

Abstract
The known function of the DEXH/D-box protein Prp43p is the removal of the U2, U5, and U6 snRNPs from the postsplicing lariat-intron ribonucleoprotein complex. We demonstrate that affinity-purified Prp43p-associated material includes the expected spliceosomal components; however, we also identify several preribosomal complexes that are specifically purified with Prp43p. Conditional prp43 mutant alleles confer a 35S pre-rRNA processing defect, with subsequent depletion of 27S and 20S precursors. Upon a shift to a nonpermissive temperature, both large and small-ribosomal-subunit proteins accumulate in the nucleolus of prp43 mutants. Pulse-chase analysis demonstrates delayed kinetics of 35S, 27S, and 20S pre-rRNA processing with turnover of these intermediates. Microarray analysis of pre-mRNA splicing defects in prp43 mutants shows a very mild effect, similar to that of nonessential pre-mRNA splicing factors. Prp43p is the first DEXH/D-box protein shown to function in both RNA polymerase I and polymerase II transcript metabolism. Its essential function is in its newly characterized role in ribosome biogenesis of both ribosomal subunits, positioning Prp43p to regulate both pre-mRNA splicing and ribosome biogenesis.
DEXH/D-box proteins comprise a family of RNA helicase and RNA helicase-like proteins that function in almost all aspects of eukaryotic gene expression. Included in these processes is RNA transcription (30, 49), pre-mRNA splicing (reviewed in reference 35), mRNA export (21, 34, 38), translation initiation (40), RNA interference (29, 41), RNA turnover (1), viral replication (31), and ribosome biogenesis (reviewed in references 13 and 44). Among this class of RNA helicases are the DEAD-box proteins and the DEAH-box proteins, which differ by subtle sequence variations in one or more of the motifs that define these proteins (reviewed in reference 42). In addition to an RNA-unwinding activity demonstrated by some of these enzymes, others exhibit an “RNPase” function that serves to displace proteins from or otherwise rearrange ribonucleoproteins (10, 20). Although the specific function and mechanism of most DEXH/D-box proteins are unknown, the processes in which many of these enzymes function have been studied in great detail.
The eukaryotic pre-mRNA splicing reaction is an ATP-intensive process in which at least eight DEXH/D-box proteins function (35). The genes for all eight Saccharomyces cerevisiae spliceosomal DEXH/D-box proteins are essential, although two have been shown to be dispensable in concert with bypass suppressor mutations (6, 23). DEXH/D-box proteins are required for virtually every known step of the pre-mRNA splicing reaction and are thought to monitor and modify RNA-RNA interactions and RNA-protein interactions in the highly dynamic process.
Among the trans-acting spliceosomal RNA helicase-like proteins is Prp43p, which was originally identified in a PCR-based screen for RNA helicase genes in yeast (2, 9). It has been shown to be a bona fide RNA-dependent ATPase, although no RNA-unwinding activity has been shown for this enzyme. Martin and colleagues have demonstrated that in vitro, Prp43p functions in the disassembly of the postcatalytic spliceosome by releasing the U2, U5, and U6 snRNPs from the intron lariat RNA, although the mechanism of this release is currently not well defined (27). Although PRP43 is an essential gene, it functions very late in the splicing cycle temporally near Dbr1p, a nonessential protein (5), after the pre-mRNA splicing reaction is completed.
A number of essential RNA helicases participate in yeast pre-rRNA processing and ribosome assembly and export. At least 17 RNA helicases have been shown to uniquely function in ribosome biogenesis (reviewed in references 13 and 42). The biogenesis of ribosomes in eukaryotes requires a symphony of proteins and RNAs acting in a rapid and highly organized manner. Dozens of yeast snoRNAs modify the ribosome by site-specific modifications including pseudouridylation and methylation, and over 120 nonribosomal proteins participate in the process (13). The possible functions of the RNA helicases in the biogenesis of ribosomes include unwinding of the snoRNAs from their substrate(s), chaperoning snoRNPs to their sites of action, rearranging intra-rRNA helices, and removing proteins from the massive preribosomal RNP as well as possibly serving as a molecular clock to monitor the fidelity of ribosome biogenesis (see Fig. 1 for the major rRNA biogenesis pathway).
FIG. 1.

The major pre-rRNA processing pathway in yeast. (A) The rRNA gene locus in yeast. Features of the rRNA gene locus are shown, and cleavage sites are noted above. Note that the 18S and 25S regions are not in proportion. (B) Major rRNA gene processing pathway in yeast. Major cleavage sites leading to the processing intermediates in the rRNA gene biogenesis pathway are noted, as are the exonucleolytic (EXO) processing steps. ETS, external transcribed spacer; ITS1, internal transcribed spacer region 1.
We initiated studies on Prp43p to address the substrate and function of this essential polypeptide and to identify factors interacting with it. Prior to a recent large-scale green fluorescent protein (GFP) screen (18), we found that Prp43p is predominantly nucleolar and is associated with several pre-rRNA species and snoRNAs and a spectrum of ribosome biogenesis factors, which are specifically affinity purified from Prp43p-TAP extracts. Two strains carrying cold-sensitive (CS) mutant alleles of PRP43 grown at the nonpermissive temperatures exhibit early pre-rRNA processing defects as shown by the accumulation of the 35S pre-rRNA precursor, depletion of 27S and 20S pre-rRNAs, accumulation of ribosomal proteins in the nucleolus, and altered polysome profiles. Pulse-chase analysis shows that while 35S rRNA gene transcription is still robust at nonpermissive temperatures, there is a profound defect in processing of 35S, 27S, and 20S pre-rRNAs to mature 25S and 18S rRNA genes. A rapid and remarkable turnover of the vast majority of pre-rRNA precursors indicates that one potential function of Prp43p may be to serve as a molecular clock in the quality control for processing both large- and small-subunit rRNA genes. These data coupled with array data demonstrating only a very mild pre-mRNA splicing defect and a severe depletion of 5.8S, 18S, and 25S rRNA signals indicate a remarkable bifunctionality of Prp43p in pre-mRNA splicing and ribosome biogenesis and that Prp43p functions in the biogenesis of both ribosomal subunits. These data also lead to the exciting possibility that Prp43p may also serve to coregulate both RNA polymerase I (Pol I) and RNA polymerase II transcript metabolism.
MATERIALS AND METHODS
Yeast strains.
Strains ySS567 (MATa ura3-52 trp1-63 his3-Δ200 leu2-1 ade2-101 lys2-801 prp43::KAN + pPRP43::TRP1-CEN), ySS568 (ySS567 but with pprp43S247A::TRP1-CEN), and ySS569 (ySS567 but with pprp43G429A:: TRP1-CEN) were kind gifts from Beate Schwer; strains BY4734 (MATα his3Δ1 leu2Δ0 met15Δ0 trp1Δ63 ura3Δ0), ySS490 (BY4734 + PRP43-GFP::HIS3), ySS491 (ySS490 + pNOP1-dsRED), ySS578 (ySS567 + pRS316-RPL25-eGFP), ySS579 (ySS567 + pRS316-RPS2-eGFP), ySS580 (ySS569 + pRS316-RPL25-eGFP), ySS581 (ySS569 + pRS316-RPS2-eGFP), ySS582 (ySS568 + pRS316-RPL25-eGFP), ySS583 (ySS568 + pRS316-RPS2-eGFP), ySS480 (BY4734 + PRP43-TAP::TRP1), and ySS482 (ySS480 + dbr1::HIS3) were from our laboratory; strain ySS575 (MATa his3 leu2 met15Δ0 ura3 YGL099W::KANMX4 + pAJ740 [lsg1-1::LEU2-CEN]) was a kind gift from Arlen Johnson. Yeast transformations were performed by using the lithium acetate procedure (16). Gene replacement of DBR1 with HIS3 in ySS480 was performed by PCR fragment transformation as previously described (45), with oligonucleotides DBR1KOA (5′-GTAAATATGTAACTAAAAATTAAGATGGGCAGACATTTATCATTTTGCTTTTAGTGCTTTACGGCACCTCGACC-3′) and DBR1KOB (5′-GGCTTGGCTTTAAGCTCTAATTCCGTCTGCATTCGTAATAGAAATATCTCAGCAGATTGTACTGAGAGTGCACC-3′), using plasmid pRS413.
