells expressing the CXCR4 receptor upon challenge with the CXCL12 chemokine (Figure 4C). The CXCR4 receptor is widely expressed in leukocytes and has been shown to be modulated by GRK2 (Orsini et al., 1999) and to stimulate the ERK pathway (Tilton et al., 2000). These results suggest that the inhibitory effect of GRK2 occurs at a level between MEK and MAPK, downstream of MEK phosphorylation, and that this phenomenon can be observed upon activation of different chemokine receptors.
Figure 4.

GRK2 expression does not inhibit CCL2-induced MEK phosphorylation. (A) HEK293 cells were transiently transfected with pcDNA3-FLAG-CCR2B and pcCDNA1-HA-ERK1 in presence of pcDNA3GRK2 or empty plasmid as indicated and stimulated with 20 nM CCL2. Analysis of ERK activation was performed as in previous figures. Analysis of MEK phosphorylation was detected by immunoblot with anti-phospho-MEK antibodies, and expression levels of the different proteins were assessed as detailed in Materials and Methods. Blots are representative of three experiments. (B) Experiments were performed as described in A, including GRK2K220R. Blots are representative of two different experiments. (C) Cells were transfected with pCDNA3-CXCR4 and the constructs specified above for stimulation during the indicated times with CXCL12. Analysis was performed as described above. Blots are representative of four experiments. Arrowheads indicate HA–ERK migration.
To further investigate this functional interaction between GRK2 and MEK, we performed coimmunoprecipitation experiments in cells transfected with these proteins using an anti-HA-MEK antibody. As can be observed in Figure 5A, both GRK2 and the GRK2K220R mutant can be found in complex with MEK, and this interaction can be detected even at endogenous levels of MEK and GRK2 expression in different cell types (Figure 5B). These results suggest that GRK2 may regulate MAPK activation through a mechanism involving the binding of GRK2 to MEK or to components of the MEK multimolecular complex.
Figure 5.
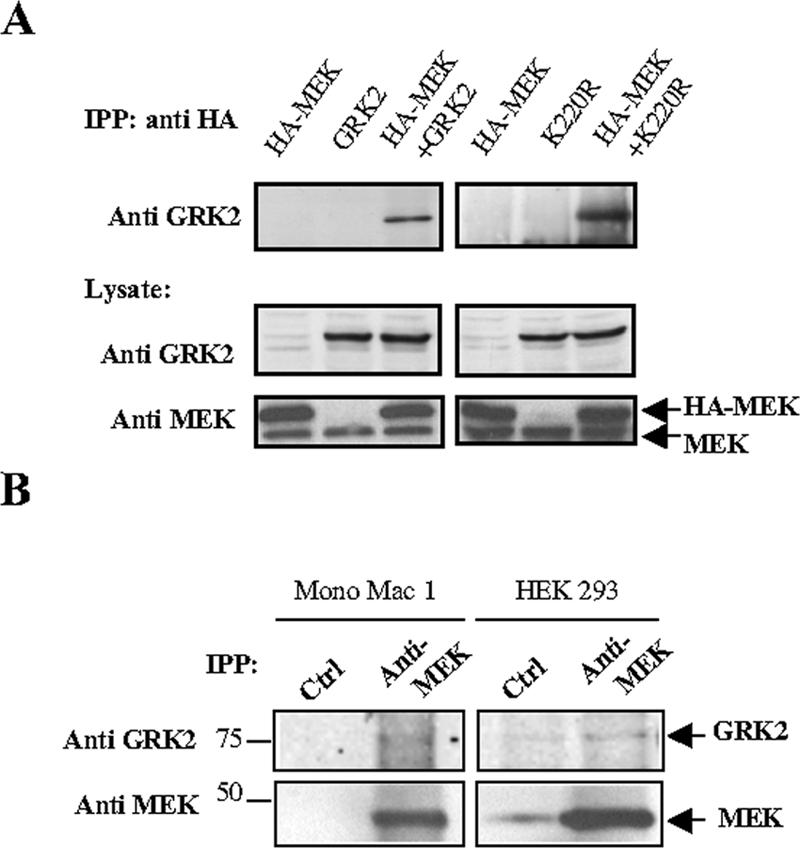
Coimmunoprecipitation of GRK2 with MEK. (A) HEK293 cells were transiently transfected with pCDNA3-HA-MEK1 and pCDNA3-GRK2 or GRK2K220R expression plasmids and kept in DMEM-10% FBS. Two days after transfection, cells were lysed and subjected to immunoprecipitation with an anti-HA antibody, and the presence of GRK2 analyzed with specific antibodies as detailed in Materials and Methods. The levels of expression of GRK2 and MEK in lysates were visualized with anti-GRK2 and anti-MEK and are shown below. Blots are representative of three experiments. (B) Endogenous MEK protein present in Mono Mac 1 monocytic cells and HEK293 cells was immunoprecipitated with an anti-MEK antibody (anti-MEK) as specified in Materials and Methods, and the presence of coprecipitated endogenous GRK2 was analyzed by Western blot. An equal amount of a rabbit preimmune serum was used as a negative control for immunoprecipitation (Ctrl). Molecular weight standards are shown to the left in kilodaltons. A representative experiment out of three performed is shown.
