

Abstract
Site-specific recombinases of the λ Int family carry out two single-strand exchanges by binding as head-to-head dimers on inverted core-type DNA sites. Each protomer may cleave its own site as a monomer in cis (as for Cre recombinase), or it may recruit the tyrosine from its partner in trans to form a composite active site (as for Flp recombinase). The crystal structure of the λ Int catalytic domain is compatible with both cleavage mechanisms, but two previous biochemical studies on λ integrase (Int) generated data that were not in agreement. Support for cis and trans cleavage came from assays with bispecific DNA substrates for λ and HK022 Ints and from functional complementation between recombination-deficient mutants, respectively. The data presented here do not provide new evidence for cis cleavage, but they strongly suggest that the previously described complementation results cannot be used in support of a trans-cleavage mechanism. We show here that IntR212Q retains some residual catalytic function but is impaired in binding to core-type DNA on linear substrates and in forming higher-order attL intasome structures. The binding-proficient mutant IntY342F can stabilize IntR212Q binding to core-type DNA through protein-protein interactions. Similarly, the formation of higher-order Int complexes with arm- and core-type DNA is boosted with both mutants present. This complementation precedes cleavage and thus precludes any conclusions about the mechanism of catalysis. Cross-core stimulation of wild-type HK022-Int cleavage on its cognate site (in cis) by mutant λ Ints on bispecific core DNA suicide substrates is shown to be independent of the catalytic tyrosine but appears to be proportional to the respective core-binding affinities of the λ Int mutants.
The large λ integrase (Int) family of site-specific recombinases exhibits two distinct mechanisms of DNA cleavage. In one, the enzymes catalyze DNA cleavage as monomers at the site where they are bound (in cis). In the other, the proteins act as dimers and cleave DNA with a composite active site in which the catalytic tyrosine is provided (in trans) by a protomer bound to a site other than the one being cleaved (5, 14, 22, 23, 31, 32, 46). The Cre recombinase of bacteriophage P1 and the yeast-derived Flp are prototypes for cis and trans cleavage, respectively, as revealed by their cocrystal structures (9, 16). Biochemical evidence from the XerC/D system shows convincingly that these recombinases fall into the cis-cleavage category (2, 4, 18). For λ Int, there are biochemical data for both types of cleavage (19, 41). In the crystal structure of the catalytic domain of λ Int, the loop containing the phenylalanine mutant of the active site Y342 is flexible, allowing the modeling of either cis or trans cleavage modes (29). In the present report we examine the potential allosteric consequences of mutating a catalytic residue and their impact on the interpretation of complementation.
The Int protein of Escherichia coli phage λ is a type I topoisomerase that carries out the conservative strand exchanges of integrative and excisive recombination between the phage and bacterial chromosomes (3, 30, 37). The signature triad Arg-His-Arg (RHR) and two additional basic amino acids (extensively studied in the Flp system) form the catalytic pocket and activate the scissile phosphate for cleavage and ligation, involving a transient, covalent bond via Tyr342 (5, 7, 8, 16, 33, 38). The basic pentad in λ Int is comprised of residues R212, K235, H308, R311 and H332. The catalytic C-terminal domain of the heterobivalent Int binds with low affinity to the inverted core-type sequences on either side of the 7-bp overlap (O) region (C and C" on phage DNA and B and B" on bacterial DNA). The smaller N-terminal domain of Int binds with high affinity to so-called arm-type sequences on phage DNA (P and P"). In combination with DNA-bending proteins, i.e., integration host factor (IHF), Int forms higher-order complexes, called intasomes, on attachment (att) site DNA. Intermolecular Int bridges lead to synapsis between the phage attP and bacterial attB during integration and between the prophage sites attL and attR during excision (24, 44). After synapsis, recombination proceeds in a prescribed manner with a top-strand (TS) exchange to form a Holliday junction intermediate and a bottom-strand (BS) exchange to resolve it to recombinant products (3, 26, 30, 37, 39).
Half-att sites are also substrates for Int binding, cleavage, and synapsis with a full partner att site to yield three-armed recombination intermediates (40). Using the analogous FRT half-sites in the Flp system, the Jayaram laboratory pioneered the mechanism of trans cleavage with a combination of mutants of catalysis at either the conserved RHR or the Tyr nucleophile. The mixing of two FRT half-sites, each preincubated with a different mutant, led to exclusive cleavage on the half-site to which the Y→F mutant Flp was bound (8). The formation of a composite active site between the basic catalytic pocket (RHR) of one monomer and the Tyr-OH nucleophile from another protomer in trans was corroborated by extensive biochemical analyses and by the cocrystal structure of wild-type (wt) Flp bound to FRT half-sites in a pseudo-Holliday junction conformation (9, 14, 22). In contrast, the cocrystal structures of Cre with half and full lox sites all show that each monomer provides the catalytic Tyr to the lox site to which it is bound in cis (13, 16, 17).
On the mechanism of cleavage by λ Int, the crystallographic data are ambiguous and the biochemical data are contradictory. The argument for cis cleavage was drawn from experiments using two closely related proteins (λ and HK022 Ints) with different core-binding specificities (41). The sites cleaved on hybrid suicide substrates, both linear core-type DNA and artificial Holliday junctions, were exclusively those that harbored an Int with an active Y342. The argument for trans cleavage was derived from experiments showing complementation of Arg mutants by IntY342F (19). The test system consisted of attLs (BOC") with either a TS or a BS nick that form higher-order attL intasome structures in the presence of Int and IHF (25, 42). Previous cleavage and nuclease protection data had indicated that Int that is bound to the arm-type site P"1 with its N-terminal domain bridges to the BS core-type site, C", with its C-terminal domain, thereby stabilizing core binding at this site (25). Furthermore, when nicked attLs were preincubated with the inactive IntY342F and IHF and then challenged with wt Int, DNA cleavage occurred at the TS site, B, with faster kinetics than at the BS site, C". This predilection was understood to reflect a preferential exchange of the Int at the B site, where it was not bivalently bound, in contrast to the doubly bound Int bridging P"1 to C".
A clever extension of this system was used to test for complementation between IntY342F and Ints with mutations of the conserved arginines involved in activating the scissile phosphate: R212Q, R311C, and R311H (19). It was assumed that Int mutants with changes in the catalytic triad would behave the same as wt Int and IntY342F in forming an intramolecular bridge on attL (P"1/C"). If so, the doubly bound mutant Int would again be less prone to be replaced at C" by a second Int than the protomer singly bound to the B site. Whereas preincubation of a TS-nicked attL with IntY342F followed by IntR212Q showed no complementation, preincubation with IntR212Q followed by IntY342F yielded TS-cleaved products. Because this nonequivalent complementation pattern was dependent on the order of addition, it was interpreted to constitute evidence for TS cleavage in trans. That is, IntR212Q, which was expected to be firmly bound to C", provided the tyrosine in trans to the IntY342F that preferentially replaced the Arg mutant at the B site, thereby activating the scissile phosphate at the TS site with its RHR triad in cis. A puzzling and troubling result at that time was the fact that the reciprocal experiment did not work. When the BS-nicked substrate was preincubated with IntY342F and then challenged with IntR212Q, the expected trans complementation by the tyrosine of IntR212Q was not observed. Indeed, with this substrate, practically no BS cleavage was obtained, irrespective of the order in which the two mutants were added (19).