Extract preparation and affinity.
Nine liters each of strains BY4734, ySS480, and ySS482 was harvested by centrifugation and resuspended in 1.5 ml A10 buffer (10 mM HEPES, 10 mM KCl) per gram of wet cells, followed by agitation with glass beads (BioSpec), extract preparation (36), and affinity purification as previously described (32).
Bulk RNA analysis.
Immunoglobulin G affinity-purified material was eluted with TEV protease, and RNA was extracted with phenol and precipitated with ethanol.
Analysis of time course and immunoprecipitated RNAs.
RNA from time course experiments was isolated using the hot acid phenol method (48). Immunoprecipitated RNAs were recovered by standard phenol-chloroform extraction and precipitated with ethanol (33).
(i) Northern blots.
For large RNAs, 10 μg of RNA from each sample was glyoxylated and separated on a 1% agarose gel made with 1× BPTE (33). Analysis of small RNAs was performed by electrophoresis of 5 μg RNA through a 7% acrylamide (19:1)-8 M urea-1× Tris-borate-EDTA polyacrylamide gel. RNA was transferred to a charged nylon membrane (Ambion) in 10× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate). 32P-labeled oligonucleotide probes D-A2 and U2 were used to detect the 35S pre-rRNA transcript and the U2 loading control RNA as previously described (24). Mature rRNAs were detected using oligonucleotides 25S (5′-AAACAACTCGACTCTTCGAAGGCACTTTACAAAGAACCGCACTCC-3′), 18S (5′-GGAAAGGCCCCGTTGGAAATCCAGTACACGAAAAAATCGGACCGG-3′), and 5.8S (5′-TTTCGCTGCGTTCTTCATC-3′).
(ii) Primer extension.
Primer extension analysis was performed with the following oligonucleotides to detect pre-rRNA, snoRNAs, snRNAs, and scR1 using a previously described primer extension procedure: 35S (5′-CGCTGCTCACCAATGG-3′), 27SA2/7S (5′-TTTCGCTGCGTTCTTCATC-3′), U14 (5′-TCCACGGTAGGAGTACGCTTACGAACCCATCGT-3′), U24 (5′-GAGATCTTGGTGATAATTGGTATGTC-3′), U3, snR3 (5′-GCAATCCACGAGTTCTTATA-3′), U1 (5′-GAATGGAAACGTCAGCAAACAC-3′), U2 (5′-CTTAAAAAGTCTCTTCCCGTC-3′), U4 (5′-ACCATGAGGAGACGGTCTGG-3′), U5 (5′-ATGTTCGTTATAAGTTCTATAGGC-3′), and U6 (5′AGGGGAACTGCTGATC-3′) (36).
(iii) RNase protection assay.
To detect the presence of 20S pre-rRNA, a terminal deoxynucleotidyltransferase/[32P]ddATP-labeled oligonucleotide probe, A2overlap (5′-GCCCAAAGAAAAAGTTGCAAAGATATGAAAACTCCACAGTGTGTTGTATTGAAACGGTTTTAATTGTCCTATAACAAAAGCACAGAAATC-3′), was annealed to 10 μg total RNA. This oligonucleotide spans the A2 cleavage site and allows discrimination of 20S pre-rRNA from 35S, 33S, and 32S precursors after RNase protection. Reactions were carried out according to the instructions of the manufacturer (Ambion), and the 45-nucleotide product resulting from RNase protection of only the 20S pre-rRNA was separated from the 35S, 33S, and 32S products (90 nucleotides) by polyacrylamide gel electrophoresis (PAGE) (10% acrylamide [19:1]-8 M urea).
Polysome analysis.
Cultures (300 ml) of PRP43, prp43S247A, prp43G429A, and lsg1-1 were grown to an OD (A600) of ∼0.3 and shifted to 16°C for 3 h. Cycloheximide was added at 150 μg/ml to lock polysomes in situ and mixed vigorously. Cells were harvested immediately in ice and washed in 1 volume of lysis buffer (10 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 6 mM β-mercaptoethanol, 150 μg/ml cycloheximide, 100 mM KCl). Cells were resuspended in 1 volume of lysis buffer, and 1 volume of glass beads was added. Cells were lysed by vortexing four times for 30 s followed by 30 s on ice. Lysates were clarified by centrifugation at 4°C at 12,000 rpm in a Beckman JA17 rotor for 10 min. Nine OD units (A260) were sedimented on a 7 to 47% sucrose gradient (Beckman SW41 rotor) at 4°C and 40,000 rpm for 2.5 h.
Rpl25-enhanced GFP (eGFP) and Rps2-eGFP reporter assays.
pRS316-Rpl25eGFP and pRS316-Rps2eGFP (both kind gifts from Ed Hurt, University of Heidelberg) were transformed into strains containing PRP43, prp43G429A, and prp43S247A. The six resulting strains described above were grown at permissive (30°C) or nonpermissive (16°C) temperatures for 5 h. The cultures were maintained below an OD (A600) of 0.4. Formaldehyde was added to 4% and mixed vigorously. After incubating at 16°C for 30 min, cells were pelleted, washed, and resuspended in phosphate-buffered saline prior to fluorescence microscopy. Images were captured using a Nikon Eclipse E600 microscope and Openlab 4.0.3 software with a Macintosh computer.
Pulse-chase analysis. (i) [3H]methyl-methionine pulse-chase analysis.
Cultures of PRP43, prp43G429A, and prp43S247A were grown to an OD (A600) of 0.3 in medium lacking methionine at 30°C. Forty OD (A600) units were pelleted and resuspended in 12 ml of medium lacking methionine. After a 2-min pulse with 175 μCi [3H]methyl-methionine, 1.5-ml samples from each culture were taken, corresponding to a 0-min time point. Aliquots were taken at 1, 3, 5, and 10 min after the addition of 150 μg cold methionine.
(ii) [5,6-3H]uracil pulse-chase analysis.
Cultures of PRP43, prp43G429A, and prp43S247A were grown to an OD (A600) of 0.3 in medium lacking uracil at 30°C. Forty OD (A600) units were pelleted and resuspended in 12 ml of medium lacking uracil. After a 2-min pulse with 100 μCi [5,6-3H]uridine, 1.5-ml samples from each culture were taken, corresponding to a 0-min time point. Aliquots were taken at 5, 15, 30, and 60 min after the addition of 150 μg cold uracil. RNA from each sample was extracted with hot acid phenol as described above, precipitated, treated with glyoxal, separated on a 1% agarose gel, and transferred to a Brightstar membrane (Ambion) prior to exposure to a 3H phosphorimager screen (Bio-Rad). Analysis and quantitation were performed using the Quantity-One software package (Bio-Rad).
Microarray analysis.
PRP43, prp43G429A, and prp43S247A yeast strains were grown to an OD (A600) of 0.6 at 30°C and shifted to a nonpermissive temperature (16°C) for 45 min. RNA preparation, probe synthesis, labeling with Cy3 and Cy5 dyes, and hybridization were carried out as previously described (8). RNA isolated from cold-shifted PRP43 was used as a reference sample in prp43G429A and prp43S247A cold-shift experiments. Experiments were conducted in duplicate using reciprocal label or dye swap experiments. Image processing and normalization were carried out as described previously (4). Intron accumulation index (IAI) values were calculated as described previously (8). Hierarchical clustering was carried out using the Genesis package (39). For splicing efficiency characterization of prp43 mutants, IAI values for prp43G429A and prp43S247A and the IAI for 76 gene expression perturbations (4) were clustered using an uncentered Pearson correlation distance metric and average linkage algorithm. For hierarchical clustering analysis of stable RNAs, normalized log ratio values from stable RNA features for prp43 mutants and 71 gene expression perturbations were clustered using the centered Pearson correlation distance metric and average linkage algorithm. PRP43 array data described in this paper are available through the gene Expression Omnibus repository at the National Center for Biotechnology Information website (http://www.ncbi.nlm.gov/geo/). A graphical representation of intron accumulation comparing prp43, prp4, and dbr1 mutants is available upon request.