Together, these results reinforce the idea of a direct role for GRK2 in the control of the activation of MAPK by chemokines that is partly independent of receptor phosphorylation and occurs downstream of MEK. To test this hypothesis in a more physiological setting, heterozygous (+/–) GRK2 mice (knockout mice are embryonic lethal; Jaber et al., 1996) were analyzed for their response to chemokines. Spleen cells from heterozygous (+/–) mice have less GRK2 protein (40% of littermate wild-type [wt] controls), whereas expression of β-arrestin or GRK6 are similar (Figure 6A). Splenocytes were then stimulated with the chemokine CXCL12 and the effect on ERK activation was studied both in control (+/+) and heterozygous (+/–) mice. As can be observed in Figure 6B, CXCL12 induces phosphorylation of endogenous ERK1 and ERK2 with a peak response between 3 and 5 min. Interestingly, heterozygous mice show a more robust response to CXCL12 than control animals (Figure 6B). This effect was not due to a different surface expression of CXCR4 receptor as assessed by FACS analysis (Figure 6C). Similar to what we found in HEK293 cells, CXCL12-induced MEK activation was unaffected by the altered GRK2 expression (Figure 6D), with a mean stimulation of 2.52-fold in (+/+) and 1.75 in (+/–) mice. Therefore, these results confirm the hypothesis that the levels of expression of GRK2 are important in the control of chemokine signaling to the ERK pathway in vivo, and that this effect also occurs at a level downstream of MEK activation.
Figure 6.

Splenocytes from GRK2 (+/–) mice display an increased chemokine-mediated ERK activation. (A) Lysates from splenocytes from GRK2 (+/+) and GRK2 (+/–) were analyzed for expression of GRK2, β-arrestin, and GRK6 as described in Materials and Methods. Bands corresponding to GRK2 were quantified by densitometry and are normalized to levels found in wt littermates expressed as the fold ± SEM from three independent experiments. **p < 0.01 versus splenocytes from (+/+) mice. (B) Comparative analysis of CXCL12 stimulation (3 min) of ERK phosphorylation between control (+/+) and (+/–) heterozygous mice. Phosphorylated ERK was quantified from three independent experiments as in previous figures. (C) FACS analysis of surface-expressed CXCR4 receptor was performed in isolated splenocytes from GRK2 (+/+) and (+/–) mice as described in Materials and Methods. The isotype antibody control is also shown (gray area). (D) Splenocytes from GRK2 (+/+) and (+/–) mice were stimulated with 20 nM CXCL12 for 3 and 5 min. Lysates were used for Western Blot analysis with anti-phospho-MEK and MEK antibodies and are representative of two independent experiments performed in duplicate.