We now show that the activation-deficient R212Q mutant is not completely devoid of activity and that it is capable of cleaving a 5" bridging phosphorothioate suicide substrate on its own. This cryptic catalytic activity had been masked in previous cleavage assays with nicked suicide substrates that depended on dissociation of a few nucleotides for trapping a cleavage product. We also show that the R212Q mutant displays multifunctional defects: it binds poorly to core-type site DNA, including linear COC" and C" half-sites; it binds weakly to Holliday junctions; it fails to form sandwich complexes when provided with both arm- and core-type site DNA (45); and it is easily displaced from core-type DNA binding sites by Ints with unimpaired binding affinities. Thus, the R212Q mutant is compromised not only for catalysis but also for higher-order complex formation, a prerequisite for a functional attL intasome. IntR212Q can be complemented for binding to core-type DNA and for sandwich formation by the presence of small quantities of IntY342F. Because the observed complementation for cleavage of nicked attL suicide substrates can be explained entirely by DNA binding and complex formation, it is not possible to draw inferences about the nature of the cleavage mechanism from these experiments. We propose that the enhancement of cleavage seen by combining the two distinct catalytic mutants might be due to cross-core cooperation, with IntY342F providing a stable scaffold that rescues the otherwise cryptic catalytic activity of IntR212Q.
MATERIALS AND METHODS
Int cloning, expression, and purification.
All Ints used in this study (full-length Int and the catalytic domain, spanning residues 65 to 356 [C65-Int]) were expressed and purified from a plasmid under the control of a T7 promoter in E. coli BL21(pLys) as previously described in detail (48). All clones of mutant Ints were made from DNA fragments amplified by PCR with the appropriate primers (Operon, New England Biolabs) by using the wt Int parent plasmids pRT101 and pRT301 as templates for the full-length and C65 Ints, respectively. These fragments were purified and ligated into the expression plasmid backbone according to standard protocols. The entire int gene of each transformed clone (E. coli strain DH5α) was sequenced to verify the desired mutation and exclude undesired changes. The concentration of purified Ints was measured by the Bradford assay and by comparative staining analysis on standard discontinuous sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels.
Structure and preparation of DNA substrates.
All core-type DNA substrates used in this study were prepared by annealing appropriate oligonucleotides. One or more single-stranded DNA molecules were radioactively labeled with [γ-32P]ATP and polynucleotide kinase (New England Biolabs), boiled, slowly annealed with the complementary partner(s), precipitated, and gel purified (41). The sequences used for COC" DNA were 5" GTTGAAGCCAGCTTA|ATTCTATAAAGTTGG 3" (TS) and 5" TTCCAACTTT|ATAGAATTAAGCTGGCTTCAAC 3" (BS). In this substrate and the one shown below, core-type recognition sequences are underlined; the vertical line indicates the position of Int nicking, as well as that of the 5" bridging phosphorothioate, when present (see below). Suicide half-site C" oligomers contained a nick (shown by a hyphen) 1 base adjacent to the Int cleavage site and a GAA hairpin to prevent dimer formation:
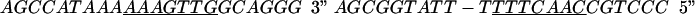 |
Bispecific hybrid core DNA substrates carrying modified HK022 and λ Int binding sites at the TS C and BS C" sites, respectively, have been described in detail (41). The sequence was designed according to the exclusive binding specificity established for the wt B" site for each recombinase (36, 50, 51).
To construct the two nicked attL suicide substrates, one full-length and two half-site fragments were prepared by PCR on templates pSN83 and pBF32, gel purified after XbaI restriction, and then denatured and annealed with each other at an optimal ratio as previously described (25, 39, 42). One strand of a half-site was radiolabeled with 32P. In a typical annealing reaction, the recovery of nicked substrates was approximately 70%, with the remainder appearing as a radiolabeled half-site. The nicks in these substrates are located at the XbaI restriction sites within the overlap region, three nucleotides 3" from the Int cleavage sites. In the presence of IHF, Int-covalent complexes are trapped with similar kinetics almost exclusively at the TS site with TS-nicked substrate and at the BS site with BS-nicked substrate (25).
Cleavage and mobility shift assays.
Reaction mixtures contained 10 mM Tris-HCl (pH 7.5 to 7.8), 1 mM EDTA, 50 to 80 mM NaCl, 0.5 to 1 mg of bovine serum albumin per ml, 2.5 mM dithiothreitol, and, for mobility shift assays only, 5% glycerol. Small 32P-labeled DNA fragments were present at 0.01 to 0.05 μM, and protein concentrations in general were 2- to 20-fold in excess of DNA (as stated in text, figures, or figure legends). Samples were incubated at 24°C and then mixed with a Ficoll (final concentration, 2%) loading solution in Tris-borate for mobility shift assays without dye, to which bromphenol blue and SDS (final concentration, 0.2%) were added for cleavage reactions only. Electrophoretic separations were carried out in 8% polyacrylamide gels, native or with SDS (SDS-PAGE), at 200 V. Experiments with nicked attL substrates were identical to those published previously (19, 25). Under these conditions, wt Int cleaved TS- and BS-nicked attLs with similar kinetics (25). Samples were analyzed on 1.2% agarose gels. Dried gels were autoradiographed on Kodak X-ray films as well as scanned and quantified with a Fuji film BAS 2500 phosphorimager.
Int sandwich formation with core- and arm-type DNA.
Sandwich formation experiments were carried out as published elsewhere, with minor changes (45). The reaction mixes (50 mM Tris-HCl [pH 7.8], 60 mM NaCl, 5 mM dithiothreitol, 0.5 mg of bovine serum albumin per ml), containing different concentrations of DNA and proteins, were incubated at 19°C overnight (∼20 h), and complexes were separated on a 7% native polyacrylamide gel run at 200 V. The sequences of the annealed arm- and core-type DNA fragments (Operon) are represented by the TS sequence of the BOC" fragment (5" CGTTTCTCGTTCTGCTTT|TTTATACTAAGTTGGCATTATAAAAAAGCTTTA 3") and the TS sequence of the P"1P"2 fragment (5" TAACGAACAGGTCACTATCAGTCAAAATACCGATAC 3").
Synthesis and purification of oligonucleotides containing 5" bridging phosphorothioates.
We used internucleotide 5" bridging phosphorothioate (also called phosphorothiolate) substitutions at the site of DNA cleavage by Int, as first described by Burgin et al. (6). The single-atom replacement of the 5" bridging oxygen with sulfur was incorporated into the DNA substrate sequence by preparing a modified adenosine monomer compatible with phosphoramidite-based automated oligonucleotide synthesis. The preparation of 5"-(S-triphenylmethyl)-mercapto-N6-benzoyl-2",5"-dideoxyadenosine-3"-O-(2-cyanoethyl N,N-diisopropyl-phosphoramidite) and its incorporation into oligonucleotides have been described (27, 28, 34). After addition of the thio monomer, the 5"-triphenylmethyl protecting group for sulfur was removed by treatment with aqueous silver nitrate. Preceding and subsequent ordinary phosphoramidite monomers were added in a conventional manner as previously described (27). Oligonucleotides, with or without a 5" bridging phosphorothioate at the position of Int cleavage, were isolated by reversed-phase high-pressure liquid chromatography (HPLC), desalted on a Sephadex G-10 column, and purified to homogeneity by reversed-phase HPLC. Each oligomer met the following criteria for purity: (i) a single peak by analytical reversed-phase HPLC, (ii) the expected nucleoside composition, as determined by enzymatic digestion and HPLC analysis, and (iii) a single band on a denaturing PAGE gel, comigrating with a control strand of equal sequence and length (1). The oligonucleotides were stored in HPLC buffer (triethylammonium acetate, pH 7, at −20°C). Appropriate oligonucleotides were annealed at equimolar concentrations by boiling and slow cooling to create thio-substitution-containing suicide COC" substrates.