GEO accession numbers for the experiments described are GSM78602, GSM78603, GSM78604, and GSM78605.
RESULTS
Prp43p-GFP localizes to the yeast nucleolus.
Yeast pre-mRNA splicing is a nuclear event (7); thus, we expected a nucleoplasmic localization of Prp43p. Cells containing Prp43-eGFP showed an intense signal in what appeared to be a subnuclear location by fluorescence microscopy (data not shown). This was confirmed by cotransformation with a Nop1p-dsRED-producing plasmid that produces a polypeptide shown to localize to the nucleolus (14). Fluorescence microscopy visualization demonstrated that the Prp43p-eGFP and the Nop1p-dsRED signals overlapped in a subnuclear structure, a hallmark of the yeast nucleolus (data available upon request). As the majority of Prp43p is nucleolar, it seems likely that a major function of Prp43p might occur in the nucleolus. Since Prp43p acts at a late stage in the pre-mRNA splicing reaction, it remains a formal possibility that intron lariat RNP metabolism may be nucleolar; however, Dbr1p, which acts after Prp43p, is a nucleoplasmic protein (18). Subsequent to our finding, a large-scale GFP screen of yeast genes also confirmed that Prp43p is nucleolar (18).
Affinity-purified Prp43p-associated complexes contain spliceosomal snRNA and pre-rRNAs.
To better understand the cellular machinery with which Prp43p associates, we affinity purified Prp43p-associated RNA and protein using the TAP tag (32). We constructed two strains to perform these studies, one containing Prp43p-TAP in the native chromosomal locus and that same strain modified by the removal of DBR1, the gene responsible for intron lariat debranching (5). Others have previously shown that in in vitro splicing reactions, intron lariat RNP accumulated to a greater degree in the absence of Dbr1p and that upon the addition of dominant-negative Prp43p, the spliceosome arrests at the disassembly step (27). We envisioned that in vivo, the kinetics of the reaction might slow sufficiently to allow accumulation of the Prp43p-associated lariat RNP for preparative purification.
RNA was harvested from the TAP-purified material from wild-type (BY4734), PRP43-TAP, and PRP43-TAP/Δdbr1 cells. When probed for actin intron sequences, only total and immunopurified RNA from the PRP43-TAP/Δdbr1 cells contained detectable amounts of that species (Fig. 2A), confirming that in vivo, Prp43p associates with a fraction of complexes containing the lariat intron RNA (19). Given the nucleolar localization of Prp43p-eGFP, we probed the TAP affinity-purified RNAs from BY4734 and PRP43-TAP for a number of preribosomal as well as spliceosomal RNA and snoRNA species. As shown in Fig. 2B, the spliceosomal snRNAs U2, U5, and U6 are specifically detected in the PRP43-TAP eluate by primer extension; all are absent from material isolated from the untagged strain. The U2, U5, and U6 snRNAs are present in the postsplicing lariat RNP complex. The U1 and U4 snRNAs are not expected to be present at this stage of the splicing reaction and were not present in our samples. We note that according to both primer extension and Northern blot analysis, the U5 snRNA is of greater abundance in our immunoprecipitation experiments than U2 and U6 snRNAs, indicating that Prp43p appears to be associated with a U5 snRNA-containing complex representing a possible postlariat RNP decomposition product.
FIG. 2.
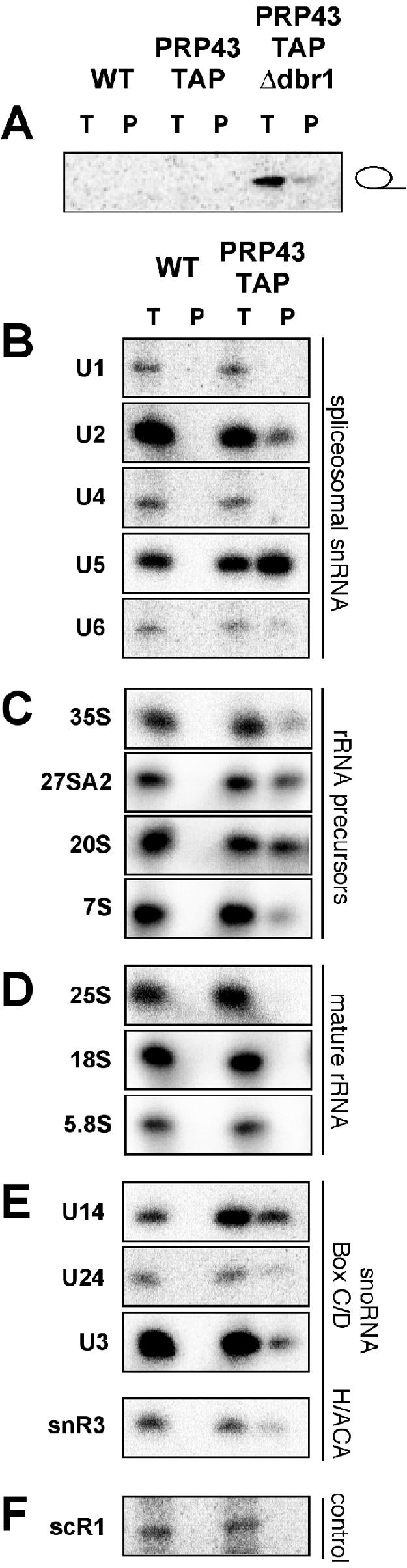
Prp43p is associated with intronic lariat RNA, spliceosomal snRNAs, several preribosomal species, and both H/ACA- and C/D-box snoRNAs. (A) Prp43p is associated with ACT1 intronic RNA in extracts lacking Dbr1p. A 1/10 equivalent of total RNA (T) and affinity-purified RNA (P) prepared from extracts of wild-type (WT), PRP43-TAP, and PRP43-TAP/Δdbr1 cells was separated by urea-PAGE, transferred to nylon membranes, and Northern blotted with an ACT1 intron oligonucleotide. (B) U2, U5, and U6 spliceosomal snRNAs are associated with Prp43p. RNA from extracts equivalent to 1/10 of the applied affinity-purified material (T) and immunopurified (P) RNAs from wild-type (WT) and Prp43p-TAP extracts were analyzed by primer extension reactions using oligonucleotides specific to the U1, U2, U4, U5, and U6 snRNAs. Primer extension products were resolved by urea-PAGE. (C) Prp43p-TAP immunopurifies several pre-rRNA processing intermediates. A 1/10 equivalent of total RNA (T) and immunoglobulin G-purified RNAs from WT and Prp43p-TAP extracts was analyzed by primer extension or RNase protection analysis as described in Materials and Methods. Products were resolved by urea-PAGE. (D) Mature rRNA genes are not associated with Prp43p. A 1/10 equivalent of total RNA from extracts (T) and immunopurified (P) RNAs from wild-type (WT) and Prp43p-TAP extracts was analyzed by primer extension analysis for the 25S, 18S, and 5.8S rRNAs. Primer extension products were resolved by urea-PAGE. (E) Box C/D and box H/ACA snoRNAs are contained in Prp43p immunoprecipitations. Primer extension analysis of U14, U24, U3, and snR3 snoRNAs was performed from total RNA (T) or immunoprecipitated RNAs (P), and primer extension products were resolved by urea-PAGE. (F) Prp43p immunoprecipitation control. scR1 small RNA was analyzed from a 1/10 equivalent of total RNA from extracts (T) and immunopurified (P) RNAs from wild-type (WT) and Prp43p-TAP extracts by primer extension analysis. Primer extension products were resolved by urea-PAGE. The above-described experiments were analyzed by using a phosphorimager.