DISCUSSION
Recent data have indicated that GRK2 can modulate GPCR signal propagation at levels different from agonist-induced receptor phosphorylation via binding to specific G protein subunits (Carman et al., 1999, 2000; Sallese et al., 2000). In this article, we define a different level of regulation by GRK2 taking place at the interface between MEK and ERK. First, we have observed that elevated levels of GRK2 induce a strong inhibition of chemokine activation of the ERK pathway. Second, we find that this effect does not require the kinase activity of GRK2 or the phosphorylation of cytoplasmic serine and threonine residues on the CCR2B receptor, thus indicating that this effect is phosphorylation independent and occurs downstream from the receptor level. Third, experiments using independent overexpression of constructs containing GRK2 RH or PH domains of GRK2 or a GRK2 mutant (D110A) unable to interact with Gαq implicate a level of control of the ERK pathway downstream from G protein subunits. Fourth, we show that phosphorylation of MEK in response to chemokine receptors is not impaired upon overexpression of GRK2 or GRK2K220R and that MEK and GRK2 can be present in the same multimolecular complex, strongly suggesting that signal regulation by GRK2 takes place at the MEK–ERK interface. Finally, experiments performed in splenocytes from heterozygous GRK2-deficient mice indicate that this modulation of chemokine-induced ERK activation by GRK2 levels can also be observed in vivo.
The finding that GRK2 overexpression has inhibitory effects on ERK/chemokine signaling could in principle be caused by desensitization of the activated receptor. In fact, several different chemokine receptors have been shown to become desensitized by GRKs (see references in Aragay et al., 1998b). However, we find that the kinase activity of GRK2 is not necessary for its inhibitory effect, and that ERK activation by CCR2BIX, a mutant receptor poorly phosphorylated by GRK2, is strongly inhibited by GRK2 expression. This is in sharp contrast with the fact that the expression of the GRK2K220R mutant with the CCR2B receptor does not produce any inhibition of the CCL2-triggered calcium response per se in HEK293 cells, neither does GRK2 inhibit the CCL2-induced CCR2BIX calcium response (Aragay et al., 1998a). Our findings suggest that changes in GRK2 protein can lead to additional regulation of GPCR signaling at levels different from receptor phosphorylation.
The RGS-like RH domain of GRK2 can tightly bind Gαq, whereas the C-terminal PH domain associates to Gβγ subunits (Carman et al., 1999, 2000; Sallese et al., 2000). In the crystal structure of GRK2 (Lodowski et al., 2003), the kinase seems capable of holding together Gαq and Gβγ protein subunits in a nonexclusive way, which supports a model of phosphorylation-independent desensitization mediated by GRK2 acting at the level of G protein subunits. The fact that the observed effect of GRK2 on ERK activation is not mimicked by domains known to interact with Gαq or Gβγ subunits and that it is independent of the ability of GRK2 to bind to Gαq, because it is also observed when using the GRK2-D110A mutant, points to a mechanism taking place downstream of G protein activation. Interestingly, two very recent articles using small interfering RNA to reduce the levels of different GRKs and test their role in the modulation of angiotensin or vasopressin-mediated activation of ERK cascade may prove related to our findings (Kim et al., 2005; Ren et al., 2005). In these reports, GRK2 was shown to be responsible for some 60% of the receptor phosphorylation, whereas GRK2 overexpression led to more than 80% reduction in ERK activation. Although the authors did not investigate in detail the level at which GRK2 inhibition of ERK signaling occurs, these data are consistent with our conclusion that GRK2 can act to downplay ERK activation at a level additional to receptor phosphorylation, and they suggest that this inhibitory mechanism could be operating, possibly to varying extents, for different GPCR-activated ERK pathways.