RESULTS
Cleavage of suicide substrates by IntR212Q.
Stock solutions of highly purified wt and mutant Ints were prepared at comparable molar concentrations. To verify the approximate “functional” concentrations relative to wt Int, each catalytically inactive mutant was carefully titrated for binding to short core-type DNA fragments and analyzed in a gel shift assay (see below). All these recombination-deficient mutants were also tested for DNA cleavage activity with core-type DNA substrates containing a suicide feature that traps the Int-DNA covalent complex (42). In a typical experiment, wt Int bound to TS- or BS-nicked attL DNA becomes trapped when the small oligomer is released due to Int cleavage (Fig. 1). On this substrate, full-length Int is stimulated for cleavage by the presence of IHF, which promotes the formation of a higher-order intasome complex (see below) (25). In contrast, on a simpler substrate, containing only the core DNA with a nick between the inverted core-type binding sites, full-length Int is less active than its isolated catalytic domain, spanning residues 65 to 356 (C65-Int). It has been shown that the N-terminal domain of Int is a context-sensitive modulator of catalysis that inhibits DNA binding and cleavage of full-length Int (45). Binding and cleavage experiments, therefore, are more efficient with C65-Ints.
FIG. 1.

A nick within the overlap region of an att site acts as a cleavage suicide feature. In the presence of IHF, a DNA-bending protein, and Int, an attL substrate forms a higher-order intasome structure. The N-terminal domains of Int (small circles) are bound to the arm-type binding sites P"1 and P"2, and the C-terminal domains are bound to the core-type binding sites B and C" (large circles). Whereas the Int bound at C" has both domains occupied, the Int bound at B has a free arm-binding domain (N). The position of the nick within the 7-bp overlap region influences the prevalence of Int cleavage at either the TS or BS sites, favoring the closer site. The nick is placed 3 bp from the TS and BS cleavage sites in TS- and BS-nicked attL substrates, respectively. This allows almost exclusive trapping of unique covalent Int-DNA complexes from cleavages of the nicked strands (rather than cleavages of the unnicked strands 4 bp removed from and opposite the nick).
All Int mutants of catalytic residues tested, including R212Q and Y342F, are defective in cleavage assays with full core-type DNA carrying a nick within the overlap region or with half-att substrates (19, 41). In these substrates, the trapping of covalent Int-DNA cleavage products depends not only on the nicking reaction per se but also on the dissociation of the small (3-base) oligonucleotide between the cleavage site and the 3" end of the same strand (i.e., removal of the 5"OH nucleophile). The amount of covalent product recovered, therefore, depends on the balance between oligomer diffusion and strand religation. Since the back reaction may mask a residual cleavage activity, we used a different suicide substrate that cannot be ligated after cleavage. Burgin et al. introduced core-type DNA substrates with an internucleotide 5" bridging phosphorothioate substitution at the site of Int cleavage (6). These substrates are nicked efficiently by Int to form a covalent Int-DNA complex at the 3" end with a sulfhydryl group on the 5" side of the nick. In contrast to the normal 5"OH, the 5"SH group is a poor nucleophile for religation in this system. These substrates accumulate covalent Int-DNA cleavage products with faster kinetics than nicked substrates because each cleavage event has a higher probability of being captured (data not shown).
Contrary to the expectation that the R212Q mutant is cleavage incompetent, assays with a TS 5" bridging phosphorothioate suicide substrate, carried out at 25°C, revealed some residual activity of C65-IntR212Q, especially at higher concentrations (Fig. 2). The mutant displayed approximately 1/10 the efficiency of wt C65-Int at 0.4 μM. The addition of C65-IntY342F into the reaction mixture did not increase, but rather decreased, the amount of covalent Int-DNA product recovered from C65-IntR212Q. It appears that the Y342F mutant with its higher affinity for core-type DNA competes for binding to this suicide substrate and inhibits the R212Q mutant. A more conservative mutation of Arg212, C65-IntR212K, tested for binding and cleavage on this substrate, exhibits an intermediate phenotype (56% cleavage at 0.4 μM). The defect of both Arg212 mutants is more apparent at lower temperature, also reflecting slower cleavage kinetics (data not shown). But the crucial point for the arguments developed here is the fact that catalysis is not totally abolished in these recombination-deficient Arg212 mutants, as was previously concluded from experiments with substrates that had the potential to be religated after cleavage, such as the nicked attL suicide substrates (19).
FIG. 2.

Cleavage of thio-substitution-containing core-type DNA. Wt and R212Q mutant C65-Ints become covalently trapped at the TS site (arrow) of a 32P-labeled (∗) COC" substrate next to a TS 5" bridging phosphorothioate (S), left of the overlap region (shaded).
Core-type DNA binding by mutant Ints.
The relative affinity of the different catalytic mutants for binding to core-type DNA was tested with mobility shift assays using C65-Ints that typically bind core-type DNA better than full-length Int (see above). In general, the proportion of faster- and slower-moving bands on COC" DNA (singly or doubly bound by Int) depends on the concentration of Int used, the molar ratio between Int and core-type DNA, and the presence or absence of heterologous carrier DNA. A comparison of C65-Int mobility shifts between the R212Q and Y342F mutants reveals that approximately fourfold more C65-IntR212Q is required for specific binding of one or two molecules to a small COC" substrate. That is, 0.2 μM C65-R212Q and 0.05 μM C65-Y342F give a comparable binding pattern with 70% singly bound and 7% doubly bound protein-DNA complexes (Fig. 3A, lanes 3 and 6). The mixing experiments in lanes 8 to 10 demonstrate that the binding of the two mutants is additive, leading to 100% substrate binding. At the lowest combined concentration tested (lane 10), the 50:50 distribution between the one- and two-Int complexes is similar to the pattern seen with each individual mutant at double the concentration (lanes 2 and 5).
FIG. 3.

Mobility shift of core-type DNA by mutant C65-Ints. COC" (36 bp) and C" (21 bp plus 3-base hairpin) fragments (0.01 μM) were incubated with different concentrations of C65-Int R212Q (Q) and Y342F (F) mutants. Titrations on COC" DNA were in the absence (A) and in the presence (B) of equimolar non-att DNA (276 bp), which functions as a nonspecific (non-spec.) binding competitor. Another recombination-deficient mutant, C65-K235A, is competent for DNA binding and is included for comparison (B, lanes 3 and 4). It displays 68% specific binding at concentrations that saturate non-att DNA. (C) Full-length Int mutants R212Q (IntQ) and Y342F (IntF) retard a half-att site (half-core C") more than their respective C65-Ints (C65Q and C65F).