Surprisingly, we also detected pre-rRNA species associated with Prp43p-TAP (Fig. 2C). The 35S, 27S, 20S, and 7S pre-rRNAs are specifically affinity purified, indicating a role for Prp43p in the biogenesis of both small- and large-subunit rRNAs. Prp43p is associated with a remarkably diverse set of pre-rRNAs, from the initial 35S pre-rRNA to the relatively late 20S and 7S pre-rRNAs (Fig. 1). We did not detect an association of Prp43p with the mature 25S, 18S, or 5.8S rRNAs (Fig. 2D), consistent with the lack of significant quantities of Prp43p in the cytoplasm (data available upon request). The U3 snoRNA is also specifically immunoprecipitated with Prp43p (Fig. 2E). U3 snoRNP is a very-early-acting RNP, indicating potential functions for Prp43p at the very earliest stages of pre-rRNA processing.
In addition to the U3 snoRNA, we detected the presence of the U14, U24, and snR3 snoRNAs (Fig. 2E). These are members of both the box C/D (U14 and U24) and box H/ACA (snR3) snoRNA families. Although we cannot determine from our data whether Prp43p is associated directly with these snoRNAs, or if their coisolation is due to their coassociation with the assembling ribosome, the incomplete recovery of the snoRNAs would suggest that Prp43p and the snoRNAs are copurifying by virtue of being independently associated with the pre-rRNA. The nonspecific small RNA scR1 was used as a control for these analyses. In Fig. 2E, scR1 is associated with the immunoprecipitated material from neither the wild-type nor Prp43p-TAP extracts, further demonstrating the specificity of the above-described immunoprecipitation experiments.
rRNA processing defect in conditional alleles of PRP43.
Since pre-mRNA splicing and ribosome biogenesis are both essential processes, the in vivo defect in one or both essential processes correlating to the growth defect should reveal the essential function of Prp43p. Upon a shift to the nonpermissive temperature (16°C), cultures of PRP43 continued growing in an exponential manner, whereas the CS alleles prp43S247A and prp43G429A exhibited a severe growth defect (Fig. 3A). Total RNA was harvested from cultures of PRP43, prp43S247A, and prp43G429A grown for 0, 0.5, 6, and 20 h at the nonpermissive temperature. We found a time-dependent accumulation of unprocessed 35S precursor in the conditional alleles prp43S247A and prp43G429A (Fig. 3B) (22). We observed no appreciable change in the 35S representation in the wild-type cultures at 16°C. The same blots were probed for U2 snRNA as a loading control. We also assayed for the presence of 27S and 20S precursors over the time course. The levels of the 27S and 20S pre-rRNAs were severely reduced between 0.5 and 6 h in both of the prp43 mutants and were completely absent at the 20-h time point (Fig. 3B). The levels of 27S and 20S pre-rRNA were essentially unchanged in the wild-type PRP43 cultures at 16°C.
FIG. 3.

Growth defects in prp43 mutants correlate to rRNA processing defects but not pre-U3 splicing defects. (A) Mutant prp43 strains are growth impaired at 16°C. Growth curves of PRP43 (open box), prp43G429A (open diamond), and prp43S247A (open circle) after the shift to the nonpermissive temperature (16°C) are shown. (B) Pre-rRNA processing defects are detected in prp43 mutant strains. Shown is Northern blot analysis of total RNA from strains in panel A over a 20-h time course. Note the accumulation of 35S pre-rRNA and the depletion of 27S and 20S pre-rRNA over time. (C) Mutant prp43 strains do not exhibit pre-mRNA splicing defects in pre-U3A or pre-U3B snoRNA introns. RNA from strains grown as described above (A) and a Δsnu66 control were subjected to primer extension analysis using oligonucleotide primers designed to detect U3 pre-snoRNA, U3 snoRNA, and U1 snRNA (loading control). (D) Mutant prp43 strains are depleted for ribosomal subunits at nonpermissive temperatures. Sucrose gradient polysome profiles are shown for PRP43, prp43S247A, prp43G429A, and the lsg1-1 control. The direction of the increasing sedimentation coefficient is from left to right. Ribosomal subunits, polysomes, and half-mers are noted.
To probe for defects in pre-mRNA splicing, we tested RNA from the cultures described above for accumulation of unspliced pre-U3A and pre-U3B pre-snoRNAs by primer extension analysis (36). With respect to the wild type, there was no increase in unspliced U3A and U3B pre-snoRNAs in the prp43S247A and prp43G429A samples, even after growth at the nonpermissive temperature for 20 h. In contrast, a Δsnu66 mutant at the nonpermissive temperature of 16°C (37) showed an increase in abundance of U3A and U3B pre-snoRNAs even after only 3 h at the nonpermissive temperature (Fig. 3C). This suggests that although the loss of PRP43 function strongly inhibits pre-rRNA processing, it does not result in a strong inhibition of U3 splicing. Because U3 snoRNA is also involved in ribosome biogenesis, it is also possible that these results are due to other factors. To determine whether this is the case, we also tested the pre-mRNA splicing efficiency of all known yeast introns (see below). Levels of mature U3 snoRNA are unchanged at nonpermissive temperatures in all cultures (Fig. 3C).
Polysome and ribosomal subunit profile analysis in prp43 mutants.
A hallmark of ribosome biogenesis defects is the reduction in abundance of ribosomal subunits in cellular extracts. We next determined whether defects in rRNA processing conferred by prp43 mutant strains correlated to defects in either 40S or 60S ribosomal subunit accumulation, or both, by sucrose gradient analysis. At 16°C, wild-type cells exhibited a 60S-to-40S subunit ratio of approximately 1:1 (Fig. 3D), with a sharp 80S peak and a relatively level polysome tail. The ratio of 60S to 40S subunits in lysates from lsg1-1, a known 60S synthesis mutant (22), is diminished in free 60S subunits at nonpermissive temperatures (Fig. 3D). The prp43S247A and prp43G429A mutants exhibited a similar altered subunit ratio as well as a characteristic “half-mer” polysome profile (Fig. 3D) indicative of a rapid loss of 60S ribosome subunit synthesis when grown at the restrictive temperature. Although the levels of 40S subunits may also be decreased, we can only comment on the relative levels of the ratio of 40S to 60S subunits, and the levels of 60S subunits are reduced more than 40S subunits.
Ribosome subunit export analysis in prp43 mutants.
If the destruction of rRNA intermediates occurs in prp43 mutants, ribosomal proteins known to bind these RNAs in the nucleolus would accumulate there upon elimination of Prp43p function. We therefore tested for large- and small-ribosomal-subunit protein retention in the nucleolus using reporter constructs. PRP43, prp43S247A, and prp43G429A strains were transformed with either pRS316-Rpl25-eGFP (15) or pRS316-Rps2-eGFP (28), grown at either permissive or nonpermissive temperatures, fixed with formaldehyde, and visualized using a fluorescence microscope (Fig. 4). At permissive temperatures, PRP43, prp43S247A, and prp43G429A cells exhibited similar, predominantly cytoplasmic localization of the Rpl25p-eGFP and Rps2p-eGFP reporter proteins (Fig. 4A to C, left panels). Upon a shift of PRP43 cells to the nonpermissive temperature, the reporter protein localization was unchanged, indicating no gross defect in the localization of these reporters at 16°C under normal circumstances (Fig. 4A, right panel). In both prp43S247A and prp43G429A strains grown at 16°C, we detected a localization defect for both the Rpl25p-eGFP and Rps2p-eGFP (Fig. 4B and C, right panels). In approximately half of the cells visualized in both strains and for both reporter constructs, the GFP reporters displayed a distinct subnuclear localization, a hallmark of the nucleolus (see Fig. 4D for enlarged panels). In the remainder of the cells visualized, the GFP signal was observed in the nucleus.