The fact that increased levels of GRK2 lead to an inhibition of ERK stimulation would suggest that, in cells with low levels of GRK2, a stronger ERK stimulation should be observed, and, in fact, we demonstrate such is the case. In splenocytes from GRK2 hemizygous mice, an enhanced ERK activation is observed in response to chemokines. These findings are in excellent agreement with recent results (Vroon et al., 2004) showing that in T-cells, reduced GRK2 expression is also associated with increased phosphorylation of protein kinase B and MAPK activation by CCL4 and to increased migration toward chemokines. This effect can have interesting physiological consequences. Proinflammatory cytokines decrease GRK2 expression and diminished activity and levels of GRKs have been found in peripheral blood mononuclear cells of patients from rheumatoid arthritis (Lombardi et al., 1999), a condition where CCL2 is produced at high levels by both infiltrated monocytes and synovial cells (Harigai et al., 1993). Also, in an animal model of multiple sclerosis, GRK2 (+/–) mice show an earlier onset of the disease with increased infiltration of leukocytes into the central nervous system, and accordingly, leukocytes from patients with multiple sclerosis show reduced levels of GRK2 (Vroon et al., 2005). In contrast, elevations in GRK2 levels have been found in lymphocytes of hypertensive patients (Gros et al., 1997, 1999), and changes in GRK2 activity have been demonstrated during T-cell activation (De Blasi et al., 1995). The fact that GRK2 levels are important both for chemokine receptor desensitization and for ERK signaling suggest that changes in GRK2 expression in such physiopathological conditions may lead to relevant alterations in chemokine signaling, and open a new avenue for investigating possible therapeutic strategies.
The question remains of what is the mechanism of GRK2 interference on ERK activation. The results herein presented argue in favor of a mechanism involving MEK. We have found that in presence of overexpressed GRK2, chemokine-mediated ERK stimulation is reduced, whereas MEK activation is not altered. We also describe that GRK2 and GRK2K220R coprecipitate with MEK and that this association can be detected with endogenous levels of expression in different cell types. Both are indications that these two proteins may form part of the same multimolecular complex and that this colocalization is independent of the kinase activity of GRK2. It is tempting to suggest that by binding to MEK and/or other components of the complex, GRK2 could interfere (at the cellular level or at defined cellular locations) with MEK association to proteins important for its subcellular compartmentation, internalization, or activity, such as MEK–ERK scaffolds.
One of the most relevant avenues of current MAPK research focuses on the influence of subcellular localization of MAPK activators or MAPK itself for the final extent, location, or duration of ERK activation. For example, a receptor/arrestin/ERK complex is described to be increased in certain cellular locations such as pseudopodia where a prolonged activation of ERK is associated with migratory responses (Ge et al., 2003). Accordingly, the location of MEK in certain submembranous vesicles whose pinching off is inhibited by a dominant negative mutant of dynamin (K44) seems to be essential for ERK activation, whereas MEK phosphorylation remains intact (Kranenburg et al., 1999). This is in striking similarity to the results we describe here and suggests that a related mechanism could be underlying GRK2-mediated inhibition of the MEK–ERK interface. In fact, we have reported that CCL2-induced MAPK activation is also sensitive to the expression of dynamin-K44, whereas MEK is still stimulated (Jiménez-Sainz et al., 2003). Notably, GRK2 is known to colocalize with certain internalized receptors, and a large fraction of intracellular GRK2 is associated with intracellular membranes (Murga et al., 1996). Thus, it is tempting to speculate that GRK2 could act to specifically target MEK to certain cell compartments away from the vesicles where it must locate to relay an efficient phosphorylation of ERK (Kranenburg et al., 1999). A better understanding of the mechanisms involved in GRK2 modulation of ERK activation by chemokines, and its relationship to the other levels of signal regulation exerted by this kinase would help to better understand its role in chemokine physiology, and the consequences of the alterations in GRK2 levels in several cardiovascular and inflammatory diseases.