Specificity of binding was further tested with mobility shift experiments in the presence of two radiolabeled substrates in the same reaction, a 36-bp specific att site DNA with two specific binding sites (C and C") and a 276-bp non-att plasmid DNA fragment. Although these two fragments were present at equimolar concentrations (approximately 5 nM each), the eightfold-larger size of the nonspecific DNA offers binding access for at least 16 molecules of Int. Indeed, 8 to 10 individual shifted protein-DNA bands can be detected on the non-att DNA fragment when it is not fully saturated by Int (Fig. 3B). The smear in the lanes is likely due to instability of the nonspecific binding complexes during electrophoresis. Whereas non-att DNA does not adversely affect specific core-type DNA binding by C65-IntY342F (95 to 98% bound in lanes 4 and 5 of Fig. 3A and lanes 1 and 2 of Fig. 3B), it acts as a strong competitor for core-specific binding by C65-IntR212Q (compare lanes 2 [97%] and 3 [77%] of Fig. 3A with lanes 5 [48%] and 6 [4%] of Fig. 3B). On the other hand, C65-IntR212Q binds relatively well to the non-att DNA, similar to the other mutants tested, as is demonstrated by the different degrees of disappearance of the two types of substrates. This observation not only reflects a lower affinity for core binding by C65-IntR212Q compared to C65-IntY342F but also may point to some aberrant binding to nonspecific DNA.
Another method for looking at relative core-binding affinities is based on the exchangeability of one mutant Int with the other on a single C" half-att substrate (Fig. 3C). Preincubation with a full-length Int followed by the C65 protein, or vice versa, allows us to distinguish two different Int-DNA complexes by size in a gel retardation assay. Typically, C65-IntY342F can displace IntY342F at equimolar concentrations, and similarly, C65-IntR212Q can displace the same full-length mutant Int (data not shown). This is consistent with previous observations that the amino-terminal domain of full-length Int inhibits its carboxy-terminal domain binding to isolated core-type DNA (45). In the reverse experiment, as expected, neither IntR212Q (Fig. 3C, lane 8) nor IntY342F (data not shown) is capable of replacing C65-IntY342F bound to the half-site C" DNA, but IntY342F partly displaces C65-IntR212Q at comparable concentrations (lanes 4 to 6), evidence of just how poorly C65-IntR212Q binds.
In summary, it appears that specific core-type DNA binding is compromised by the R212Q mutation on linear COC" and C" substrates. Furthermore, the presence of two inverted core-type binding sites, which invite wt or Y342F Ints to interact as dimers, does not appear to enhance the binding affinity of the R212Q mutant through possible cross-core cooperativity (compare Fig. 3A and C). Similarly, incubation of an artificial Holliday junction with IntR212Q allowed a mobility shift equal only to a dimer species, in contrast to the formation of a tetramer with wt Int or IntY342F. Increasing the protein concentration did not allow stable tetramer formation but rather just produced protein-DNA aggregates (data not shown). However, on an SDS-PAGE gel, traces of Holliday junction cleavage and resolution products can be seen with IntR212Q (data not shown). This is consistent with the above results showing that IntR212Q retains some DNA cleavage activity but is severely compromised in its ability to form specific complexes with att site DNA.
Int cleavage of TS- or BS-nicked attL substrates after preincubation with mutant Ints.
At low concentrations of wt Int, cleavage of a nicked suicide attL is dependent on the IHF-induced formation of a looped intasome structure (Fig. 1). Under these conditions, the formation of covalent Int-attL complexes occurs exclusively on the nicked strand, due in part to the position of the nick within the overlap region (see the legend to Fig. 1). In this attL intasome, the binding properties of Int at the two core-type sites, B and C", are not equivalent. The heterobivalent Int bridging the P"1 arm-type and the C" core-type sites has both DNA-binding domains satisfied. It is therefore expected to exchange with an Int in solution at a lower rate than the B-bound Int, which has an unsatisfied arm-binding domain. On the basis of this prediction, two sets of preincubation experiments with TS- and BS-nicked attL substrates led to different interpretations. The faster kinetics of TS versus BS cleavages by wt Int displacing the inactive IntY342F in an attL complex paralleled the predicted faster exchange rates at the B versus C" sites, which suggested cis cleavage (25). On the other hand, complementation between the R212Q and Y342F mutants for TS cleavage of a TS-nicked attL depended on the order of addition, i.e., IntR212Q preceding IntY342F, which suggested trans cleavage, as described above (19).
The two different conclusions drawn from the above preincubation experiments are based on the same assumption that all Ints tested have equal binding properties and are equally stable in an attL intasome complex. In order to validate such a presumption, we tested the exchangeability of different mutant Int molecules within a higher-order complex on the same suicide attL substrates, carrying either a TS or a BS nick within the overlap region. After preincubation of these substrates with IHF and an inactive Int mutant, wt Int was added as the second enzyme. The rates of TS and BS cleavages by wt Int reflect the exchangeability of the mutant proteins bound at the B and C" sites. If the mutant protein forms normal attL intasomes, wt Int is expected to displace a TS-bound mutant Int (with only one DNA-binding domain satisfied) more rapidly than a BS-bound one (which has both DNA-binding domains occupied).
Since IntR212Q binds poorly to core DNA, it may also be deficient for the formation of the attL intasome. In that case, it might be expected to lose or diminish the differential between TS and BS exchangeability. A typical time course of cleavage by wt Int after preincubation of a TS- or BS-nicked attL suicide substrate with either IntY342F or IntR212Q is shown in Fig. 4A. We confirm the previous observation that, after preincubation with IntY342F, the initial rate of wt Int covalent complex formation from TS cleavage at the B site is substantially faster than that from BS cleavage at the C" site (25). For IntR212Q, in contrast, that difference was much smaller. IntR212Q is exchanged by wt Int faster than IntY342F, at both TS- and BS-binding sites. In particular, the “slower” BS cleavage by wt Int after IntR212Q preincubation is faster than the “fast” TS cleavage after IntY342F preincubation (Fig. 4A). The percent TS and BS cleavage as a function of time was analyzed and summarized from multiple experiments. The calculated ratios of TS over BS cleavages by wt Int are significantly smaller for samples preincubated with IntR212Q than for those preincubated with IntY342F (Fig. 4B). This suggests that IntR212Q does not readily form stable attL intasome structures with an intramolecular bridge between P"1 and C".
FIG. 4.

Cleavage of nicked suicide attL substrates by wt Int after preincubation by mutant Ints. (A) In a typical experiment, TS-nicked and BS-nicked attL substrates (S) were preincubated for 15 min with IHF and either IntR212Q or IntY342F. Then, at time zero, wt Int was added to the reaction mixtures to measure the appearance of covalent wt Int-DNA products (P). The 32P label at the 5" side of the nick (∗) was derived from the half-site (1/2 att) that cannot be cleaved by wt Int (when not annealed to a full-length strand; see Material and Methods). (B) The results from several preincubation experiments were compiled and expressed for IntR212Q (Q) and IntY342F (F) as ratios of TS cleavage of the TS-nicked attL to BS cleavage of the BS-nicked attL (t/b).
Formation of ternary Int complexes with core- and arm-type DNA.