FIG. 4.

Both small- and large-ribosomal-subunit proteins exhibit nucleolar and nuclear accumulation in prp43 mutant cells at nonpermissive temperatures. (A) Phase-contrast (PC), GFP, DNA strain (DAPI [4′,6′-diamidino-2-phenylindole]), and merged images of GFP plus DAPI (MERGE) from PRP43 cells grown at the permissive temperature (30°C; left panel) or nonpermissive temperature (16°C; right panel) and containing the Rpl25p-eGFP reporter construct (top row) or the Rps2p-eGFP reporter construct (bottom row). Images similar to those described above (A) using a strain containing the prp43G429A mutation are shown in panel B, and those using a strain containing the prp43S247A mutation are shown in panel C. (D) The merged magnified images of the Rpl25p-eGFP and Rps2p-eGFP reporter strains are shown from the nonpermissive temperature conditions for PRP43 (left panels) and prp43S247A (right panels) to highlight the nucleolar and nuclear accumulation of the ribosomal subunit proteins.
Pulse-chase analysis of rRNA precursors in prp43 mutants.
Prp43p function in ribosome biogenesis was further assessed by pulse-chase analysis of rRNAs in PRP43 cells and prp43S247A cells at 23°C, a nonpermissive temperature condition. In PRP43 cells, 35S pre-rRNA is rapidly and nearly quantitatively converted into mature 25S and 18S rRNAs within 10 min at 23°C as measured by [3H]methyl-methionine pulse-chase (Fig. 5A and B). In prp43S247A cells, 35S pre-rRNA transcription at 0 min appeared robust, indicating that rRNA transcription is not impaired and that subsequent methylation by snoRNPs is occurring (Fig. 5B). As quantitated by phosphorimage analysis, the 35S signal in prp43 samples at the zero time point was twice that of the wild type. Indeed, total counts for the combined signal of 35S, 27S, and 20S pre-rRNAs are 1.3-fold higher in the mutant than in the wild type. This suggests not only that there is no gross methylation defect but also that there is a delay in the kinetics of processing of 35S pre-rRNA in prp43 mutant cells. The 1.3-fold effect in the prp43S247A mutant was reproducible but not seen at permissive temperatures (data not shown), confirming our interpretation of the 35S processing delay. These results are consistent with the accumulation of 35S precursors in steady-state experiments followed by rapid turnover of the 35S, 27S, and 20S pre-rRNA species (Fig. 3B and 5A). In comparison to the wild type, conversion of 35S pre-rRNA into 27S and 20S pre- rRNAs in prp43S247A was significantly weaker at 3 min (∼19% and ∼18%, respectively), with the balance of 35S precursor presumably being degraded. Strikingly, there was no detectable conversion of 27S into mature 25S rRNAs, indicating that in mutant prp43 cells, 27S rRNA intermediates fail to be correctly processed and that both 35S and 27S rRNAs appear unstable. The small amounts of 20S pre-rRNA produced in prp43S247A cells were converted to 18S at ∼40% efficiency, but the total levels of 18S rRNAs at 10 min after chase were <8% of that of the identically handled PRP43 cultures (Fig. 5B).
FIG. 5.

Pulse-chase analysis shows early defects in pre-rRNA processing in prp43S247A mutant cells. (A) PRP43 cells process pre-rRNA normally at nonpermissive temperatures. (B) prp43S247A cells synthesize 35S pre-rRNA and process less 27S and 20S pre-rRNA into mature 25S and 18S rRNAs than the wild type. Wild-type PRP43 and prp43S247A cultures were pulsed with [3H]methyl-methionine and chased with cold methionine for the times indicated above the lanes. Pre-rRNA (35S, 27S, and 20S) and mature rRNA (25S and 18S) gene species are noted. (C) [5,6-3H]uracil pulse-chase analysis of pre-rRNA processing in PRP43 cells. (D) [5,6-3H]uracil pulse-chase analysis of pre-rRNA in prp43S247A cells. Wild-type PRP43 and prp43S247A cultures were pulsed with [5,6-3H]uracil and chased with cold uracil for the times indicated above the lanes.
As discussed above, Prp43p functions in the release of the intron lariat RNA from the U2, U5, and U6 snRNPs. Since there are a number of yeast rRNA gene methylation snoRNAs encoded in intronic sequences, we repeated the pulse-chase analysis in PRP43 and prp43S247A cultures using [5,6-3H]uracil to demonstrate that the results presented in Fig. 5B were not due to deficiencies in methylation which may be directed from intronic snoRNAs (Fig. 5C and D). Similar to the [3H]methyl-methionine experiment, the kinetics of 35S, 27S, and 20S pre-rRNA processing were delayed compared to that of the wild-type culture. The amount of 25S and 18S mature rRNAs in the prp43S247A cultures after 60 min of chase were 10% and 18% respectively. We believe that the increased signal in the [5,6-3H]uracil experiments is due to the relative differences in the number of methylation sites compared to the number of uridine residues in the transcripts; however, in both experiments, the kinetics of processing of 35S, 27S, and 20S pre-rRNAs were severely delayed, and the amount of both mature rRNA species produced was significantly decreased.
Microarray analysis of pre-mRNA splicing and rRNA processing in prp43 mutants.
To determine whether the lack of a pre-U3 splicing phenotype shown in Fig. 3C was a pre-mRNA splicing effect specific to pre-U3 splicing or a general pre-mRNA splicing defect, we analyzed RNA from cultures shifted to nonpermissive temperatures by microarray analysis for the accumulation of any of the ∼240 introns/pre-mRNAs in yeast (4, 8). In Fig. 6A, we show that the two prp43 mutants cluster together in their intron accumulation profiles and that they exhibit only a slight increase in relative intron levels for some genes with respect to a wild-type control. Figure 6B shows the effect on the IAI value for the ACT1 intron when essential and early splicing factors are perturbed. Consistent with the splicing defect previously observed by others when actin pre-mRNA was used (2), ACT1 intron accumulates to some degree in cold-shifted prp43 mutants. The splicing efficiency of the ACT1 intron is also strongly affected by essential splicing mutants; however, the ACT1 intron is much less affected when early splicing factors are perturbed. Indeed, both prp43 mutants cluster separately from the essential pre-mRNA splicing factors (e.g., PRP2, PRP3, and PRP4) (Fig. 6A). This global intron accumulation analysis suggests that the behavior of two independent CS mutants of PRP43 do not confer their conditional growth phenotype through the splicing-related function of Prp43p as suggested by the fact that Prp43p exhibits an intron accumulation phenotype similar in degree to that of nonessential pre-mRNA splicing factors.
FIG. 6.

Microarray analysis of total RNA from prp43 cells shows only a mild splicing defect but a strong reduction of rRNAs. (A) A segment of a hierarchical cluster comparing prp43 mutants with yeast gene expression perturbations. Gene expression perturbations are labeled on the y axis, and the IAI value for each of ∼240 yeast introns is shown on the x axis. The structure of the hierarchical cluster tree is displayed on the right of the panel. Yellow bars denote essential splicing factors and early splicing factors. Tree nodes comprising early pre-mRNA splicing factors and essential splicing factors are colored yellow. Arrows indicate the positions of ACT1 intron. A color scale representing a severalfold change in IAI value is shown at the top left of panel. (B) Detail for ACT1 intron. IAI values for the ACT1 intron for each perturbation are displayed. Gene expression perturbations are labeled on the y axis. The color scale for panel B is the same as that described for panel A. (C) Levels of 25S, 18S, and 5.8S rRNA sequences are decreased in prp43 cells. Hierarchical cluster analysis of the severalfold change for stable RNAs in each of 73 gene expression perturbations is presented. Gene expression perturbations are labeled on the horizontal axis, and stable RNAs are indicated on the vertical axis. For panels A, B, and C, the position of prp43 mutants is highlighted using red text.