Acknowledgments
We thank Drs. M. G. Caron, J. Moscat, J. L. Benovic, S. Cotecchia, and P. Crespo for experimental tools, and Susana Rojo and Ingrid Gavlend for excellent technical assistance. This work was supported by grants from the Ministerio de Educación y Ciencia (SAF2002-0408), Fondo de Investigaciones Sanitarias (FIS) (PI030543), the Cardiovascular Network (RECAVA) of the Instituto de Salud Carlos III, Comunidad de Madrid (08.4/009.1/2003), the Norwegian Research Council, the Norwegian Melzer Foundation, and the European MAIN Network of Excellence. M.J.P. holds a predoctoral fellowship from the FIS (Ministerio de Sanidad), and C. M. is recipient of a “Ramón y Cajal” contract. The Centro de Biología Molecular holds an institutional grant from the Fundación Ramón Areces.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05–05–0399) on October 12, 2005.
Abbreviations used: ERK, extracellular signal-regulated kinase; GPCR, G protein-coupled receptor; GRK, G protein-coupled receptor kinase; MAPK, mitogen-activated protein kinase; MEK, mitogen-activated protein kinase kinase.
References
- Aragay, A. M., Mellado, M., Frade, J. M., Martin, A. M., Jiménez-Sainz, M. C., Martinez, A. C., and Mayor, F., Jr. (1998a). Monocyte chemoattractant protein-1-induced CCR2B receptor desensitization mediated by the G protein-coupled receptor kinase 2. Proc. Natl. Acad. Sci. USA 95, 2985–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragay, A. M., Ruiz-Gomez, A., Penela, P., Sarnago, S., Elorza, A., Jiménez-Sainz, M. C., and Mayor, F., Jr. (1998b). G protein-coupled receptor kinase 2 (GRK2): mechanisms of regulation and physiological functions. FEBS Lett. 430, 37– 40. [DOI] [PubMed] [Google Scholar]
- Ashida, N., Arai, H., Yamasaki, M., and Kita, T. (2001). Distinct signaling pathways for MCP-1-dependent integrin activation and chemotaxis. J. Biol. Chem. 276, 16555–16560. [DOI] [PubMed] [Google Scholar]
- Carman, C. V., Barak, L. S., Chen, C., Liu-Chen, L. Y., Onorato, J. J., Kennedy, S. P., Caron, M. G., and Benovic, J. L. (2000). Mutational analysis of Gβγ and phospholipid interaction with G protein-coupled receptor kinase 2. J. Biol. Chem. 275, 10443–10452. [DOI] [PubMed] [Google Scholar]
- Carman, C. V., Parent, J. L., Day, P. W., Pronin, A. N., Sternweis, P. M., Wedegaertner, P. B., Gilman, A. G., Benovic, J. L., and Kozasa, T. (1999). Selective regulation of Gα(q/11) by an RGS domain in the G protein-coupled receptor kinase, GRK2. J. Biol. Chem. 274, 34483–34492. [DOI] [PubMed] [Google Scholar]
- De Blasi, A., Parruti, G., and Sallese, M. (1995). Regulation of G protein-coupled receptor kinase subtypes in activated T lymphocytes. Selective increase of β-adrenergic receptor kinase 1 and 2. J. Clin. Investig. 95, 203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franci, C., Gosling, J., Tsou, C. L., Coughlin, S. R., and Charo, I. F. (1996). Phosphorylation by a G protein-coupled kinase inhibits signaling and promotes internalization of the monocyte chemoattractant protein-1 receptor. Critical role of carboxyl-tail serines/threonines in receptor function. J. Immunol. 157, 5606 –5612. [PubMed] [Google Scholar]
- Ge, L., Ly, Y., Hollenberg, M., and DeFea, K. (2003). A β-arrestin-dependent scaffold is associated with prolonged MAPK activation in pseudopodia during protease-activated receptor-2-induced chemotaxis. J. Biol. Chem. 278, 34418 –34426. [DOI] [PubMed] [Google Scholar]