We recently showed that the presence of DNA fragments carrying arm-type Int-binding sites helps full-length Int to bind to a separate DNA fragment containing only core-type binding sites (45). The formation of such ternary sandwich complexes also stimulates full-length wt Int cleavage of a nicked suicide core-type DNA substrate. Since the Int binding in the ternary complexes resembles an arm-core bridge, analogous to that predicted to exist between P"1 and C" in an attL intasome, we tested both IntR212Q and IntY342F for their ability to form sandwich complexes. Different concentrations of mutant Ints were incubated with 32P-labeled BOC" (analogous to attB) and unlabeled P"1P"2 oligomers, and complex formation was monitored on a native polyacrylamide gel (Fig. 5B, left). Whereas IntY342F, like wt Int (Fig. 5A), almost exclusively formed sandwich complexes, even at low concentrations (0.15 μM), IntR212Q produced only simple adducts between Int and core DNA. Even at high concentrations of IntR212Q (0.7 μM), only one shifted band was present, displaying the same mobility as wt Int binding to core DNA in the absence of P"1P"2-arm oligomers (Fig. 5A).
FIG. 5.

Mobility shift assays showing sandwich formation by Int, binding simultaneously to core-type and arm-type DNA sequences. (A) Whereas full-length wt Int (0.2 μM) produces a single adduct band in a mobility shift assay with 0.02 μM 32P-labeled core-type DNA (COC"*), the addition of a second unlabeled DNA fragment (0.1 μM) containing arm-type sequences (P"1,2), leads to a supershift of all the Int-bound DNA. (B) Mixtures of 32P-labeled core-type DNA (0.05 μM) and arm-type DNA (BOC"* + P"1,2) were incubated with IntR212Q (Q) and IntY342F (F) individually (left) or combined (right) and yielded either an adduct (Int/BOC"*) or a sandwich band (P"1,2/Int/BOC"*). The P"1,2 fragment was present at half-molar amounts of Int and at 0.15 μM in the left and right panels, respectively.
In a different experiment using low Int concentrations insufficient for stable DNA binding, the ternary complex was detected when both mutants were combined in equimolar amounts (Fig. 5B, right). We infer from the approximately twofold stimulation of ternary complex formation between the reaction containing only IntY342F and the reaction containing both proteins that IntR212Q is part of the sandwich complex. In summary, IntR212Q is deficient in all binding functions involving core-type DNA alone and in the formation of bridged complexes when arm- and core-type-binding sequences are present, either on an attL substrate or on two separate molecules. Significantly, a partial rescue of these functions is seen when IntY342F is added to the reaction mixtures. In other words, there is complementation between IntY342F and IntR212Q at the level of binding and complex formation.
Noncatalytic stimulation of Int cleavage through cooperative binding.
In contrast to the nicked attL intasomes described above, which are preferentially cut by Int on the nicked strand, the simpler COC" DNA with a nick in the overlap region serves as a suicide substrate for TS and BS cleavage by Int. Therefore, two different covalent Int-DNA complexes can be recovered from a TS-nicked COC" substrate, depending on the location of Int cleavage at the TS or BS. Whereas TS cleavage leads to a gapped product with Int covalently attached to the TS after dissociation of the oligomer, BS cleavage creates a half-site C" with Int attached to the BS as a consequence of the double-strand break (41). The two products can be separated by size on a SDS-PAGE gel (Fig. 6). In earlier experiments with a bispecific, TS-nicked suicide substrate, we observed that a wt HK022 Int, bound to an HK022-specific TS site, stimulated BS cleavage by wt λ Int, bound to its cognate BS site, but could not activate or complement the inactive λ Int Y342F and H308I mutants (41). The reverse cross-core stimulation of HK022 Int-mediated TS cleavage by a λ Int was also reported, which was independent of the presence of a Tyr342 or His308 in λ Int.
FIG. 6.

Stimulation of TS cleavage at the HK022-specific core-type site by λ C65-Ints. A hybrid suicide substrate with a TS nick 4 bp from the HK022-specific TS site (HK-C) yields more TS-covalent Int-DNA product by HK022 C65-Int (HK) in the presence of mutant λ C65-Ints, without appreciable cleavage at the λ-specific BS site (λ-C") that is readily cleaved by the wt λ equivalent (342Y).
The cross-core stimulation experiment on bispecific COC" substrates was extended here by using additional λ C65-Int mutants and the λ-HK022 hybrid C65-Int previously described (10, 36, 41, 50, 51). This hybrid differs from λ Int only in the six residues responsible for core-binding specificity and therefore is expected to interact with λ Int as well as with itself. Alone, the hybrid HK022 C65-Int produces a small amount of single TS-covalent complex (Fig. 6). The addition of catalytically inactive mutants of λ C65-Int leads to an increase of cleavage at this HK022-specific TS site. This stimulation is independent of the presence or absence of a catalytic Tyr342 in the complementing λ protein. Instead, what stands out in this experiment is the relative amount of stimulation seen with different inactive mutants, which appears to be approximately proportional to their respective core-binding affinities. Stimulation of TS cleavage by the weakest binder, C65-IntR212Q, is 1.8-fold, and that by the strongest binder, C65-IntY342F, is 4.6-fold. The R212K and K235A mutants are somewhat intermediate, both in their binding affinities and in their cleavage stimulation properties (2.5- and 3.5-fold, respectively). As had been described previously with wt HK022 Int, the hybrid HK022 C65-Int again did not donate its tyrosine to any of the inactive λ mutants for BS cleavage in trans (41).
The wt λ C65-Int, added to the reaction mixture with hybrid HK022 C65-Int, also increases the amount of TS cleavage product (approximately twofold). However, the main covalent complex seen with λ C65-Int is a double-strand cleavage product due to BS cleavage. This half-site product is seen with or without HK022 C65-Int (41). Because both the original TS-nicked substrate and the gapped TS-covalent product serve as substrates for catalysis at the BS site, the actual TS cleavage enhancement by a cross-core wt λ C65-Int is underestimated. The unlabeled, TS-cleaved half-att site is invisible and removed from the potential pool of HK022 C65-Int derived products.
Although each of the related wt C65-Ints (λ and HK022) gives only covalent cleavage products at their respective cognate sites, trans cleavage via a tyrosine recruited from a third, free Int out of solution cannot be excluded when wt proteins are used. However, the comparison of the relative cross-core stimulation by the mutant λ C65-Ints speaks against a solution Int as the source of a trans tyrosine. At the same limiting concentrations of wt HK022 C65-Int in all assays, none of the tyrosine-positive λ proteins stimulated TS cleavage as much as the Y342F mutant of λ C65-Int. Therefore, we conclude that the cross-core stimulation of hybrid HK022 C65-Int by different λ C65-Ints is noncatalytic, as it is independent of a catalytic Tyr342 in the complementing λ protein. It is reasonable to apply this observation of noncatalytic cross-core cooperation between λ and HK022 Ints to the complementation data between the Y342F and R212Q mutants. The binding and conformational defects of the latter may be overcome through noncatalytic Int-Int interactions with a neighboring high-affinity protomer of IntY342F, thereby unmasking the otherwise cryptic cleavage activity of R212Q.