We next analyzed the steady-state levels of stable RNAs in prp43 mutants at the nonpermissive temperatures and compared the relative levels to those of several other experiments with deletions of or mutations in RNA metabolism-related genes (4). In prp43 CS mutants, the levels of 25S, 18S, and 5.8S rRNAs were markedly decreased relative to those of a wild-type culture (Fig. 6C). Importantly, levels of 5S rRNA, which is not processed from the same 35S transcript, were not affected in these strains. This decrease in rRNAs is not an indirect effect on ribosome biogenesis due to the inability to splice the ribosomal protein gene pre-mRNAs, as the essential pre-mRNA splicing mutants in this analysis did not show reduced levels of rRNAs (Fig. 6C). This indicates that the decrease in rRNAs is a specific and direct effect of mutant prp43 on the processing of pre-rRNA. Note that the prp43 alleles cluster next to Δrnt1 and rat1-1, known rRNA gene processing factors.
Bioinformatic analysis correlating PRP43 to pre-rRNA processing.
Further evidence of Prp43p functioning in pre-rRNA processing has been provided by computational means in a recent probabilistic functional network of yeast genes (25). In this analysis, functional protein networks were predicted with high accuracy by integration of data from numerous experimental means. An analysis of PRP43 is available upon request. The top 10 genes directly linked to PRP43 ranked by relative strength are shown (12). Of the top 10 correlated genes, 6 (DBP3, PWP1, NUG1, ERB1, NSA1, and RRB1) have been experimentally confirmed to be ribosome biogenesis factors. Interestingly, LHP1 encodes a polypeptide, the La-homologous protein, which has been shown to associate with Prp43p by immunoprecipitation from human cell extracts (11), indicating that these connections reflect a biological reality which has been conserved from yeast to humans.
DISCUSSION
We began experimentation on Prp43p to learn more about its role in the pre-mRNA splicing reaction and concluded that Prp43p is essential for the biogenesis of both ribosomal subunits. Specific association of Prp43p with a number of pre-rRNA species, from the very early 35S pre-rRNA to the relatively late 20S and 7S pre-rRNAs, highlights the relatively long time frame in which Prp43p is associated with the maturing preribosomal subunits. Molecular analyses showed that cold-sensitive mutants of prp43 accumulate 35S pre-rRNA precursors, indicating an inability to efficiently process this species in these strains. Additionally, prp43 mutant strains are depleted for 27S and 20S pre-rRNAs after incubation at low temperatures. Mutant prp43 strains behave similarly to nonessential pre-mRNA splicing factors in their intron accumulation patterns. Conversely, depletion of the Pol I transcripts 25S, 18S, and 5.8S are severe. Pulse-chase analysis of rRNA synthesis demonstrates that in an ATPase-deficient prp43 mutant, 35S, 27S, and 20S pre-rRNAs fail to be processed efficiently and are likely turned over. Consequently, the ribosomal proteins Rpl25p and Rps2p accumulate in the nucleolus and nucleus, as their means of transportation to the cytoplasm are no longer available.
We demonstrate that there is processing inhibition of both large and small ribosomal subunits. In our experimentation, biogenesis defects for the large subunit were reproducibly slightly greater than that of the small subunit in prp43 mutants. This may explain why there are greater levels of 40S subunits and half-mers in mutant extracts than 60S subunits at nonpermissive temperatures. Additionally, prp43 mutant cultures contained slightly larger numbers of cells with a nucleolar accumulation of Rpl25-eGFP than Rps2-eGFP, and pulse-chase analysis showed a greater defect in 25S accumulation than in 18S accumulation. These data are consistent with a role in the biogenesis of both subunits but show a more severe defect in large-subunit biogenesis.
Although our data and previous work showed that Prp43p is also a factor participating in pre-mRNA splicing, we demonstrate that its apparent essential role is its function in ribosome biogenesis. As pre-mRNA splicing and ribosome biogenesis are both essential processes, it is possible that the rRNA processing defect conferred by prp43 mutants results in cell death before a pre-mRNA splicing defect is detectable. Our data suggest, however, that Prp43p behaves like a nonessential pre-mRNA splicing factor with an essential role in biogenesis of both ribosomal subunits. Another CS prp43 mutant from the Staley laboratory (25a) and data from the Henry laboratory (24a) corroborate our results.
Possible mechanism of action of Prp43p in ribosome biogenesis.
An obvious potential function of Prp43p in both pre-mRNA splicing and pre-rRNA processing is the unwinding of snRNA:intron base pairing and snoRNA:rRNA base pairing, respectively. Although this remains a possibility, the inability to detect RNA-unwinding activity in vitro suggests that the mechanism of Prp43p action may not be duplex unwinding. Additionally, evidence of DEXH/D-box proteins functioning as RNPases (10, 20) and molecular clocks (35) expands the potential functional range of these proteins.
Our data support a model describing the function of Prp43p in ribosome biogenesis (Fig. 7). Affinity purification data show that Prp43p joins the maturing ribosome early in the process. Proper Prp43p function sets in motion the dissociation of the 90S rRNP into pre-40S and pre-60S RNPs, and the presence of 7S pre-rRNA in Prp43p immunoprecipitates implies additional functions for Prp43p in large-subunit biogenesis relatively late in the process (Fig. 1 and 7). The failure of Prp43p to properly function initiates a discard pathway that destroys the majority of 35S, 27S, and 20S pre-rRNAs (Fig. 3B and 5) and leads to the accumulation of both large and small structural ribosomal proteins in the nucleolus. Our model was developed from the following criteria: (i) the earliest detectable association of Prp43p in the ribosome biogenesis pathway is with the 35S pre-rRNA transcript, yet the relatively late 7S pre-rRNA is also associated with Prp43p; (ii) at nonpermissive temperatures, a mutant of prp43 exhibits delayed 35S processing kinetics, although there is robust Pol I transcription and methylation; (iii) 35S, 27S, and 20S pre-rRNAs fail to be efficiently processed, which leads to their subsequent degradation in prp43 mutants at nonpermissive temperatures; and (iv) prp43 mutants arrest the Rpl25p and Rps2p reporter proteins in both the nucleus and the nucleolus, indicating nucleolar and nuclear functions for Prp43p in the biogenesis of both ribosome subunits.
FIG. 7.

Model for the time and mechanism of action of Prp43p in preribosome biogenesis. Association of Prp43p occurs early, at the 35S stage, in the U3-snoRNP-containing 90S rRNP. In prp43 mutants, the failure of Prp43p to function initiates a discard pathway in which 35S, 27S, and 20S processing defects lead to the degradation of these pre-rRNAs. Red and green spheres indicate transiently interacting large- and small-subunit factors respectively. Dark blue and light blue spheres represent stably interacting large- and small-subunit factors, respectively. A red ring represents Prp43p.
It is interesting that the prp43S247A mutant retains 65% of wild-type Prp43p ATPase activity in vitro (27). This is unlike dominant-negative prp43 mutants, which generally possess ≤6% of wild-type ATPase activity and do not allow cell growth. The two CS mutants in our study are point mutations in DEAH-box motifs III and VI, which are thought to couple ATP hydrolysis with DEAH-box protein function (42). Although the precise molecular details of this model are not currently understood, that Prp43p is a bifunctional DEAH-box protein will further illuminate the mechanism of these polypeptides by allowing the examination of Prp43p function of both ribosome biogenesis and pre-mRNA splicing to dissect the functions unique to each process and common to both processes.
Evolutionary conservation of Prp43p bifunctionality?
The putative human orthologue for the yeast Prp43p is DDX15/hPrp43p, which has been shown to be associated with the pre-mRNA splicing machinery in HeLa extracts (17) and has been localized to nuclear speckles (a site of concentrated pre-mRNA splicing factors) as well as to human nucleoli (11). Although this is not evidence for hPrp43p function in human ribosome biogenesis, nucleolar localization of hPrp43p strongly implies this functionality. It will be interesting to see if there are other pre-mRNA splicing factors with evolutionarily conserved multiple functions which may allow for coordinated regulation of multiple processes in the highly organized program of eukaryotic gene expression.