- Gerard, C., and Rollins, B. J. (2001). Chemokines and disease. Nat. Immunol. 2, 108 –115. [DOI] [PubMed] [Google Scholar]
- Gros, R., Benovic, J. L., Tan, C. M., and Feldman, R. D. (1997). G-protein-coupled receptor kinase activity is increased in hypertension. J. Clin. Investig. 99, 2087–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros, R., Tan, C. M., Chorazyczewski, J., Kelvin, D. J., Benovic, J. L., and Feldman, R. D. (1999). G-protein-coupled receptor kinase expression in hypertension. Clin. Pharmacol. Ther. 65, 545–551. [DOI] [PubMed] [Google Scholar]
- Harigai, M., Hara, M., Yoshimura, T., Leonard, E. J., Inoue, K., and Kashiwazaki, S. (1993). Monocyte chemoattractant protein-1 (MCP-1) in inflammatory joint diseases and its involvement in the cytokine network of rheumatoid synovium. Clin. Immunol. Immunopathol. 69, 83–91. [DOI] [PubMed] [Google Scholar]
- Jaber, M., Koch, W. J., Rockman, H., Smith, B., Bond, R. A., Sulik, K. K., Ross, J., Jr., Lefkowitz, R. J., Caron, M. G., and Giros, B. (1996). Essential role of β-adrenergic receptor kinase 1 in cardiac development and function. Proc. Natl. Acad. Sci. USA 93, 12974 –12979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Sainz, M. C., Fast, B., Mayor, F., Jr., and Aragay, A. M. (2003). Signaling pathways for monocyte chemoattractant protein 1-mediated extracellular signal-regulated kinase activation. Mol. Pharmacol. 64, 773–782. [DOI] [PubMed] [Google Scholar]
- Kim, J., Ahn, S., Ren, X. R., Whalen, E. J., Reiter, E., Wei, H., and Lefkowitz, R. J. (2005).. Functional antagonism of different G protein-coupled receptor kinases for β-arrestin-mediated angiotensin II receptor signaling. Proc. Natl. Acad. Sci. USA 102, 1442–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong, G., Penn, R., and Benovic, J. L. (1994). A β-adrenergic receptor kinase dominant negative mutant attenuates desensitization of the β2-adrenergic receptor. J. Biol. Chem. 269, 13084 –13087. [PubMed] [Google Scholar]
- Kranenburg, O., Verlaan, I., and Moolenaar, W. H. (1999). Dynamin is required for the activation of mitogen-activated protein (MAP) kinase by MAP kinase kinase. J. Biol. Chem. 274, 35301–35304. [DOI] [PubMed] [Google Scholar]
- Kuziel, W. A., Morgan, S. J., Dawson, T. C., Griffin, S., Smithies, O., Ley, K., and Maeda, N. (1997). Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc. Natl. Acad. Sci. USA 94, 12053–12058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodowski, D. T., Pitcher, J. A., Capel, W. D., Lefkowitz, R. J., and Tesmer, J. J. (2003). Keeping G proteins at bay: a complex between G protein-coupled receptor kinase 2 and Gβγ. Science 300, 1256 –1262. [DOI] [PubMed] [Google Scholar]
- Lombardi, M. S., Kavelaars, A., Schedlowski, M., Bijlsma, J. W., Okihara, K. L., Van de Pol, M., Ochsmann, S., Pawlak, C., Schmidt, R. E., and Heijnen, C. J. (1999). Decreased expression and activity of G-protein-coupled receptor kinases in peripheral blood mononuclear cells of patients with rheumatoid arthritis. FASEB J. 13, 715–725. [DOI] [PubMed] [Google Scholar]
- Luttrell, L. M., Ostrowski, J., Cotecchia, S., Kendall, H., and Lefkowitz, R. J. (1993). Antagonism of catecholamine receptor signaling by expression of cytoplasmic domains of the receptors. Science 259, 1453–1457. [DOI] [PubMed] [Google Scholar]