DISCUSSION
The originally surprising finding that the λ Int-family recombinases are capable of both cis and trans mechanisms of DNA cleavage has been fully explained by the subtle folding differences seen in the cocrystal structures of Cre (a cis-cleaving enzyme) and Flp (a trans-cleaving enzyme) (9, 16). What remained unclear, due to seemingly conflicting results, is the question of whether λ Int has the option of using either of the two mechanisms depending on the condition of the experiment and whether one mechanism or the other is prevalent in the normal reaction (19, 41). Both models are compatible with the crystal structure of the λ Int catalytic domain because the 342F residue, substituted for the wt tyrosine nucleophile, is on a flexible loop close to the carboxy terminus (29). The experiments reported here addressed this question by using several biochemical assays and different suicide substrates to test the binding, exchangeability, cooperativity, and catalytic activity of wt and mutant Ints.
We have duplicated the original complementation between IntY342F and one of the Arg mutants involved in the activation of the scissile phosphate that was observed in cleavage assays with nicked attL intasomes (19). However, the trans-cleavage interpretation of these results relies on the assumption that the complementing mutant proteins are cleavage impaired but have similar binding properties and can be mutually exchanged by each other with the same preference for the TS-binding site. We argue that this assumption is not supported by the data presented here. IntY342F is a good example of a catalytic mutant that is not compromised in any of the steps preceding catalysis; it behaves the same as wt Int in all noncatalytic functions tested. This includes binding to core- and arm-type DNA, individually and in a sandwich complex bridging both types (Fig. 3 and 5), formation of Holliday junction tetramers, and formation of IHF-dependent, higher-order attL intasomes (12, 25, 35).
On the other hand, IntR212Q was found not only to be a poor binder to core type DNA but also to be defective in higher-order complex formation and in appropriate protein-protein interactions. This is reflected in the high concentrations of this mutant required to form dimer complexes on COC" (Fig. 3), in the lack of tetramer formation on artificial Holliday junctions (data not shown) and in the inability to form sandwich complexes between core- and arm-type DNA (Fig. 5). Given these aberrant properties, it is questionable whether IntR212Q can form the typical IHF-dependent attL-intasome structure by itself. In keeping with our in vitro results, IntR212Q also does not form attL complexes in the in vivo challenge phage assay (20). The high propensity for arm-core bridging interactions by IntY342F in an attL intasome is responsible for the observed preferential exchange of the unsatisfied protomer, singly bound at the TS site B, by wt Int (25). The same is not true for IntR212Q, which was displaced from nicked attL suicide substrates by wt Int with similar kinetics at the TS and BS cleavage sites (Fig. 4). The lack of behavioral equality between these two catalytic mutants (a basic assumption for the original trans-cleavage model) invites other interpretations of the attL complementation experiments.
Since the R212Q mutant is capable of cleaving an internucleotide 5" bridging phosphorothioate-substitution-containing, core-type DNA substrate on its own (Fig. 2), the defect of activation of the scissile phosphate is only partial. Therefore, IntR212Q may cleave an attL suicide substrate in cis, when aided by another Int with normal DNA binding and interaction properties. We propose that the TS-nicked-attL-IntR212Q preincubation mixture does not lead to the formation of a proper or stable attL intasome structure. Rather, IntR212Q may bind loosely to the core region of the attL fragment, shaped like a hairpin due to the IHF-induced bend at H" (Fig. 7). When IntY342F is added to this mixture, its high propensity for arm-core bridging is responsible for displacement of IntR212Q from the C" BS site, thereby forming an attL intasome. Although the IntR212Q is equally exchangeable at the TS B site and at the BS C" site, intasome stabilization evidently occurs only in one configuration, namely, when IntY342F forms a bridge between arm- and core-type DNA. In so doing, IntY342F also complements IntR212Q for correct binding at the TS B site, where it now can cleave the TS-nicked suicide substrate in cis. This scenario, based on the notion that IntY342F preferentially forms an intasome structure whereas IntR212Q does not, explains not only the unique order of addition required for positive complementation and TS cleavage of a TS-nicked attL by “activated” TS-bound IntR212Q in cis but also the previously puzzling negative results with a BS-nicked attL that is not a substrate for TS cleavage (see the introduction and Fig. 7). Whereas IntY342F easily displaces IntR212Q out of an attL hairpin, IntR212Q cannot displace IntY342F out of a preformed attL intasome when added as the second enzyme, regardless of the position of the nick.
FIG. 7.

Models for the formation of different higher-order attL structures with IntY342F or IntR212Q. On the left, the previously described model for attL intasome formation is shown after preincubation with IHF and IntY342F (25). The Int bound to the P"2 site (dotted circles), important for intasome formation, is not implicated in cleavage. In this model, the first inactive Int (white circles) bound at the TS site can be replaced by a second Int (dark circles), i.e., wt Int, at a higher rate than the BS-bound Int. This is reflected in a fast-versus-slow cleavage pattern of nicked attL substrates (column 1) (25). Because IntR212Q binds core-type DNA with a lower affinity than IntY342F, it is not expected to displace it at either site when added as the second enzyme (NO cut, column 2) (19). On the right, a different model is shown for an attL that is preincubated with IHF and IntR212Q. Because IntR212Q is severely defective in sandwich formation between arm-type and core-type DNA, it is unlikely that it can form a true intasome complex, although it might bind to all DNA-binding sites. Addition of wt Int quickly leads to cleavages at both the TS and the BS sites, because IntR212Q can easily be exchanged by an Int with a higher affinity (column 3) (Fig. 4). Similarly, IntY342F added as the second enzyme is expected to displace IntR212Q quickly at the TS site, the BS site, or both. Because of the high propensity for simultaneous binding of arm-type and core-type DNA by IntY342F and its high affinity for core-type DNA, displacement at the BS site is likely to be concomitant with the formation of an intramolecular C"-P"1 bridge by IntY342F. In one of these configurations, a sandwiched IntY342F may stimulate a remaining TS-bound IntR212Q to cleave the TS-nicked attL in cis (column 4) (19).
The aberrant phenotype of IntR212Q can be understood on the basis of the specific location of this mutation. Arg212 is located in the conserved box I region immediately preceding one of the helices forming the helix-turn-helix motif that has been likened to that of Cap protein (21, 47). In the Cre-DNA cocrystal, this motif involves α-helices H and J, which contribute to the site-specific recognition of the lox sites (16). Whereas the conserved Arg173 (equivalent to the λ Arg212) hydrogen-bonds to a nonbridging oxygen of the scissile phosphate, the adjacent Ile174 and Ala175 on α-helix H directly contact the opposite strand in the major groove, approximately 5 bp from the cleavage site (16). It is possible that an Arg-to-Gln mutation on this DNA-binding surface will negatively influence the binding properties of the adjacent residues. As a consequence, secondary long-range effects on the conformation of this mutant would not be unexpected. Interestingly, mutations at the equivalent box I Arg in XerC/D leads to altered interactions between the mutated proteins and their wt partners that influences an isomeric switch in catalysis within Holliday junction complexes (2). Another example of an Arg-to-Gln DNA contact mutation that significantly perturbs nearby structural elements has recently been described for the p53 core domain (49).
The behavior of the other two mutants, R311C and R311H that were complemented by Y342F (19), could be explained by a similar mechanism, namely by complementation at the level of DNA binding and complex formation. Indeed, in another Int family member, the integron IntI1, point mutations at either of the two conserved arginines abolish substrate recognition by these mutants (15). The equivalent of Arg311 in Cre (R292) also contacts a nonbridging oxygen of the scissile phosphate and is part of a DNA-binding surface, involving α-helix K and three residues in close proximity: S287, G288, and H289 (16). Most significantly, the equivalent Arg308 mutants of Flp form weaker protein-DNA complexes but can be stabilized on the DNA substrate through cooperativity with a tightly binding partner Flp (43).