Coordinate regulation of RNA Pol I and RNA Pol II metabolism?
Another hypothesis we put forth from these data is the possibility of coordinate regulation of both Pol I and Pol II RNA processing. Bioinformatic analyses of yeast intron-containing genes demonstrate that 70% of structural ribosomal subunit protein genes contain introns (26) and that ∼45% of intron-containing genes encode structural ribosomal subunit proteins (RPs) (3). Indeed, owing to their abundant transcription, the RPs account for ∼90% of pre-mRNA splicing events (3, 46). A factor that could bridge the two processes may be responsible for coordinate regulation of both. Although the CS mutants of prp43 in this study do not confer an essential pre-mRNA splicing phenotype, the correspondence of intron-containing genes and RPs may represent the last remnant of pre-mRNA splicing relevance in budding yeast, an organism which has eliminated most of its introns compared to other eukaryotes. A possible mechanism of this regulation is sequestration of Prp43p in a compartment (the nucleoplasm or the nucleolus) which prevents the processing of either pre-mRNA or pre-rRNA. Preventing either pre-mRNA splicing or pre-rRNA processing will eliminate ribosome biogenesis. The direction in which this regulation may occur is currently under investigation.
In metazoa, where the vast majority of genes contain introns, it will be interesting to determine if Prp43p is coordinately regulating both processes and the mechanism of such coregulation. Another compelling connection between the late stage of pre-mRNA splicing, in which Prp43p acts, and ribosome biogenesis is the function of Dbr1p. Many pre-rRNA processing-related snoRNAs are processed from introns (43), which are debranched by Dbr1p, representing another functional coordinate connection between late pre-mRNA splicing stages and pre-rRNA processing that may exist.
Acknowledgments
We acknowledge the contributions and support of the members of the Stevens lab; Jon Staley and Yves Henry for communicating results prior to publication; Susan Baserga for helpful conversations; Arlen Johnson and the members of the Johnson laboratory, especially Matthew West, for their contributions of plasmids, strains, techniques, equipment, and advice; and Edward Marcotte for the use of a mass spectrometer and computational analysis. We are grateful to Beate Schwer and Ed Hurt for contributions of plasmids and to Beate Schwer for many thoughtful discussions.
This work was supported by grants to S.W.S. from the Welch Foundation (grant F-1564), the American Cancer Society (grant RSG-05-137-01), and the National Science Foundation (grant MCB-0448556) and by funds from the University of Texas at Austin College of Natural Science and Institute for Cellular and Molecular Biology. Microarray work in the Ares lab was funded by grant GM040478 and the W. M. Keck Foundation.
REFERENCES
- 1.Anderson, J. S. J., and R. Parker. 1998. The 3′ to 5′ degradation of yeast mRNAs is a general mechanism for mRNA turnover that requires the SKI2 DEVH box protein and 3′ to 5′ exonucleases of the exosome complex. EMBO J. 17:1497-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arenas, J. E., and J. N. Abelson. 1997. Prp43: an RNA helicase-like factor involved in spliceosome disassembly. Proc. Natl. Acad. Sci. USA 94:11798-11802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ares, M., L. Grate, and M. H. Pauling. 1999. A handful of intron-containing genes produces the lion's share of yeast mRNA. RNA 6:1138-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burckin, T., R. Nagel, Y. Mandel-Gutfreund, L. Shiue, T. A. Clark, J. L. Chong, T. H. Chang, S. Squazzo, G. Hartzog, and M. Ares. 2005. Exploring functional relationships between components of the gene expression machinery. Nat. Struct. Mol. Biol. 12:175-182. [DOI] [PubMed] [Google Scholar]
- 5.Chapman, K. B., and J. D. Boeke. 1991. Isolation and characterization of the gene encoding yeast debranching enzyme. Cell 65:483-492. [DOI] [PubMed] [Google Scholar]
- 6.Chen, J. Y. F., L. Stands, J. P. Staley, R. R. Jackups, L. J. Latus, and T. H. Chang. 2001. Specific alterations of U1-C protein or U1 small nuclear RNA can eliminate the requirement of Prp28p, an essential DEAD box splicing factor. Mol. Cell 7:227-232. [DOI] [PubMed] [Google Scholar]
- 7.Clark, M. W., S. Goelz, and J. Abelson. 1988. Electron-microscopic identification of the yeast spliceosome. EMBO J. 7:3829-3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clark, T. A., C. W. Sugnet, and M. Ares. 2002. Genomewide analysis of mRNA processing in yeast using splicing-specific microarrays. Science 296:907-910. [DOI] [PubMed] [Google Scholar]
- 9.Company, M., J. Arenas, and J. Abelson. 1991. Requirement of the RNA helicase-like protein PRP22 for release of messenger RNA from spliceosomes. Nature 349:487-493. [DOI] [PubMed] [Google Scholar]
- 10.Fairman, M. E., P. A. Maroney, W. Wang, H. A. Bowers, P. Gollnick, T. W. Nilsen, and E. Jankowsky. 2004. Protein displacement by DExH/D “RNA helicases” without duplex unwinding. Science 304:730-734. [DOI] [PubMed] [Google Scholar]
- 11.Fouraux, M. A., M. J. M. Kolkman, A. van der Heijden, A. S. de Jong, W. J. van Venrooij, and G. J. M. Pruijn. 2002. The human La (SS-B) autoantigen interacts with DDX15/hPrp43, a putative DEAH-box RNA helicase. RNA 8:1428-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fraser, A. G., and E. M. Marcotte. 2004. A probabilistic view of gene function. Nat. Genet. 36:559-564. [DOI] [PubMed] [Google Scholar]
- 13.Fromont-Racine, M., B. Senger, C. Saveanu, and F. Fasiolo. 2003. Ribosome assembly in eukaryotes. Gene 313:17-42. [DOI] [PubMed] [Google Scholar]
- 14.Gadal, O., D. Strauss, J. Braspenning, D. Hoepfner, E. Petfalski, P. Philippsen, D. Tollervey, and E. Hurt. 2001. A nuclear AAA-type ATPase (Rix7p) is required for biogenesis and nuclear export of 60S ribosomal subunits. EMBO J. 20:3695-3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gadal, O., D. Strauß, J. Kessl, B. Trumpower, D. Tollervey, and E. Hurt. 2001. Nuclear export of 60S ribosomal subunits depends on Xpo1p and requires a nuclear export sequence-containing factor, Nmd3p, that associates with the large subunit protein Rpl10p. Mol. Cell. Biol. 21:3405-3415.11313466 [Google Scholar]
- 16.Gietz, R. D., and R. A. Woods. 2002. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method, Methods Enzymol. 350:87-96. [DOI] [PubMed] [Google Scholar]
- 17.Hartmuth, K., H. Urlaub, H. P. Vornlocher, C. L. Will, M. Gentzel, M. Wilm, and R. Lührmann. 2002. Protein composition of human prespliceosomes isolated by a tobramycin affinity-selection method. Proc. Natl. Acad. Sci. USA 99:16719-16724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huh, W. K., J. V. Falvo, L. C. Gerke, A. S. Carroll, R. W. Howson, J. S. Weissman, and E. K. O'Shea. 2003. Global analysis of protein localization in budding yeast. Nature 425:686-691. [DOI] [PubMed] [Google Scholar]