- Murga, C., Ruiz-Gomez, A., Garcia-Higuera, I., Kim, C. M., Benovic, J. L., and Mayor, F., Jr. (1996). High affinity binding of β-adrenergic receptor kinase to microsomal membranes. Modulation of the activity of bound kinase by heterotrimeric G protein activation. J. Biol. Chem. 271, 985–994. [DOI] [PubMed] [Google Scholar]
- Oppermann, M., Mack, M., Proudfoot, A. E., and Olbrich, H. (1999). Differential effects of CC chemokines on CC chemokine receptor 5 (CCR5) phosphorylation and identification of phosphorylation sites on the CCR5 carboxyl terminus. J. Biol. Chem. 274, 8875– 8885. [DOI] [PubMed] [Google Scholar]
- Orsini, M. J., Parent, J. L., Mundell, S. J., Benovic, J. L., and Marchese, A. (1999). Trafficking of the HIV coreceptor CXCR4. Role of arrestins and identification of residues in the c-terminal tail that mediate receptor internalization. J. Biol. Chem. 274, 31076 –31086. [DOI] [PubMed] [Google Scholar]
- Penela, P., Ribas, C., and Mayor, F., Jr. (2003). Mechanisms of regulation of the expression and function of G protein-coupled receptor kinases. Cell Signal 15, 973–981. [DOI] [PubMed] [Google Scholar]
- Ren, X. R., Reiter, E., Ahn, S., Kim, J., Chen, W., and Lefkowitz, R. J. (2005). Different G protein-coupled receptor kinases govern G protein and β-arrestin-mediated signaling of V2 vasopressin receptor. Proc. Natl. Acad. Sci. USA 102, 1448 –1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi, D., and Zlotnik, A. (2000). The biology of chemokines and their receptors. Annu. Rev. Immunol. 18, 217–242. [DOI] [PubMed] [Google Scholar]
- Sallese, M., Mariggio, S., D'Urbano, E., Iacovelli, L., and De Blasi, A. (2000). Selective regulation of Gq signaling by G protein-coupled receptor kinase 2, direct interaction of kinase N terminus with activated Gαq. Mol. Pharmacol. 57, 826–831. [PubMed] [Google Scholar]
- Sterne-Marr, R., Tesmer, J. J., Day, P. W., Stracquatanio, R. P., Cilente, J. A., O'Connor, K. E., Pronin, A. N., Benovic, J. L., and Wedegaertner, P. B. (2003). G. protein-coupled receptor kinase 2/G αq/11 interaction. A novel surface on a regulator of G. protein signaling homology domain for binding Gα subunits. J. Biol. Chem. 278, 6050–6058. [DOI] [PubMed] [Google Scholar]
- Tilton, B., Ho, L., Oberlin, E., Loetscher, P., Baleux, F., Clark-Lewis, I., and Thelen, M. (2000). Signal transduction by CXC chemokine receptor 4. Stromal cell-derived factor 1 stimulates prolonged protein kinase B and extracellular signal-regulated kinase 2 activation in T lymphocytes. J. Exp. Med. 192, 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vroon, A., Heijnen, C. J., Lombardi, M. S., Cobelens, P. M., Mayor, F., Jr., Caron, M. G., and Kavelaars, A. (2004). Reduced GRK2 level in T cells potentiates chemotaxis and signaling in response to CCL4. J. Leukoc. Biol. 75, 901–909. [DOI] [PubMed] [Google Scholar]
- Vroon, A., Kavelaars, A., Limmroth, V., Lombardi, M. S., Goebe, M. U., Van Dam, A. M., Caron, M. G., Schedlowski, M., and Heijnen, C. J. (2005). G protein-coupled receptor kinase 2 in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Immunol. 174, 4400–4406. [DOI] [PubMed] [Google Scholar]
- Willets, J. M., Challiss, R. A., and Nahorski, S. R. (2003). Non-visual GRKs: are we seeing the whole picture? Trends Pharmacol. Sci. 24, 626–633. [DOI] [PubMed] [Google Scholar]