We propose that the loss of function of IntR212Q is due at least in part to decreased or altered protein-DNA interactions and secondary allosteric consequences. Some of these may be overcome when the structural integrity of the catalytic pocket is restored through cross-core interactions with IntY342F. This idea is borne out by the observation that, at low concentrations, IntY342F and IntR212Q interact with each other to form two-Int complexes on COC" DNA, and sandwich complexes with core and arm DNA (Fig. 3A and 5B). The IntY342F molecule, with its normal DNA-binding properties, seems to coax IntR212Q to undergo a conformational change that restores appropriate protein-DNA and/or protein-protein interactions and concomitantly some catalytic function. The parallel between the extent of cleavage of a thio-substitution-containing att site DNA by IntR212Q/K and the degree of cross-core stimulation of HK022 Int by IntR212Q/K suggests that the integrity of the active catalytic pocket is important for the overall conformation of the protein.
Whereas allosteric effects on an active site by distant residues are a common phenomenon (11, 49), the R212Q mutation is an example of a change at the active site with allosteric consequences for functions that precede catalysis. These precatalytic defects preclude any inferences about the catalytic mechanism based on the complementation between IntR212Q and IntY342F. We have shown here that the R212Q mutant retains some catalytic activity and is deficient in its interactions with itself and with DNA. Int-Int interactions with a neighboring IntY342F, which strongly binds to core, may be sufficient for IntR212Q to overcome this defect, bind DNA, and cleave it in cis. Although our data do not provide additional evidence for cis cleavage, they suggest that the observed complementation is at the level of binding and complex formation and that IntY342F-dependent structural rearrangements enable the defective R212Q mutant to become catalytically active on its own. This resembles the stimulation between λ and HK022 Ints on bispecific substrates, which is at the level of cross-core interactions and is independent of an active tyrosine in the complementing protein.
Acknowledgments
We thank Margaret Kovach and Jeffrey J. Liu for the construction of plasmids carrying the R212Q and K235A full-length int genes, respectively. We thank Jeffrey Gardner and Richard Gumport for helpful comments reviewing the manuscript. We thank Marco Azaro for input pertaining to Holliday junctions, Gregg Gariepy and Tina Oliveira for technical assistance, and Joan Boyles for help with the final preparation of the manuscript.
This work was supported by NIH grants GM33928 and GM62723.
REFERENCES
- 1.Andrus, A., and R. G. Kuimelis. 2000. Overview of purification and analysis of synthetic nucleic acids, unit 10.3. In S. Beaucage, D. E. Bergstrom, G. D. Glick, and R. A. Jones (ed.), Current protocols in nucleic acid chemistry. John Wiley and Sons, New York, N.Y. [DOI] [PubMed]
- 2.Arciszewska, L. K., R. A. Baker, B. Hallet, and D. J. Sherratt. 2000. Coordinated control of XerC and XerD catalytic activities during Holliday junction resolution. J. Mol. Biol. 299:391-403. [DOI] [PubMed] [Google Scholar]
- 3.Azaro, M. A., and A. Landy. 2002. λ Int and the λ Int family, p. 118-148. In N. L. Craig, R. Craigie, M. Gellert, and A. Lambowitz (ed.), Mobile DNA II. ASM Press, Washington, D.C.
- 4.Blakely, G., G. May, R. McCulloch, L. K. Arciszewska, M. Burke, S. T. Lovett, and D. J. Sherratt. 1993. Two related recombinases are required for site-specific recombination at dif and cer in E. coli K12. Cell 75:351-361. [DOI] [PubMed] [Google Scholar]
- 5.Blakely, G. W., and D. J. Sherratt. 1996. cis and trans in site-specific recombination. Mol. Microbiol. 20:234-237. [DOI] [PubMed] [Google Scholar]
- 6.Burgin, A. B., Jr., B. N. Huizenga, and H. A. Nash. 1995. A novel suicide substrate for DNA topoisomerases and site-specific recombinases. Nucleic Acids Res. 23:2973-2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, J.-W., B. R. Evans, S.-H. Yang, H. Araki, Y. Oshima, and M. Jayaram. 1992. Functional analysis of box I mutations in yeast site-specific recombinases Flp and R: pairwise complementation with recombinase variants lacking the active-site tyrosine. Mol. Cell. Biol. 12:3757-3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, J.-W., J. Lee, and M. Jayaram. 1992. DNA cleavage in trans by the active site tyrosine during Flp recombination: switching protein partners before exchanging strands. Cell 69:647-658. [DOI] [PubMed] [Google Scholar]
- 9.Chen, Y., U. Narendra, L. E. Iype, M. M. Cox, and P. A. Rice. 2000. Crystal structure of a Flp recombinase-Holliday junction complex: assembly of an active oligomer by helix swapping. Mol. Cell 6:885-897. [PubMed] [Google Scholar]
- 10.Dorgai, L., E. Yagil, and R. A. Weisberg. 1995. Identifying determinants of recombination specificity: construction and characterization of mutant bacteriophage integrases. J. Mol. Biol. 252:178-188. [DOI] [PubMed] [Google Scholar]
- 11.Fersht, A. 1999. Structure and mechanisms in protein science, p. 289-323. W. H. Freeman and Co., New York, N.Y.