- 19.James, S. A., W. Turner, and B. Schwer. 2002. How Slu7 and Prp18 cooperate in the second step of yeast pre-mRNA splicing. RNA 8:1068-1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jankowsky, E., C. H. Gross, S. Shuman, and A. M. Pyle. 2001. Active disruption of an RNA-protein interaction by a DExH/D RNA helicase. Science 291:121-125. [DOI] [PubMed] [Google Scholar]
- 21.Jensen, T. H., J. Boulay, M. Rosbash, and D. Libri. 2001. The DECD box putative ATPase Sub2p is an early mRNA export factor. Curr. Biol. 11:1711-1715. [DOI] [PubMed] [Google Scholar]
- 22.Kallstrom, G., J. Hedges, and A. Johnson. 2003. The putative GTPases Nog1p and Lsg1p are required for 60S ribosomal subunit biogenesis and are localized to the nucleus and cytoplasm, respectively. Mol. Cell. Biol. 23:4344-4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kistler, A. L., and C. Guthrie. 2001. Deletion of MUD2, the yeast homolog of U2AF65, can bypass the requirement for Sub2, an essential spliceosomal ATPase. Genes Dev. 15:42-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krogan, N. J., W. T. Peng, G. Cagney, M. D. Robinson, R. Haw, G. Q. Zhong, X. H. Guo, X. Zhang, V. Canadien, D. P. Richards, B. K. Beattie, A. Lalev, W. Zhang, A. P. Davierwala, S. Mnaimneh, A. Starostine, A. P. Tikuisis, J. Grigull, N. Datta, J. E. Bray, T. R. Hughes, A. Emili, and J. F. Greenblatt. 2004. High-definition macromolecular composition of yeast RNA-processing complexes. Mol. Cell 13:225-239. [DOI] [PubMed] [Google Scholar]
- 24a.Lebaron, S., C. Froment, M. Froment-Racine, J.-C. Rain, B. Montsarrat, M. Caizergues-Ferrer, and Y. Henry. 2005. The splicing ATPase Prp43p is a component of multiple preribosome particles. Mol. Cell. Biol. 25:9269-9282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee, I., S. V. Date, A. T. Adai, and E. M. Marcotte. 2004. A probabilistic functional network of yeast genes. Science 306:1555-1558. [DOI] [PubMed] [Google Scholar]
- 25a.Leeds, N. B., E. C. Small, S. L. Hiley, T. R. Hughes, and J. P. Staley. 2006. The splicing factor Prp43p, a DEAH box ATPase, functions in ribosome biogenesis. Mol. Cell. Biol. 26:513-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lopez, P. J., and B. Séraphin. 1999. Genomic-scale quantitative analysis of yeast pre-mRNA splicing: implications for splice-site selection. RNA 5:1135-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin, A., S. Schneider, and B. Schwer. 2002. Prp43 is an essential RNA-dependent ATPase required for release of lariat-intron from the spliceosome. J. Biol. Chem. 277:17743-17750. [DOI] [PubMed] [Google Scholar]
- 28.Milkereit, P., D. Strauss, J. Bassler, O. Gadal, H. Kuhn, S. Schutz, N. Gas, J. Lechner, E. Hurt, and H. Tschochner. 2003. A noc complex specifically involved in the formation and nuclear export of ribosomal 40 S subunits. J. Biol. Chem. 278:4072-4081. [DOI] [PubMed] [Google Scholar]
- 29.Mourelatos, Z., J. Dostie, S. Paushkin, A. Sharma, B. Charroux, L. Abel, J. Rappsilber, M. Mann, and G. Dreyfuss. 2002. miRNPs: a novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev. 16:720-728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakajima, T., C. Uchida, S. F. Anderson, C. G. Lee, J. Hurwitz, J. D. Parvin, and M. Montminy. 1997. RNA helicase A mediates association of CBP with RNA polymerase II. Cell 90:1107-1112. [DOI] [PubMed] [Google Scholar]
- 31.Pang, P. S., E. Jankowsky, P. J. Planet, and A. M. Pyle. 2002. The hepatitis C viral NS3 protein is a processive DNA helicase with cofactor enhanced RNA unwinding. EMBO J. 21:1168-1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Puig, O., F. Caspary, G. Rigaut, B. Rutz, E. Bouveret, E. Bragado-Nilsson, M. Wilm, and B. Séraphin. 2001. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24:218-229. [DOI] [PubMed] [Google Scholar]
- 33.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Plainview, N.Y.
- 34.Schmitt, C., C. von Kobbe, A. Bachi, N. Pante, J. P. Rodrigues, C. Boscheron, G. Rigaut, M. Wilm, B. Seraphin, M. Carmo-Fonseca, and E. Izaurralde. 1999. Dbp5, a DEAD-box protein required for mRNA export, is recruited to the cytoplasmic fibrils of nuclear pore complex via a conserved interaction with CAN/Nup159p. EMBO J. 18:4332-4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Staley, J. P., and C. Guthrie. 1998. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell 92:315-326. [DOI] [PubMed] [Google Scholar]
- 36.Stevens, S. W., and J. Abelson. 2002. Yeast pre-mRNA splicing: methods, mechanisms, and machinery, Methods Enzymol. 351:200-220. [DOI] [PubMed] [Google Scholar]
- 37.Stevens, S. W., I. Barta, H. Y. Ge, R. E. Moore, M. K. Young, T. D. Lee, and J. Abelson. 2001. Biochemical and genetic analyses of the U5, U6 and U4/U6·U5 small nuclear ribonucleoproteins from Saccharomyces cerevisiae. RNA 7:1543-1553. [PMC free article] [PubMed] [Google Scholar]
- 38.Strasser, K., and E. Hurt. 2001. Splicing factor Sub2p is required for nuclear mRNA export through its interaction with Yra1p. Nature 413:648-652. [DOI] [PubMed] [Google Scholar]
- 39.Sturn, A., J. Quackenbush, and Z. Trajanoski. 2002. Genesis: cluster analysis of microarray data. Bioinformatics 18:207-208. [DOI] [PubMed] [Google Scholar]
- 40.Svitkin, Y. V., A. Pause, A. Haghighat, S. Pyronnet, G. Witherell, G. J. Belsham, and N. Sonenberg. 2001. The requirement for eukaryotic initiation factor 4A (eIF4A) in translation is in direct proportion to the degree of mRNA 5′ secondary structure. RNA 7:382-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tabara, H., E. Yigit, H. Siomi, and C. C. Mello. 2002. The dsRNA binding protein RDE-4 interacts with RDE-1, DCR-1, and a DExX-box helicase to direct RNAi in C. elegans. Cell 109:861-871. [DOI] [PubMed] [Google Scholar]
- 42.Tanner, N. K., and P. Linder. 2001. DExD/H box RNA helicases: from generic motors to specific dissociation functions. Mol. Cell 8:251-262. [DOI] [PubMed] [Google Scholar]
- 43.Tycowski, K. T., M. D. Shu, and J. A. Steitz. 1996. A mammalian gene with introns instead of exons generating stable RNA products. Nature 379:464-466. [DOI] [PubMed] [Google Scholar]
- 44.Venema, J., and D. Tollervey. 1999. Ribosome synthesis in Saccharomyces cerevisiae. Annu. Rev. Genet. 33:261-311. [DOI] [PubMed] [Google Scholar]
- 45.Wach, A., A. Brachat, C. Alberti-Segui, C. Rebischung, and P. Philippsen. 1997. Heterologous HIS3 marker and GFP reporter modules for PCR-targeting in Saccharomyces cerevisiae. Yeast 13:1065-1075. [DOI] [PubMed] [Google Scholar]
- 46.Warner, J. R. 1999. The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci. 24:437-440. [DOI] [PubMed] [Google Scholar]
- 47.Reference deleted.
- 48.Wise, J. A. 1991. Preparation and analysis of low-molecular weight RNAs and small ribonucleoproteins. Methods Enzymol. 194:405-415. [DOI] [PubMed] [Google Scholar]
- 49.Yan, X. M., J. F. Mouillet, Q. L. Ou, and Y. Sadovsky. 2003. A novel domain within the DEAD-box protein DP103 is essential for transcriptional repression and helicase activity. Mol. Cell. Biol. 23:414-423. [DOI] [PMC free article] [PubMed] [Google Scholar]