- 12.Franz, B., and A. Landy. 1990. Interactions between λ Int molecules bound to sites in the region of strand exchange are required for efficient Holliday junction resolution. J. Mol. Biol. 215:523-535. [DOI] [PubMed] [Google Scholar]
- 13.Gopaul, D. N., F. Guo, and G. D. Van Duyne. 1998. Structure of the Holliday junction intermediate in Cre-loxP site-specific recombination. EMBO J. 17:4175-4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grainge, I., and M. Jayaram. 1999. The integrase family of recombinase: organization and function of the active site. Mol. Microbiol. 33:449-456. [DOI] [PubMed] [Google Scholar]
- 15.Gravel, A., N. Messier, and P. H. Roy. 1998. Point mutations in the integron integrase IntI1 that affect recombination and/or substrate recognition. J. Bacteriol. 180:5437-5442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo, F., D. N. Gopaul, and G. D. Van Duyne. 1997. Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature 389:40-46. [DOI] [PubMed] [Google Scholar]
- 17.Guo, F., D. N. Gopaul, and G. D. Van Duyne. 1999. Asymmetric DNA-bending in the Cre-loxP site-specific recombination synapse. Proc. Natl. Acad. Sci. USA 96:7143-7148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hallet, B., L. K. Arciszewska, and D. J. Sherratt. 1999. Reciprocal control of catalysis by the tyrosine recombinases XerC and XerD: an enzymatic switch in site-specific recombination. Mol. Cell 4:949-959. [DOI] [PubMed] [Google Scholar]
- 19.Han, Y. W., R. I. Gumport, and J. F. Gardner. 1993. Complementation of bacteriophage lambda integrase mutants: evidence for an intersubunit active site. EMBO J. 12:4577-4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han, Y. W., R. I. Gumport, and J. F. Gardner. 1994. Mapping the functional domains of bacteriophage lambda integrase protein. J. Mol. Biol. 235:908-925. [DOI] [PubMed] [Google Scholar]
- 21.Hayes, F., and D. J. Sherratt. 1997. Recombinase binding specificity at the chromosome dimer resolution site dif of Escherichia coli. J. Mol. Biol. 266:525-537. [DOI] [PubMed] [Google Scholar]
- 22.Jayaram, M. 1994. Phosphoryl transfer in FLp recombination: a template for strand transfer mechanisms. Trends Biochem. Sci. 19:78-82. [DOI] [PubMed] [Google Scholar]
- 23.Jayaram, M. 1997. The cis-trans paradox of integrase. Science 276:49-51. [DOI] [PubMed] [Google Scholar]
- 24.Kim, S., and A. Landy. 1992. Lambda Int protein bridges between higher order complexes at two distant chromosomal loci attL and attR. Science 256:198-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim, S., L. Moitoso de Vargas, S. E. Nunes-Düby, and A. Landy. 1990. Mapping of a higher order protein-DNA complex: two kinds of long-range interactions in λ attL. Cell 63:773-781. [DOI] [PubMed] [Google Scholar]
- 26.Kitts, P. A., and H. A. Nash. 1988. Bacteriophage λ site-specific recombination proceeds with a defined order of strand-exchanges. J. Mol. Biol. 204:95-108. [DOI] [PubMed] [Google Scholar]
- 27.Kuimelis, R. G., and L. W. McLaughlin. 1995. Cleavage properties of an oligonucleotide containing a bridged internucleotide 5"-phosphorothioate RNA linkage. Nucleic Acids Res. 23:4753-4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuimelis, R. G., and L. W. McLaughlin. 1996. Ribozyme-mediated cleavage of a substrate analogue containing an internucleotide bridging 5"-phosphorothioate. Biochemistry 35:5308-5317. [DOI] [PubMed] [Google Scholar]
- 29.Kwon, H. J., R. S. Tirumalai, A. Landy, and T. Ellenberger. 1997. Flexibility in DNA recombination: structure of the λ integrase catalytic core. Science 276:126-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Landy, A. 1989. Dynamic, structural and regulatory aspects of lambda site-specific recombination. Annu. Rev. Biochem. 58:913-949. [DOI] [PubMed] [Google Scholar]
- 31.Landy, A. 1993. Mechanistic and structural complexity in the site-specific recombination pathways of Int and FLP. Curr. Biol. 3:699-707. [DOI] [PubMed] [Google Scholar]
- 32.Landy, A. 1999. Coming or going it's another pretty picture for the λ-Int family album. Proc. Natl. Acad. Sci. USA 96:7122-7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee, J., M. C. Serre, S.-H. Yang, I. Whang, H. Araki, Y. Oshima, and M. Jayaram. 1992. Functional analysis of Box II mutations in yeast site-specific recombinases FLP and R. J. Mol. Biol. 228:1091-1103. [DOI] [PubMed] [Google Scholar]
- 34.Mag, M., S. Luking, and J. W. Engels. 1992. Synthesis and selective cleavage of an oligodeoxynucleotide containing a bridged internucleotide 5"-phosphorothioate linkage. Nucleic Acids Res. 19:1437-1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moitoso de Vargas, L., S. Kim, and A. Landy. 1989. DNA looping generated by the DNA-bending protein IHF and the two domains of lambda integrase. Science 244:1457-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagaraja, R., and R. A. Weisberg. 1990. Specificity determinants in the attachment sites of bacteriophage HK022 and λ. J. Bacteriol. 172:6540-6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nash, H. A. 1996. Site-specific recombination: Integration, excision, resolution, and inversion of defined DNA segments, p. 2363-2376. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella, 2nd ed. ASM Press, Washington, D.C.
- 38.Nunes-Düby, S., R. S. Tirumalai, H. J. Kwon, T. Ellenberger, and A. Landy. 1998. Similarities and differences among 105 members of the Int family of site-specific recombinases. Nucleic Acids Res. 26:391-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nunes-Düby, S. E., L. Matsumoto, and A. Landy. 1987. Site-specific recombination intermediates trapped with suicide substrates. Cell 50:779-788. [DOI] [PubMed] [Google Scholar]
- 40.Nunes-Düby, S. E., L. Matsumoto, and A. Landy. 1989. Half-att site substrates reveal the homology independence and minimal protein requirements for productive synapsis in λ excisive recombination. Cell 59:197-206. [DOI] [PubMed] [Google Scholar]
- 41.Nunes-Düby, S. E., R. S. Tirumalai, L. Dorgai, R. Yagil, R. Weisberg, and A. Landy. 1994. λ integrase cleaves DNA in cis. EMBO J. 13:4421-4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pargellis, C. A., S. E. Nunes-Düby, L. Moitoso de Vargas, and A. Landy. 1988. Suicide recombination substrates yield covalent λ integrase-DNA complexes and lead to identification of the active site tyrosine. J. Biol. Chem. 263:7678-7685. [PubMed] [Google Scholar]
- 43.Parsons, R. L., B. R. Evans, L. Zheng, and M. Jayaram. 1990. Functional analysis of Arg-308 mutants of Flp recombinase. Possible role of Arg-308 in coupling substrate binding to catalysis. J. Biol. Chem. 265:4527-4533. [PubMed] [Google Scholar]
- 44.Richet, E., P. Abcarian, and H. A. Nash. 1988. Synapsis of attachment sites during lambda integrative recombination involves capture of a naked DNA by a protein-DNA complex. Cell 52:9-17. [DOI] [PubMed] [Google Scholar]
- 45.Sarkar, D., M. Radman-Livaja, and A. Landy. 2001. The small DNA binding domain of λ Int is a context-sensitive modulator of recombinase functions. EMBO J. 20:1203-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stark, W. M., and M. R. Boocock. 1995. Gatecrashers at the catalytic party. Trends Genet. 11:121-123. [DOI] [PubMed] [Google Scholar]
- 47.Subramanya, H. S., L. K. Arciszewska, R. A. Baker, L. E. Bird, D. J. Sherratt, and D. B. Wigley. 1997. Crystal structure of the site-specific recombinase, XerD. EMBO J. 16:5178-5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tirumalai, R. S., H. Kwon, E. Cardente, T. Ellenberger, and A. Landy. 1998. The recognition of core-type DNA sites by λ integrase. J. Mol. Biol. 279:513-527. [DOI] [PubMed] [Google Scholar]
- 49.Wong, K.-B., B. S. DeDecker, S. M. V. Freund, M. R. Proctor, M. Bycroft, and A. R. Fersht. 1999. Hot-spot mutants of p53 core domain evince characteristic local structural changes. Proc. Natl. Acad. Sci. USA 96:8438-8442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yagil, E., S. Dolev, J. Oberto, N. Kislev, N. Ramaiah, and R. A. Weisberg. 1989. Determinants of site-specific recombination in the lambdoid coliphage HK022: an evolutionary change in specificity. J. Mol. Biol. 207:695-717. [DOI] [PubMed] [Google Scholar]
- 51.Yagil, E., L. Dorgai, and R. Weisberg. 1995. Identifying determinants of recombination specificity: construction and characterization of chimeric bacteriophage integrases. J. Mol. Biol. 252:163-177. [DOI] [PubMed] [Google Scholar]